
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransamidation vitrimers enabled by neighbouring fluorine atom activation†
Dimitri
Berne
a,
Gwendal
Tanguy
a,
Sylvain
Caillol
 a,
Rinaldo
Poli
bc,
Vincent
Ladmiral
*a and
Eric
Leclerc
*a
a,
Rinaldo
Poli
bc,
Vincent
Ladmiral
*a and
Eric
Leclerc
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: eric.leclerc@enscm.fr; vincent.ladmiral@enscm.fr
bCNRS, LCC (Laboratoire de Chimie de Coordination), UPS, INP, Université de Toulouse, 31077 Toulouse, Cedex 4, France
cInstitut Universitaire de France, 75231 Paris, France
First published on 30th June 2023
Abstract
The activation of associative transamidation via neighbouring fluorine atom activation has been unveiled, quantified and applied to the construction of catalyst-free polyamide vitrimers. The inherent thermal and chemical stability of the amide functionality usually renders this linkage unsuitable for chemical exchange. The present study demonstrated that the positioning of a CF2 group α to the amide group strongly activates this functionality, effectively turning on the dynamic transamidation. This effect was initially studied through combined DFT and kinetic investigations on the transamidation of small model molecules, prior to its application to polyamide networks. A range of fluorinated transamidation vitrimers were easily and rapidly synthesized by the reaction of α,α-difluoroesters with amines. Different amide/free amine ratios were employed, leading to materials that feature a range of relaxation and mechanical properties. Hence, flow activation energies ranging from 88.5 to 126 kJ mol−1 were obtained, allowing the facile modulation of the vitrimers’ dynamic properties. Recycling tests of these additive-free transamidation vitrimers were performed at temperatures ranging from 150 to 200 °C and the mechanical and thermal properties of the reshaped materials were compared to those of the initial ones. Thanks to this approach, transamidation vitrimers with tuneable reshaping temperature, Young's modulus and glass transition temperature have become accessible, paving the way for further applications of these materials.
Introduction
Polyamides (PAs) feature high chemical resistance and excellent mechanical properties due to the high cohesive energy linked to the strong hydrogen bonds formed between amide functions of different chains.1 A wide range of monomers are available to prepare PAs, providing access to a large structural diversity.2,3 PAs have thus been used for a broad range of applications including automotive, clothing and electronics.4,5 Since the commercialization of nylon 6,6 in 1938, numerous other thermoplastic PAs have been developed.6–9 The synthesis of biobased thermoplastic PAs obtained from renewable resources such as plant oils enables the reduction of the environmental impact of the PA family.10–13 The synthesis of PA networks is generally performed in two steps. First, PA pre-polymers are prepared by polycondensation between a diacyl chloride and a diamine or by ring-opening polymerization. These conditions are considered “harsh” as they require either the synthesis of moisture-sensitive chloride compounds or high temperatures when using diacid synthons. In the second step, the prepolymers are cross-linked by thermal- or photo-curing of double or triple bonds.14–21 In any case, PA networks are often not crystalline and thus do not feature the remarkable properties of PA thermoplastics.17,22,23In the current context of global waste reduction, the concept of Covalent Adaptable Networks (CANs) has emerged as a new manufacturing process for thermosets and as a promising solution for the recycling of cross-linked polymers. CANs refer to polymer networks composed of covalent bonds capable of exchanging under a specific stimulus (often the elevation of temperature), enabling reshaping while still retaining the characteristic properties of thermosets such as their insolubility.24–26 Numerous exchange reactions27–38 have been developed since the pioneering work on disulfide exchange,39,40 allowing the preparation of a broad range of CANs and vitrimers. Nevertheless, there are only a few examples of CANs based on a PA matrix.23,41–43
Based on the previous research work, it appears of great interest to improve on the one hand the efficiency of PA synthesis and, on the other hand, endow crosslinked PAs with dynamic properties to allow their reprocessing. A few examples of PA networks showcasing CAN properties that rely on the use of other linkages such as disulfide41 or imine bonds42 as exchangeable functions have been reported. Using the amide bond itself as the dynamic linkage is difficult due to its thermal stability and chemical inertness. Indeed, even if an amide group may theoretically exchange by reaction with an amine, with a carboxylic acid, or with another amide (amide metathesis), the reported methods for such reactions on model molecules generally require long reaction times, high temperatures (above 250 °C) or highly air- and moisture-sensitive catalysts.44–47 Most of the developed catalysts to achieve fast transamidation at low temperatures are metal complexes (e.g. Fe(III) salts, Ni(cod)2, zirconocene dichloride, scandium triflate, AlCl3, Ti(NMe2)4, and lanthanides).48–57 Only a few reports have presented metal-free transamidation of amides with amines, but they all relied on the use of co-reactants or catalysts such as boric acid, hydroxylamine hydrochloride, selenium dioxide or potassium persulfate.58–61 Transamidation is however unavoidable during melt processing, resulting in the formation of segmented block copolyamides when two chemically different homopolyamides are processed as a blend.62,63 The main mechanism for this transamidation is thought to involve hydrolysis-recombination and is thus largely affected by moisture and acidity.64
Starting from these observations, the concept of neighbouring group participation (NGP)65,66 seems particularly suitable to promote transamidation. Indeed, NGP enables the activation of covalent bonds towards exchange reactions through the presence of specific functional groups in direct proximity to the targeted bonds.65,66 So far, NGP has mostly been used for the activation of transesterification. Hence, additional hydroxyl67,68 or carboxylic acid69,70 functions placed in epoxy networks accelerate the exchange process via carbonyl group activation by H-bonding. Moreover, phthalate compounds were used to activate transesterification and transthioesterification via the formation of cyclic intermediates.71–75 Finally, an increase of the transesterification rate in epoxy-acid networks induced by neighbouring fluorine atoms was recently demonstrated by our group.76,77 This ester function activation is due to the strong electron-withdrawing effect of the fluorine atoms, which dramatically increases the electrophilicity of the ester carbonyl function.78 It is important to underline that fluorine activation maintained the associative character of the exchange reaction, whereas the formation of cyclic intermediates observed with other neighbouring groups led to a dissociative mechanism.71,79
Following the development of the NGP concept in CANs, Du Prez et al. recently highlighted that amide bonds could exchange via the formation of an imide intermediate (Scheme 1a).23 This exchange was made possible by the proximity of the amide groups (six or fewer carbon atoms between the two amide groups), enabling cyclization with the release of a free exchangeable amine under a thermal stimulus. This specific exchange was later used by Sijbesma et al. in a polymer network produced via the addition of diamines to polyimide.43 In contrast to these dissociative CANs, transamidation vitrimers prepared from renewable resources were recently reported and presumed to involve an exchange between an amide and a free amine promoted by a boric acid catalyst.80 Mechanistic considerations were not discussed in this study but the exchange seemed to occur through an associative pathway, in opposition to the previously reported dissociative CANs proceeding through the imide formation.
 | ||
| Scheme 1 Transamidation by (a) the formation of an intermediate imide23 and (b) the formation of a tetrahedral intermediate. | ||
In the present study, our aim was to develop catalyst-free transamidation vitrimers by activating the associative amine exchange reaction with the use of neighbouring fluorine atoms (Scheme 1b). The fluorine effect on the amide formation and the transamidation exchange was first highlighted on model compounds and rationalized by DFT calculations. These preliminary studies prompted us to prepare additive-free transamidation vitrimers based on this fluorine activation. The use of different amide/free amine ratios indeed led to crosslinked materials that feature a range of relaxation and mechanical properties. Rheological experiments and reprocessability tests highlighted the impact of this ratio on the dynamic properties of the resulting vitrimers and provided insights into the key transamidation parameters.
Results and discussion
Model molecules
In order to compare their transamidation reactivity and quantify the reaction rates, difluorinated amides and their non-fluorinated counterparts were synthesized in two steps (Scheme 2). First, difluorinated (1, Fig. S1–S3†) and non-fluorinated (2, Fig. S4 and S5†) esters were obtained by the nucleophilic substitution of bromide in ethyl bromodifluoroacetate and bromoacetate by n-octanethiol (Scheme 2A), respectively. This reaction was more efficient for the non-fluorinated bromoacetate, providing a cleaner reaction mixture and a higher isolated yield. One rationalizing hypothesis is that, in terms of the classical SN2 mechanism, the C–Br bond of ethyl bromoacetate is more polarized than that of ethyl bromodifluoroacetate, due to the powerful electron-withdrawing effect of the two fluorine atoms. However, it is also well known that substitution reactions of fluoroalkyl bromides with thiolates may proceed through alternative mechanisms involving halophilic substitution or single electron-transfer steps.81 Therefore, care should be taken when comparing halogen substitution in the fluorinated and non-fluorinated series. In contrast, the subsequent solvent-free amidation with benzylamine (Scheme 2B) occurred at 25 °C in 10 min on the α,α-difluorinated ester 1 to yield amide 3 (conversion > 99%), whereas 48 h at 100 °C were required to achieve 91% conversion with the non-fluorinated analogue 2 to yield amide 4. The resulting benzylamides 3 and 4 were characterized by 1H, 19F and 13C-NMR (Fig. S6–S8† for 3 and Fig. S9, S10 for 4†). These observations clearly show that the two fluorine atoms dramatically increased the electrophilicity of the carbonyl group and enhanced the reaction rate.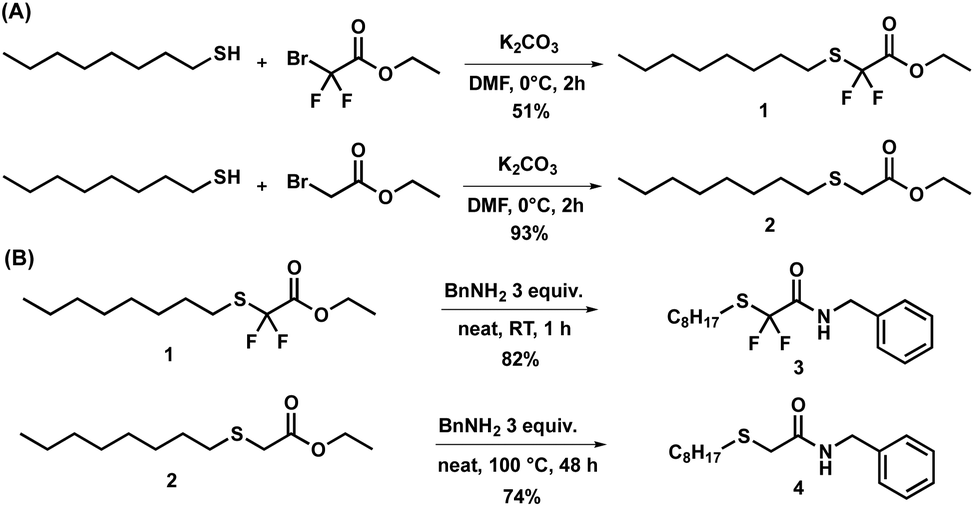 | ||
| Scheme 2 (A) Syntheses of fluorinated and non-fluorinated esters. (B) Syntheses of fluorinated and non-fluorinated benzylamides with their respective yields. | ||
In order to examine the effect of the fluorine atoms on transamidation, the fluorinated and non-fluorinated amides (3 and 4) were reacted with an excess (10 eq.) of octylamine (5) at 150 °C under neat conditions. After 72 h, 85% (determined by 1H-NMR) of 3 had been converted into fluorinated octylamide (6, Scheme 3A), while no conversion was observed for the non-fluorinated substrate 4 (Scheme 3). It should be noted that 6 was neither isolated nor purified.
 | ||
| Scheme 3 Transamidation of (A) 3 and (B) 4 with 5 under neat conditions at 150 °C. | ||
These preliminary results were corroborated by DFT calculations. The computations used the same methodology (functional, basis sets and various corrections, see details in the Experimental section) as our previously published study of the F substitution effect on the transesterification rate.78 The literature reports only a few DFT investigations of the transamidation reaction mechanism, mostly restricted to metal-catalyzed53,82,83 or organocatalyzed84–86 processes. It seems that only one report has addressed the non-catalyzed transamidation of a special amide (RCO-NMeOMe, a “Weinreb” amide), in comparison with the CO2-catalyzed process.86 As in this literature precedent, a second amine molecule was introduced in our calculations to serve as a proton shuttle, leading to a tetrahedral intermediate (Scheme 4). Our investigation has addressed three model systems, as shown in Scheme 4. The first two (A and B) serve to assess the effect of F substitution at the amide α-C atom on the activation barrier, while the comparison of systems A and C provides information on the additional effect of the sulfur atom. The alkyl chain was simplified to either a CH3S group for A and B or an ethyl group for C, while the attacking and leaving amines were simplified to methylamine, yielding a symmetric tetrahedral intermediate. The formation of the new amide product by amine elimination is thus the microscopic reverse of the initial amine addition.
 | ||
| Scheme 4 Non-catalyzed transamidation mechanism and model systems used in the present DFT investigation. | ||
The results of these calculations are summarized in Fig. 1. Starting with the separate reagents CH3ZCX2CONHCH3 and CH3NH2⋯NH2CH3 (system I at the zero Gibbs energy reference point), the process starts with the formation of an H-bonded adduct (II), in which the amide NH is a proton donor and one N atom of the CH3NH2⋯NH2CH3 dimer is the proton acceptor. This interaction is exoergic, with the enthalpic gain dominating over the entropic penalty. The stabilization is greater in the order A > B > C, correlating with the N–H bond polarity, which is regulated by the inductive effects of the X and Z groups (see optimized geometries in Fig. S19†). Along the path leading to the transition state, another local minimum (III) was identified, in which the interaction between the reactants involves an H-bond between the amine dimer as a proton donor and the amide carbonyl function as a proton acceptor. For each system, this intermediate was located and optimized using, as a guess geometry, the optimized TS structure modified along the imaginary frequency normal mode. The higher G of III relative to II is essentially of enthalpic nature (no significant entropic contribution), due to the weaker NH⋯O bond (lower acidity of the amine vs. amide NH bond; lower basicity of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vs. amine). The optimized geometries of these intermediates (Fig. S20†) also reveal an electrostatic interaction between the incoming amine N atom and the activated carbonyl C atom in IIIA and IIIC, whereas this N atom prefers to accept the amide NH proton in H-bonding in the structure of IIIB. The TS energies (optimized geometries in Fig. S21†) increase in the order A < C < B, highlighting the highly beneficial effect of the two F substituents (A ≪ B), but also a beneficial effect of the β-S atom (A < C). The overall barriers from the lowest G hydrogen-bonded intermediate II to the TS are in the order A (20.0 kcal mol−1) < C (21.0 kcal mol−1) < B (26.4 kcal mol−1). Finally, the tetrahedral intermediates (geometries shown in Fig. S22†) follow the same relative G trend as the transition states. The Gibbs energies of all molecules were also recalculated at 403 K (150 °C), yielding greater barriers because of the contribution of a negative activation entropy. The major entropic penalty is in the formation of the reactant H-bonded adduct, which is endoergic relative to the separate reagents at 150 °C. The activation barriers remain in the same relative order (A, 23.6 < C, 26.6 < B, 33.2 kcal mol−1), see profiles in Fig. S23.† The Cartesian coordinates of all optimized molecules are available in Table S1.†
O vs. amine). The optimized geometries of these intermediates (Fig. S20†) also reveal an electrostatic interaction between the incoming amine N atom and the activated carbonyl C atom in IIIA and IIIC, whereas this N atom prefers to accept the amide NH proton in H-bonding in the structure of IIIB. The TS energies (optimized geometries in Fig. S21†) increase in the order A < C < B, highlighting the highly beneficial effect of the two F substituents (A ≪ B), but also a beneficial effect of the β-S atom (A < C). The overall barriers from the lowest G hydrogen-bonded intermediate II to the TS are in the order A (20.0 kcal mol−1) < C (21.0 kcal mol−1) < B (26.4 kcal mol−1). Finally, the tetrahedral intermediates (geometries shown in Fig. S22†) follow the same relative G trend as the transition states. The Gibbs energies of all molecules were also recalculated at 403 K (150 °C), yielding greater barriers because of the contribution of a negative activation entropy. The major entropic penalty is in the formation of the reactant H-bonded adduct, which is endoergic relative to the separate reagents at 150 °C. The activation barriers remain in the same relative order (A, 23.6 < C, 26.6 < B, 33.2 kcal mol−1), see profiles in Fig. S23.† The Cartesian coordinates of all optimized molecules are available in Table S1.†
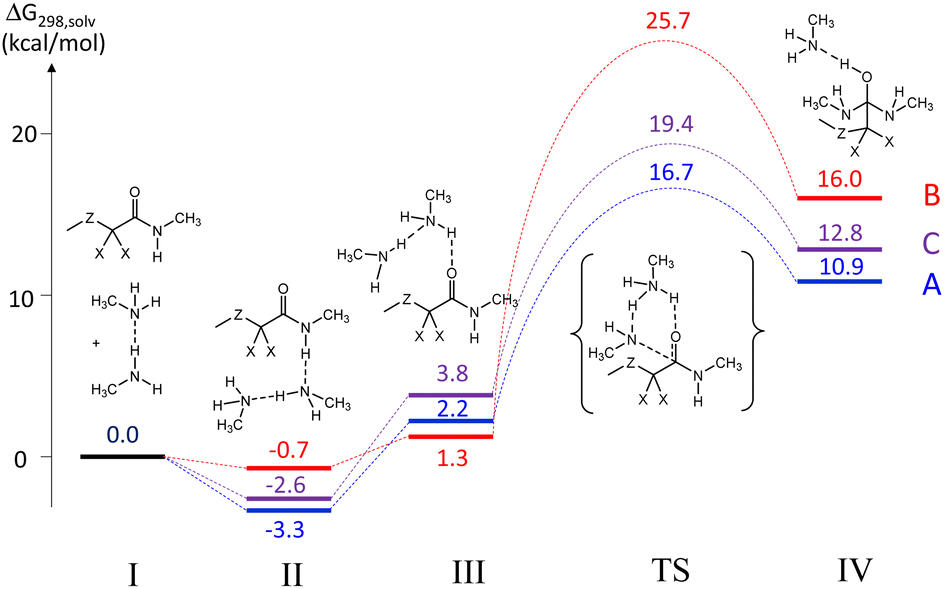 | ||
| Fig. 1 Gibbs energy profile (T = 298 K) for the transamidation process of the model molecules defined in Scheme 4. | ||
In addition to the DFT calculations, a kinetic study was carried out to determine the activation enthalpy and entropy of the transamidation of 3 using dodecylamine (8) (Scheme 5). The reaction was monitored by GC-MS, which allowed the accurate measurement of the product concentrations, especially at low conversions. Dodecylamine 8 was chosen as the competing amine over octylamine 5, used for the preliminary tests, as the resulting amide 9 was well separated from 3 by GC-MS, whereas 3 and 6 had similar retention times. The isolated fluorinated amide based on dodecylamine (9) was characterized by 1H, 19F and 13C-NMR (Fig. S11–S13†).
 | ||
| Scheme 5 Transamidation of 3 with 8 in mesitylene at temperatures ranging from 110 to 140 °C, x varied from 10 to 20. | ||
The kinetic investigation started with the determination of the rate law for this transamidation reaction, which was anticipated to have a positive reaction order for both reagents, i.e. the amide (first-order) and the competing amine (first-order or more). Mesitylene (MST, bp = 165 °C) was used as the solvent due to its high boiling point. Using different [8]0/[3]0 ratios (10, 15 and 20) at 130 °C, all kinetic profiles were in agreement with a pseudo-first-order rate law in amide, exhibiting very good linear fits when plotting ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ([8]0/[8]0 − [9]) = f(t) (Fig. S24†). The rate constants (kobs) deduced from the three reactions showed a good linear dependence vs. [8], as expected for a first-order reaction in amine (Table S2 and Fig. S25†). Hence, these first kinetic investigations confirmed the expected rate law for an associative exchange mechanism. Three additional kinetic measurements were carried out at 110, 120 and 140 °C using an [8]0/[3]0 ratio of 10. The k values obtained for these reactions are reported in Table S3.† These experiments allowed the determination of the experimental activation parameters of the transamidation reaction through the Eyring–Polanyi relationship (Fig. 2). A good linear fit was obtained and the following activation parameters were extracted: ΔH‡ = 7.78 ± 0.63 kcal mol−1 and ΔS‡ = −65.7 ± 1.6 cal mol−1 K−1 (Fig. 2B). The negative activation entropy is in agreement with the associative character of the reaction (ordered transition state).
([8]0/[8]0 − [9]) = f(t) (Fig. S24†). The rate constants (kobs) deduced from the three reactions showed a good linear dependence vs. [8], as expected for a first-order reaction in amine (Table S2 and Fig. S25†). Hence, these first kinetic investigations confirmed the expected rate law for an associative exchange mechanism. Three additional kinetic measurements were carried out at 110, 120 and 140 °C using an [8]0/[3]0 ratio of 10. The k values obtained for these reactions are reported in Table S3.† These experiments allowed the determination of the experimental activation parameters of the transamidation reaction through the Eyring–Polanyi relationship (Fig. 2). A good linear fit was obtained and the following activation parameters were extracted: ΔH‡ = 7.78 ± 0.63 kcal mol−1 and ΔS‡ = −65.7 ± 1.6 cal mol−1 K−1 (Fig. 2B). The negative activation entropy is in agreement with the associative character of the reaction (ordered transition state).
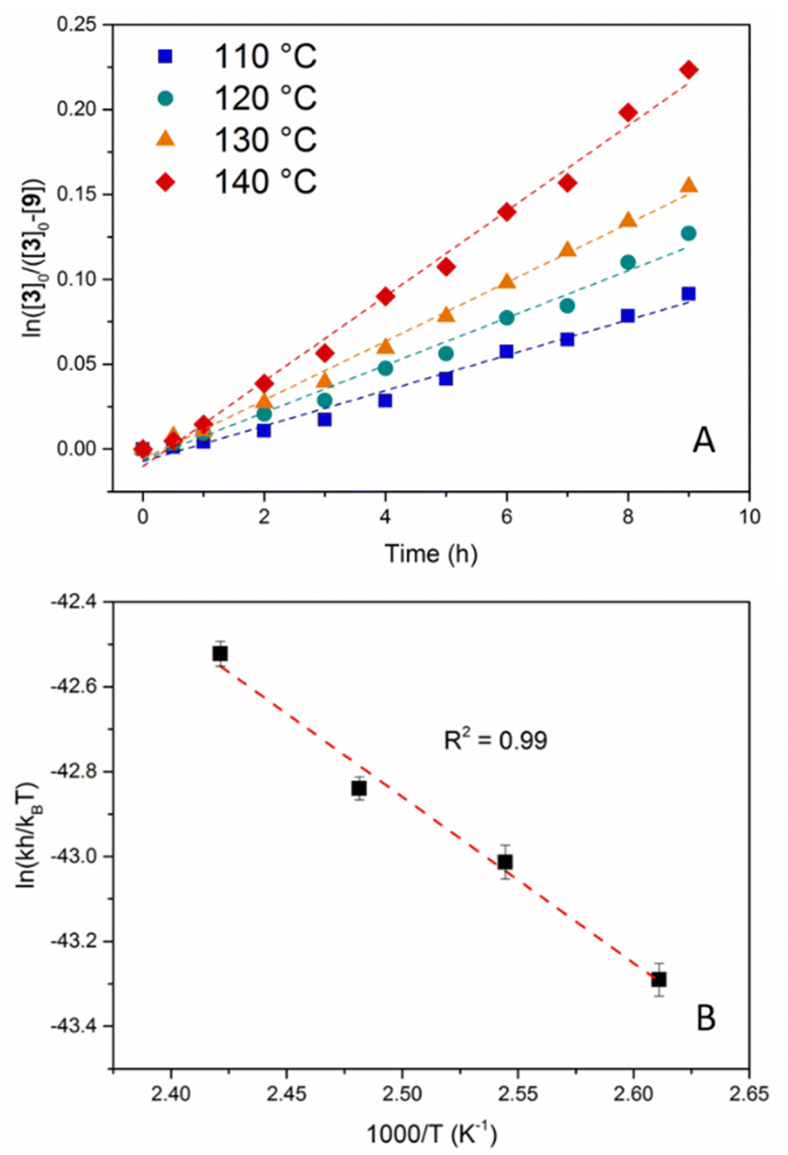 | ||
| Fig. 2 (A) Monitoring of the transamidation of 3 with 8 in MST at 110, 120, 130 and 140 °C. (B) Eyring plot of the rate constants in the 110–140 °C range. | ||
Polyamide network synthesis
Based on the promising results obtained with model molecules, dynamic fluorinated polyamide (FPA) networks constituted of α,α-difluoroamide exchangeable groups were synthesized. A new bis-(α,α-difluoro)ester (10) and its non-fluorinated analogue (11) were synthesized by substitution of, respectively, ethyl bromodifluoroacetate and bromoacetate with 1,6-hexanedithiol in DMF in the presence of K2CO3 (Scheme S1†). The desired products 10 and 11 were purified by column chromatography in order to remove the monoaddition product and the unreacted dithiol and their structures were confirmed by 1H, 19F and 13C-NMR (Fig. S14–S16† for 10 and Fig. S17, S18† for 11). It is important to note that these monomers were specifically designed so that the formation of an imide from the two resulting amides is strongly disfavoured as this would involve the formation of a 13-membered ring.Previous studies demonstrated that the degrees of cross-linking and of reactive functions (represented by the free amine group in FPA) have an influence on the relaxation time and the activation energy of the associated CANs.66 Hence, to highlight the influence of the fraction of free amine in the network, a series of FPAs with different degrees of free amine functionalities were synthesized. It should be noted that a 3D structure was maintained, even at the highest free amine/amide ratio. 10 was thus mixed with tris(2-aminoethyl)amine (TREN) as the cross-linker at different ratios (10/TREN = 3/2, 2.75/2, 2.5/2 and 2.25/2) leading to materials containing 0, 8, 17 and 25 mol% of free amine groups, respectively, and named FPA-0, FPA-8, FPA-17 and FPA-25 accordingly. The non-fluorinated equivalent of FPA-25 (named NFPA-25) was also synthesized by the reaction of 11 and TREN in a 2.25/2 ratio (Fig. 3).
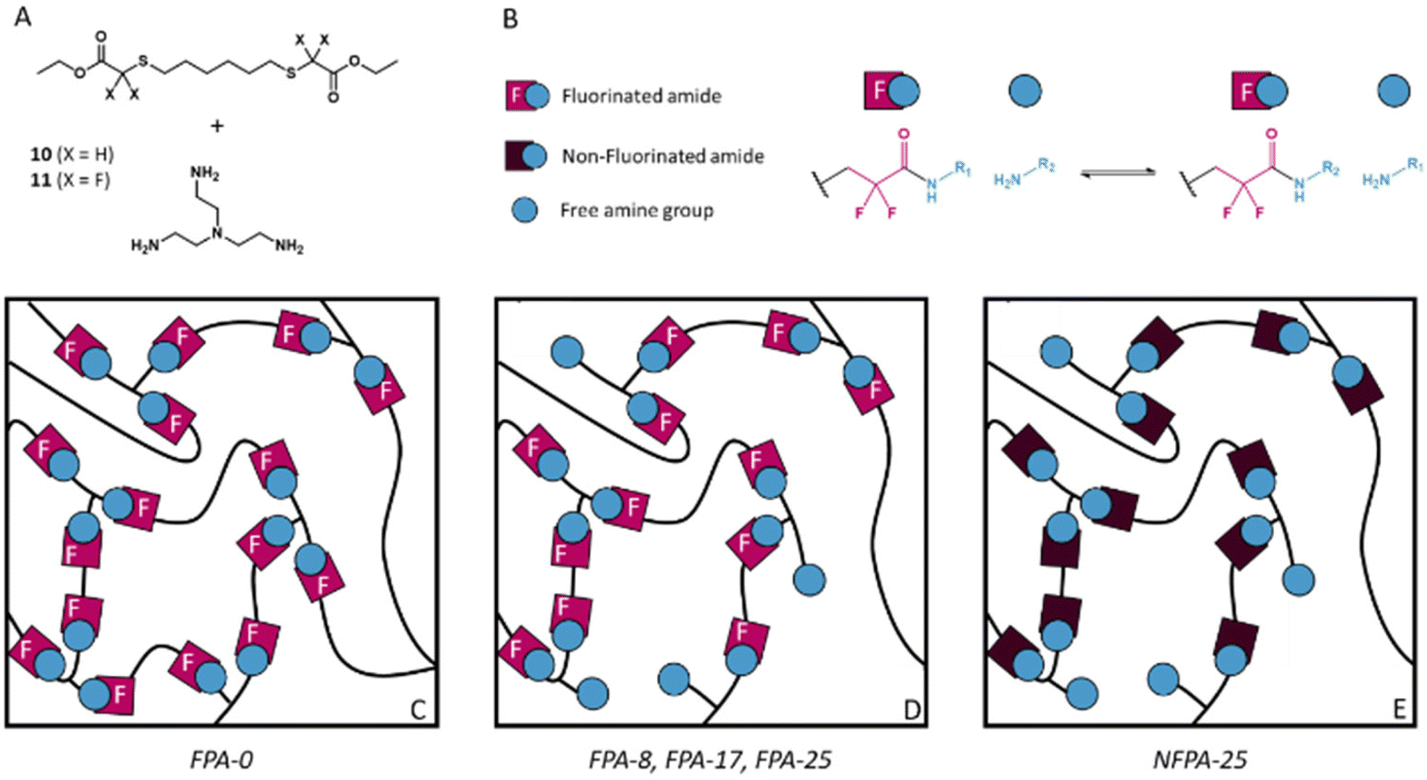 | ||
| Fig. 3 (A) Di-esters and tri-amine used for the synthesis of the PA networks. Schematic representation of (B) the associative transamidation, (C) the fluorinated PA network without free amine groups (FPA-0), (D) the fluorinated PA networks containing free amine groups (FPA-7, FPA-8 and FPA-25), and (E) the non-fluorinated PA network containing free amine groups (NFPA-25). | ||
As already observed in the synthesis of the model molecules, the fluorine atoms clearly accelerated the amidation reaction leading to the network formation. In the case of the FPA materials, gelation readily occurred 2 min after mixing the two monomers and a fully cured material was obtained after only 2 hours at 100 °C. In contrast, 30 hours at 100 °C were required to complete the synthesis of NFPA-25. Subsequently, preliminary reshaping tests using a hot press were carried out on all materials to quickly assess the influence of the free amine group fraction and the fluorine activation. The temperature required for reshaping decreased as the percentage of free amine groups increased in the FPA series (Table 1). The transamidation exchange rate thus seems to correlate with the percentage of free amine groups. FPA-0 failed to (re)shape even after a thermal treatment at 220 °C under a 3-ton load for two hours. This suggests that the mechanism of transamidation involved in these FPA materials is associative (as it requires free amine groups). Finally, the fluorine effect on transamidation was evidenced by the required temperature for shaping NFPA-25 (200 °C) which was 50 °C above that of FPA-25.
| Material | GCa (%) |
SIa (%) |
T
d
5%b (°C) |
T
gc (°C) |
T
αd (°C) |
E′glassye (GPa) |
E′rubberyf (MPa) |
T
(re)shapeg (°C) |
|---|---|---|---|---|---|---|---|---|
| a Gel content and swelling indexes measured after 24 h immersion in THF. b Temperature of degradation corresponding to 5% weight loss determined by TGA. c Glass transition temperature determined by DSC analysis. d α transition temperature determined by DMA at E′′ maxima. e Determined at a Tα of −50 °C. f Determined at a Tα of +50 °C. g (Re)shaping was carried out at T(re)shape under 3 tons in 1 h. h Shaping failed at 220 °C. | ||||||||
| FPA-0 | 93 ± 2 | 62 ± 5 | 301 | — | — | — | — | —h |
| FPA-8 | 95 ± 1 | 60 ± 7 | 298 | 28 | 15 | 2.1 | 1.6 | 200 |
| FPA-17 | 99 ± 1 | 69 ± 2 | 287 | 20 | 7 | 1.3 | 1.3 | 180 |
| FPA-25 | 96 ± 1 | 75 ± 7 | 284 | 12 | 3 | 1.2 | 0.8 | 150 |
| NFPA-25 | 97 ± 2 | 105 ± 2 | 292 | 8 | — | — | — | 200 |
The formation of a cross-linked network for all the PA materials was confirmed by several analyses (Table 1). First, all the synthesized materials demonstrated a high gel content (GC) above 93%. The percentage of free amine groups did not considerably impact the solubility properties of the PA networks, which all showed similar gel contents and swelling indexes (SI) in THF. ATR-FTIR analyses also confirmed the high conversion of the reactive functions as the initial absorption band of the ester bond of 10 at 1681 cm−1 was completely replaced by the amide peak at 1760 cm−1 in the FPA networks (Fig. S26†). These analyses did not enable differentiating the N–H bands of the amine from those of the amide, as their characteristic wavenumbers are too close. Finally, no residual exothermic peak was observed during the DSC analyses, confirming the complete curing of the materials (Fig. S27†). The glass transition temperature (Tg) determined from these DSC analyses increased as the percentage of free amine groups decreased, in agreement with the corresponding lower cross-linking density.
All the PA networks demonstrated high thermal stabilities with Td5% (temperatures corresponding to 5% degradation) above 280 °C (Fig. S28† and Table 1). Higher free amine fractions slightly decreased the thermal resistances as this chemical function is known to undergo degradation reactions at high temperatures. The NFPA-25 network also demonstrated high cross-linking and high thermal stability. Its Tg was slightly lower than that of its fluorinated analogue, which may be rationalized by the higher steric hindrance induced by the presence of the fluorine atoms.87
FPA-8, FPA-17 and FPA-25 were evaluated by dynamic mechanical analyses (Fig. 4). As expected, the Tα measured by DMA followed the same trend as the Tg measured by DSC: as the free amine group content increases, the chain mobility increases and the glassy/rubbery transition temperature decreases. Moreover, the storage modulus value slightly decreases with an increase of the free amine group content, as expected.
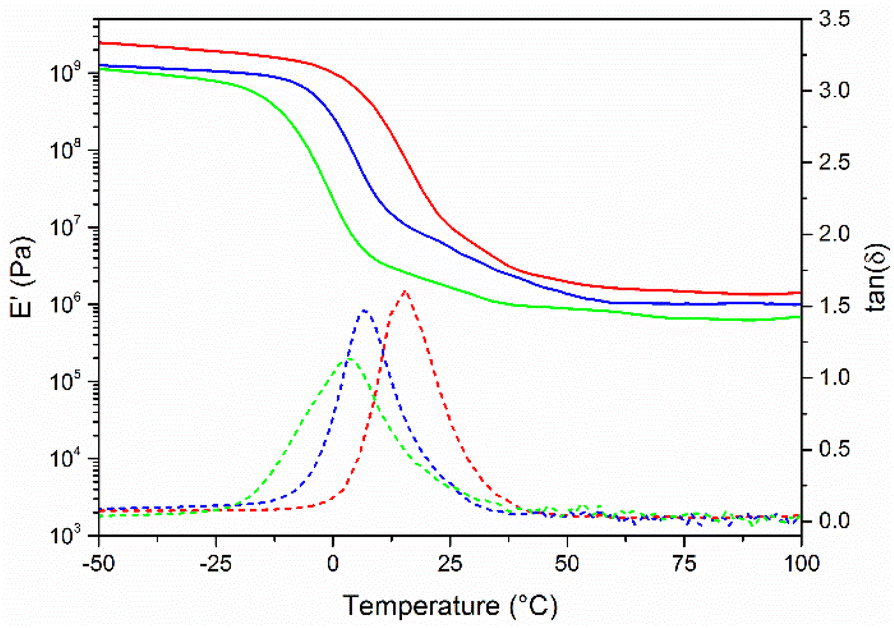 | ||
| Fig. 4 Storage modulus (plain lines) and tan(δ) (dashed lines) of FPA-8 (red), FPA-17 (blue) and FPA-25 (green). | ||
Dynamic properties
The dynamic nature of the FPA networks and the influence of the free amine group content were evaluated via frequency sweeps, relaxation tests and creep/recovery experiments. Frequency sweep analyses were performed on the FPAs at temperatures ranging between 120 °C and 180 °C (Fig. S29–S31†) and showed that the storage moduli did not vary significantly with temperature. This observation is an additional indication that an associative transamidation exchange takes place in these FPA networks.79 Indeed, for dissociative CANs, a decrease of the storage modulus with increasing T caused by a loss of connectivity is generally observed.79,88 Moreover, the increase of the loss modulus observed at low frequencies and high temperatures is related to the stress relaxation of the material.89 This increase, particularly visible for FPA-17 and FPA-25, indicates that these networks likely relax stress faster than FPA-8.Stress-relaxation experiments were then performed on the FPA-X networks in the [140–180 °C] temperature window for X = 17 or 25 and in the [160–200 °C] temperature window for X = 8 (Fig. 5). The initial relaxation modulus did not decrease with increasing temperature (Fig. S32–S34†), confirming the frequency sweep observations that the exchange reaction does not induce a loss of connectivity and thus likely does not proceed via a dissociative mechanism. The normalized stress relaxation curves were fitted with a stretched exponential, G(t) = G0exp(−(t/τ))β (Fig. 5). These stress relaxation experiments show that the relaxation time of the FPA networks at any given temperature is shortened by an increase of the free amine group content. This result can be rationalized as follows: as previously demonstrated for transesterification vitrimers,90 the stress relaxation time decreases with the increasing content of hydroxyl groups in the network. Hence, the transamidation rate increased from FPA-8 to FPA-25 as the free amine group content in the network increased. In addition, the material cross-link density plays a role in the relative mobility, diffusion and availability of the exchangeable functions within the polymer network. Several studies (dealing with dioxaborolane chemistry,91 transesterification92,93 or vinylogous urethane exchange94) have demonstrated that an increase of the cross-link density is associated with a slower stress relaxation. Accordingly, FPA-25 relaxes stress faster than FPA-17, which in turn relaxes stress faster than FPA-8.
 | ||
| Fig. 5 Normalized stress relaxation for (A) FPA-25, (B) FPA-17 and (C) FPA-8. (D) Arrhenius plot of FPA-X networks (X = 8, 17 or 25). | ||
The Arrhenius plots of the FPA relaxation times (Fig. 5D) are also consistent with an associative exchange. Indeed, even though a few dissociative CANs were shown to follow an Arrhenius behaviour within a certain T range, this property is always observed in associative CANs. The flow activation energies of 88.5, 105 and 126 kJ mol−1 obtained for FPA-25, FPA-17 and FPA-8 (Fig. 5D), respectively, can be correlated to the increasing cross-link density, as described in previous studies.91–93 It is also interesting to note that the activation energy is inversely proportional to the percentage of free amine groups, even though this trend was observed within a limited range of material compositions (Fig. S35†). In conclusion, an increase of free amine group content in the presented FPA networks translates into a faster relaxation and a lower sensitivity of the material viscosity to temperature.
The preliminary kinetics of model molecules and the DFT study already demonstrated that the transamidation reaction rate is considerably accelerated by the neighbouring fluorine atoms. This effect was further demonstrated by the comparison of the relaxation curves at 180 °C of NFPA-25 and FPA-25 (Fig. S36†). Both systems were able to relax stress at this temperature, but the relaxation time of FPA-25 was three times shorter than that of NFPA-25 (586 vs. 1703 s). The relatively fast relaxation of the non-fluorinated system is somewhat surprising. Indeed, according to previous studies,43,88 only materials in which the formation of an imide was possible allowed transamidation via dissociative exchange and were therefore able to relax stress. Here, the formation of imide is highly unlikely, compared to the associative exchange with a free amine, due to the number of carbon atoms between the amide and amine functions. In addition, FTIR measurements at the reprocessing temperature failed to detect the imide characteristic bands in any of the tested materials. This further supports the hypothesis of the transamidation reaction occurring through an associative mechanism strongly accelerated by the high electron-withdrawing effect of the fluorine atoms, which increase the electrophilicity of the amide carbonyl functions. The transamidation reaction occurring in NFPA-25, though not activated by fluorine, is probably slightly favoured by the presence of the sulfur atom on the α-carbon of the amide. This moderate influence was highlighted in the DFT study. Another possible way to rationalize this unexpected result is the high concentration of tertiary amines (from the TREN monomer) in these polyamide networks. Indeed, tertiary amines have been shown to promote transesterification in epoxy-acid vitrimers and may have the same effect on transamidation.95
In order to assess the flow properties of the FPA systems, creep/recovery measurements were also performed at 50, 100 and 150 °C (Fig. 6). At 50 °C, none of the FPA networks demonstrated any critical deformation, allowing their use at service temperatures under 50 °C. The higher temperature creep results were in good agreement with the stress-relaxation properties: FPA-25 demonstrated higher creep at 150 °C than FPA-17 or FPA-8. The 100 °C data show that FPA-8 and FPA-17 have almost no dynamic behaviour (no deformation), suggesting that these materials could be used up to 100 °C without significant flow.
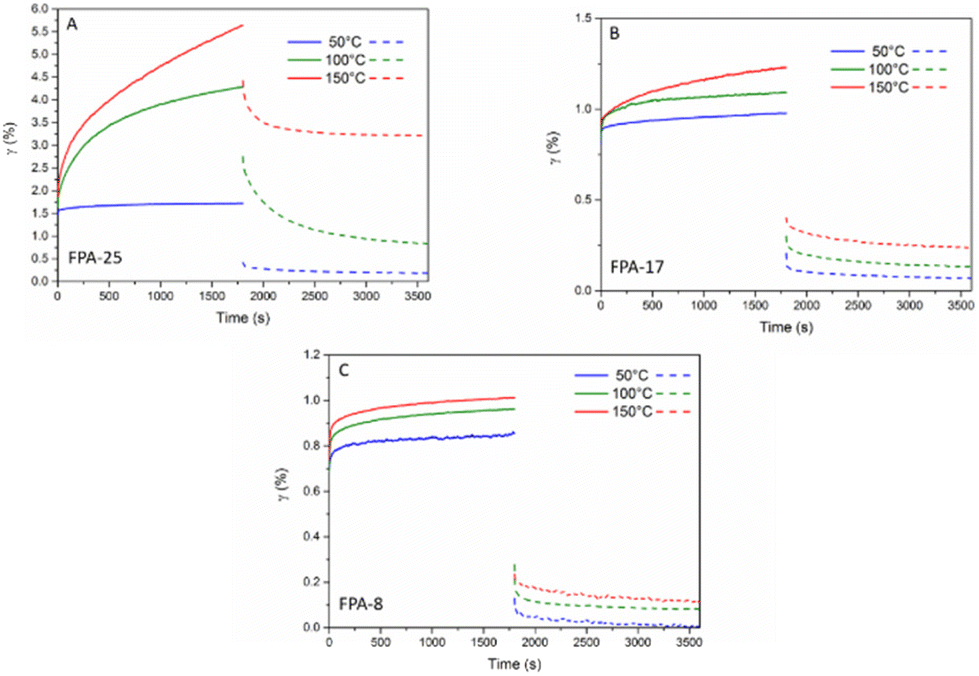 | ||
| Fig. 6 Creep and recovery behaviour at 50, 100 and 150 °C for an applied stress of 3 kPa for (A) FPA-25, (B) FPA-17 and (C) FPA-8. | ||
Recyclability
One of the main advantages of vitrimers compared to thermosets is their capability to be reprocessed/reshaped. To test this property, the FPA networks were cut into small pieces (ca. 1 mm2) and placed in a hot press for 1 h under a 3-ton load. The temperature of reshaping was first optimized for each sample by visual observation, before assessing the reprocessing quality by TGA, DSC, FTIR, insolubility test, frequency sweep and tensile analyses. Slower-relaxing materials require higher reprocessing temperatures; hence, FPA-25, FPA-17 and FPA-8 were respectively reprocessed at 150, 180 and 200 °C.The properties of the reprocessed materials were evaluated after each processing cycle up to three cycles for FPA-17 and FPA-25. For FPA-25, no significant difference was observed between the DSC traces of the initial and reshaped materials (Fig. S37†), whereas a slight increase of Tg was observed for FPA-17 and FPA-8, indicating an increase of cross-link density, probably related to slight material degradation (Fig. S38 and S39†). The retention of high thermal stability by the reprocessed samples was also indicated by thermogravimetric analyses for FPA-25 and FPA-17, whereas a slight thermal stability decrease was observed for FPA-8 (Fig. S40–S42†). The FTIR spectra were nearly identical before and after reshaping for all CANs (Fig. S43–S45†), further indicating that secondary reactions did not significantly occur during the hot press treatment and that the chemical integrity of the networks was maintained. Despite this slight material alteration with temperature, reuse is still possible, depending on the targeted secondary application.
Frequency sweep measurements of FPA-25 and FPA-17 at 180 °C (Fig. S46 and S47†) also confirmed that the cross-link density was not significantly impacted by the reprocessing step, as the storage modulus did not increase significantly for the recycled materials compared to that of the initial one. Moreover, a loss modulus increase at low frequencies could still be observed at 180 °C, demonstrating that the vitrimers’ dynamic behaviour was not lost in the reshaped materials.
The reshaping efficiency was also evaluated by performing tensile tests on the initial and reshaped samples. The Young moduli and stress/strain at break are reported in Table 2. The Young moduli of the initial FPAs are comparable to those obtained by DMA. Focusing on the FPA-25 results, no significant change of the characteristic properties was observed over three reshaping cycles. For FPA-17, on the other hand, the Young modulus and stress and strain at break were slightly decreased after one reshaping cycle. The evolution of these properties with the number of reshaping cycles was expected. Indeed, even though it did not evolve significantly, the glass transition temperature of FPA-17 was raised above the analysis temperature (25 °C) by the reshaping process. Hence, after 2 or 3 cycles, FPA-17 is in a state closer to the glassy state rather than the rubbery state of the initial and once-reshaped materials. The same phenomenon justifies the difference between the pristine and once-reshaped FPA-8. Nevertheless, these results are consistent with the other analyses and confirm an efficient reshaping protocol for each material.
| Material |
T
d5%a (°C) |
T
gb (°C) |
SIc (%) |
GCc (%) |
Young's modulusd (MPa) | Stress at breakd (MPa) | Strain at breakd (%) |
|---|---|---|---|---|---|---|---|
| a Temperature of degradation corresponding to 5% weight loss determined by TGA. b Glass transition temperature determined by DSC analysis. c Gel content and swelling indexes measured after 24 h immersion in THF. d Tensile tests were performed on dog bones of 1 mm thickness with a tensile speed of 0.1 mm s−1. | |||||||
| FPA-25 | |||||||
| Initial | 284 | 12 | 75 ± 7 | 96 ± 1 | 0.38 ± 0.03 | 0.38 ± 0.05 | 218 ± 21 |
| Reshape 1 | 283 | 13 | 74 ± 1 | 98 ± 2 | 0.41 ± 0.05 | 0.40 ± 0.04 | 202 ± 30 |
| Reshape 2 | 286 | 18 | 64 ± 7 | 94 ± 1 | 0.33 ± 0.03 | 0.24 ± 0.05 | 158 ± 29 |
| Reshape 3 | 284 | 17 | 74 ± 1 | 99 ± 1 | 0.40 ± 0.06 | 0.38 ± 0.01 | 219 ± 25 |
| FPA-17 | |||||||
| Initial | 287 | 20 | 69 ± 2 | 99 ± 1 | 2.19 ± 0.14 | 1.34 ± 0.41 | 86.7 ± 23.1 |
| Reshape 1 | 291 | 25 | 65 ± 1 | 99 ± 1 | 1.71 ± 0.16 | 0.78 ± 1.40 | 56.6 ± 17.2 |
| Reshape 2 | 291 | 30 | 61 ± 2 | 99 ± 2 | 7.10 ± 3.12 | 4.76 ± 2.22 | 10.5 ± 2.3 |
| Reshape 3 | 288 | 33 | 61 ± 1 | 99 ± 2 | 216 ± 25 | 8.26 ± 3.01 | 6.8 ± 1.9 |
| FPA-8 | |||||||
| Initial | 298 | 28 | 60 ± 7 | 95 ± 1 | 3.37 ± 0.63 | 1.68 ± 0.85 | 55.1 ± 12.7 |
| Reshape 1 | 271 | 33 | 56 ± 1 | 97 ± 1 | 106 ± 24 | 7.78 ± 4.03 | 13.4 ± 5.1 |
Conclusion
In this study, additive-free transamidation vitrimers could be developed thanks to neighbouring fluorine atom activation. The potential of α,α-difluorination on the transamidation rate was highlighted by combined DFT and kinetic investigations on model molecules. The use of α,α-difluoroesters enables the rapid and facile synthesis of FPA bulk materials. The modulation of the free amine fraction appears to be a facile approach to control the dynamic behaviour of these FPA networks, as the reprocessing temperature of these materials could be tuned from 150 °C to 200 °C. The mechanical and thermal properties of these materials are also dependent on the free amine content; hence, the Young modulus ranged from 0.38 to 3.4 MPa and the glass transition temperature from 12 to 28 °C. Considering the large amount of available amine-functionalised molecules, the synthetic simplicity and the structural flexibility, these FPAs may find many practical applications, such as composites or reversible adhesives.The polyamide vitrimers presented in this study did not demonstrate crystallinity as classical polyamides do. Hence, these materials should rather be considered as an extension of dynamic materials to polyamide networks and transamidation exchange reactions. Further work may focus on combining the crystallinity of polyamides with such dynamic properties in order to obtain ultra-resistant recyclable materials.
Materials
1,6-Hexanedithiol (purity > 96%), 1-octanethiol (98.5%), ethyl bromodifluoroacetate (98%), 2-bromoethyl acetate (97%), tris(2-aminoethyl)amine (96%), benzylamine (99%), dodecylamine (99%), octylamine (98%) and mesitylene (MST, 98%) were purchased from Sigma-Aldrich (Darmstadt, Germany). Pentane, ethyl acetate (EtAc), DMF and CDCl3 (99.5% D) were provided by Eurisotop (Saint-Aubin, France). All materials were used as received.Characterization
Nuclear magnetic resonance
The 1H, 13C and 19F NMR analyses were carried out in CDCl3 using a Bruker Avance III 400 MHz NMR spectrometer at 25 °C. For 1H NMR (400 MHz), 13C NMR (100 MHz) and 19F NMR (377 MHz), CHCl3 served as an internal standard (δ = 7.26 ppm) and data are reported as follows: chemical shift (in ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant (in Hz), and the number of nuclei from the integration. For 13C and 19F NMR (100 MHz), spectra were obtained with complete proton decoupling.Gas chromatography-mass spectrometry
Gas chromatography-mass spectra (GC-MS) were recorded on a Shimadzu QP2012-SE with a Zebron ZB-5MS (20 m × 0.18 mm) capillary apolar column (stationary phase: 0.18 μm film). GC-MS method: initial temperature: 50 °C; initial time: 2 min; ramp: 22 °C min−1; final temperature: 280 °C; final time: 15 min. MS spectra were recorded from 2.5 min to 4.4 min and 5.3 min to 15 min to avoid saturation with mesitylene (MST, 4.5 min) and octylamine (5.2 min).Mass spectrometry (MS)
Mass spectrometry (MS) analyses were recorded on a Bruker Daltonics micrOTOF-Q with an ESI source and positive ion polarity.Fourier transform infrared spectroscopy
The IR spectra were recorded on a Nicolet 210 spectrometer, equipped with an ATR accessory. The characteristic absorptions mentioned in the text are reported in cm−1.Thermogravimetric analyses
The thermogravimetric analyses (TGA) were carried out using a TG 209F1 apparatus (Netzch). Approximately 10 mg of sample were placed in an aluminum crucible and heated from room temperature to 580 °C at a heating rate of 20 °C min−1 under a nitrogen atmosphere (60 mL min−1).Differential scanning calorimetry
The differential scanning calorimetry (DSC) analyses were carried out using a Netzsch DSC200F3 calorimeter, which was calibrated using indium, n-octadecane and n-octane standards. Nitrogen was used as a purge gas. Approximately 10 mg of sample were placed in a perforated aluminum pan and the thermal properties were recorded between −100 °C and 150 °C at 20 °C min−1 heating/cooling rates to observe the glass transition temperature. The Tg values were measured on the second heating ramp to erase the thermal history of the polymer.Dynamic mechanical analyses
The dynamic mechanical analyses (DMA) were carried out on a Metravib DMA 25 with Dynatest 6.8 software. The samples were tested in the uniaxial tension mode at a frequency of 1 Hz with a fixed strain of 10−5 m (0.1% of material height), while applying a temperature ramp at a rate of 3 °C min−1 from −100 °C to +150 °C. The Tα was determined as the maximum of the loss modulus E′′.Rheology
The rheology measurements were carried out on a Thermo Scientific Haake Mars 60 rheometer equipped with a lower electrical temperature module and an active upper heating system, with a textured 8 mm plane–plane geometry. For all rheology experiments, the selected applied stress was within the linear viscoelastic region. A 1 N axial force was applied to ensure appropriate contact between the plates and the samples for all experiments. For the stress-relaxation experiments, 0.5% torsional strain was applied to the samples, and the rubbery modulus evolution with time was monitored at different temperatures. The obtained characteristic relaxation time (τ) was used to calculate the activation energy. Creep recovery experiments were carried out at 50, 100 and 150 °C by applying 3 kPa shear stress for a duration of 1800 s, followed by a recovery period of 1800 s.Swelling index
Three samples from the same batch of the material, of ca. 20 mg each, were separately immersed in THF for 24 h. The swelling index (SI) was calculated using eqn (1), where m2 is the mass of the swollen material and m1 is the initial mass. The reported swelling index is the average value of the three samples.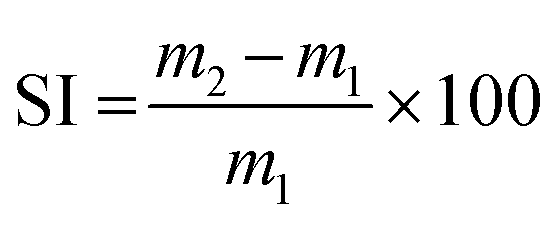 | (1) |
Gel content
Three samples from the same material, of around 20 mg each, were separately immersed in THF for 24 h. The samples were then filtered and dried in a ventilated oven at 70 °C for 24 h. The gel content (GC) was calculated using eqn (2), where m2 is the mass of the dried material and m1 is the initial mass. The reported gel content is the average value of the three samples.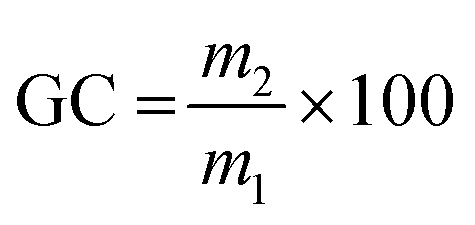 | (2) |
Tensile tests
The material tensile tests were executed on an Instron 3366L5885 mechanical tester. Dog-bone samples of 10 × 2 × 1 mm were used for these analyses. The tensile speed was set at 0.1 mm s−1 with a 100 ms sampling time. The Young modulus and the stress and strain at break were measured on five samples.Procedure for the kinetic experiments
A flame-dried 10 mL scintillation vial was charged with the appropriate excess molar amount of dodecylamine and the amine was stirred at the appropriate temperature for 2 h to ensure amine decarboxylation. Compound 3 (0.25 mmol, 1 eq.), dissolved in mesitylene (MST, the volume of which was adjusted so that the total reaction volume is 2.5 mL), was then added quickly and the reaction was monitored by GC of periodically withdrawn aliquots (ca. 100 μL of the reaction mixture diluted in approximately 5 mL of chloroform). The reaction temperature was varied from 110 °C to 140 °C. The reactions were carried out in the presence of TMB (trimethoxybenzene, 0.3 eq.) as an internal standard for the GC quantification.Preparation of model molecules and monomers
Synthetic protocols used for the preparation of fluorinated molecules are followed by those used for the preparation of their non-fluorinated counterparts.Ethyl 2,2-difluoro-2-(n-octylthio)acetate 1
In a 250 mL round-bottom flask were placed n-octanethiol (8.4 g, 57.6 mmol, 1 eq.) and ethyl 2-bromo-2,2-difluoroacetate (17.5 g, 86.4 mmol, 1.5 eq.) dissolved in 100 mL of DMF. Potassium carbonate (7.9 g, 57.6 mmol, 1 eq.) was then added slowly at 0 °C. The mixture was then stirred and allowed to warm to room temperature. After 2 h, the reaction was quenched with water (100 mL). The mixture was extracted with diethyl ether (3 × 100 mL) and the organic layers were washed with a saturated sodium bicarbonate solution (3 × 100 mL), then with water (3 × 100 mL) and finally dried over MgSO4. The solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (from 100/0 up to 80/20 pentane/AcOEt). The product was obtained as a colourless oil (9.57 g, 62%).

1H NMR (400 MHz, CDCl3): δ (ppm) 4.38 (q, 3J = 7.2 Hz, 2H, CH3–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–O–CO), 2.89 (t, 3J = 7.4 Hz, 2H, S–C2–CH2), 1.75–1.62 (m, 2H, S–CH2–C2–(CH2)5), 1.48–1.20 (m, 13H, C3–CH2–O–CO–CF2–S–CH2–CH2–(C2)5–CH3), 0.97–0.81 (m, 3H, S–CH2–CH2–(CH2)5–C3).
2–O–CO), 2.89 (t, 3J = 7.4 Hz, 2H, S–C2–CH2), 1.75–1.62 (m, 2H, S–CH2–C2–(CH2)5), 1.48–1.20 (m, 13H, C3–CH2–O–CO–CF2–S–CH2–CH2–(C2)5–CH3), 0.97–0.81 (m, 3H, S–CH2–CH2–(CH2)5–C3).
19F NMR (377 MHz, CDCl3): δ (ppm) −82.9 (s).
13C NMR (101 MHz, CDCl3): δ (ppm) 162.0 (t, 2J = 33.0 Hz, O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O–CF2–S), 120.7 (t, 1J = 285 Hz, O–CO–F2–S), 63.6 (s, CH3–H2–O–CO), 28.8 (t, 3J = 3.0 Hz, S–H2–CH2), 28.7, 29.0, 29.1, 29.6 and 31.8 (s, S–CH2–H2–H2–H2–H2–H2–CH2–CH3), 22.6 (s, S–CH2–CH2–CH2–CH2–CH2–CH2–H2–CH3), 14.1 (s, H3–CH2–O–CO), 13.9 (s, S–CH2–CH2–(CH2)5–H3).
O–CF2–S), 120.7 (t, 1J = 285 Hz, O–CO–F2–S), 63.6 (s, CH3–H2–O–CO), 28.8 (t, 3J = 3.0 Hz, S–H2–CH2), 28.7, 29.0, 29.1, 29.6 and 31.8 (s, S–CH2–H2–H2–H2–H2–H2–CH2–CH3), 22.6 (s, S–CH2–CH2–CH2–CH2–CH2–CH2–H2–CH3), 14.1 (s, H3–CH2–O–CO), 13.9 (s, S–CH2–CH2–(CH2)5–H3).
N-Benzyl-2,2-difluoro-2-(n-octylthio)acetamide 3
In a test tube were added compound 1 (0.50 g, 1.86 mmol, 1eq) and benzylamine (0.60 g, 5.58 mmol, 3 eq.). The mixture was stirred for 10 minutes at 25 °C. The crude product was purified by column chromatography on silica gel (from 95/5 up to 70/30 pentane/AcOEt). The product was obtained as a colourless oil (0.50 g, 82%).
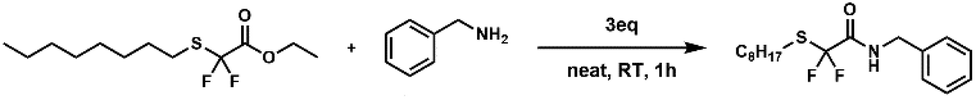
1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.30 (m, 5H, aromatics), 4.53 (d, 3J = 5.8 Hz, 2H, CO–NH–C2–Ar), 2.92 (t, 3J = 7.4 Hz, 2H, CH3–(CH2)5–CH2–C2–S), 1.78–1.61 (m, 2H, CH3–(CH2)5–C2–CH2–S), 1.49–1.20 (m, 10H, CH3–(C2)5–CH2–CH2–S), 0.96–0.87 (m, 3H, C3–(CH2)5–CH2–CH2–S).
19F NMR (377 MHz, CDCl3): δ (ppm) −82.5 (s).
13C NMR (101 MHz, CDCl3): δ (ppm) 162.1 (t, 2J = 28.9 Hz, S–CF2–O–NH), 136.6 (s, aromatic carbon), 128.92 (s, 2 aromatic carbons), 128.02 (s, aromatic carbon), 127.86 (s, 2 aromatic carbons), 123.29 (t, 2J = 286.4 Hz, S–F2–CO–NH), 43.78 (s, CO–NH–H2–Ar), 28.7, 29.0, 29.1, 29.6 and 31.8 (s, S–CH2–H2–H2–H2–H2–H2–CH2–CH3), 28.6 (t, 3J = 2.8 Hz, CH3–(CH2)5–CH2–H2–S), 22.7 (s, CH3–H2–CH2–CH2–CH2–CH2–CH2–CH2–S), 14.1 (s, H3–(CH2)5–CH2–CH2–S).
HRMS (ESI+): calc. m/z for [M + H]+: 330.1698, measured: 330.1694.
N-Dodecyl-2,2-difluoro-2-(n-octylthio)acetamide 9
In a test tube were added compound 1 (0.5 g, 1.86 mmol, 1 eq.) and dodecylamine (1.03 g, 5.58 mmol, 3 eq.). The mixture was stirred for 1 h. The crude product was purified by column chromatography on silica gel (from 95/5 up to 70/30 pentane/AcOEt). The product was obtained as a colourless oil (0.61 g, 80%).

1H NMR (400 MHz, CDCl3): δ (ppm) 3.35 (q, 3J = 6.9 Hz, 2H, CO–NH–C2–CH2), 2.91 (t, 3J = 7.4 Hz, 2H, CH2–C2–S–CO–NH), 1.79–1.52 (m, 4H, CH2 on alkyl chains), 1.47–1.22 (m, 28H, CH2 on alkyl chains), 1.01–0.84 (m, 6H, CH3 at the end of both alkyl chains).
19F NMR (377 MHz, CDCl3): δ (ppm) −82.6 (s).
13C NMR (101 MHz, CDCl3): δ (ppm) 162.0 (t, 3J = 28.4 Hz, S–CF2–O–NH), 123.3 (t, 1J = 286 Hz, S–F2–CO–NH), 28.7, 29.0, 29.1, 29.2, 29.2, 29.4, 29.5, 29.6, 29.6, 29.6, 31.8, 31.9 and 39.9 (s, C in alkyl chains), 28.6 (t, 3J = 2.9 Hz, CH3–(CH2)5–CH2–H2–S), 14.1, 14.1, 22.6, 22.7, 26.7 (s, C in alkyl chains).
HRMS (ESI+): calc. m/z for [M + H]+: 408.3182, measured: 408.3189.
Synthesis of ethyl 2-(n-octylthio)acetate 2
In a 50 mL round-bottom flask were placed octanethiol (3.3 g, 23 mmol, 1 eq.) and ethyl 2-bromoacetate (5.8 g, 34.5 mmol, 1.5 eq.), dissolved in 25 mL of DMF. Then, potassium carbonate (3.2 g, 23 mmol, 1 eq.) was added slowly at 0 °C. The mixture was then stirred and allowed to warm to room temperature. After 2 h, the reaction was quenched with water (50 mL). The mixture was extracted with diethyl ether (3 × 50 mL) and the organic layers were washed with a saturated sodium bicarbonate solution (3 × 50 mL), then with water (3 × 50 mL) and finally dried over MgSO4. The solvent was removed under reduced pressure. The resulting product (4.97 g, 93%) was used without further purification.

1H NMR (400 MHz, CDCl3): δ (ppm) 4.20 (q, 3J = 7.1 Hz, 2H, CH3–C2–O–CO), 3.21 (s, 2H, O–CO–C2–S), 2.64 (t, 3J = 7.3 Hz, 2H, S–C2–CH2–(CH2)5–CH3), 1.67–1.55 (m, 2H, S–CH2–C2–(CH2)5–CH3), 1.43–1.22 (m, 13H, C3–CH2–O–CO–CH2–S–CH2–CH2–(C2)5–CH3), 0.92–0.86 (m, 3H, S–CH2–CH2–(CH2)5–C3).
13C NMR (101 MHz, CDCl3): δ (ppm) 170.6 (s, O–O–CH2–S), 61.3 (s, CH3–H2–O–CO), 33.7 (s, O–CO–H2–S), 28.8, 29.0, 29.1, 29.2, 31.8 and 32.7 (s, S–CH2–H2–H2–H2–H2–H2–CH2–CH3), 22.6 (s, S–CH2–CH2–CH2–CH2–CH2–CH2–H2–CH3), 14.2 (s, H3–CH2–O–CO), 14.1 (s, S–CH2–CH2–CH2–CH2–CH2–CH2–CH2–H3).
N-Benzyl-2-(n-octylthio)acetamide 4
In a test tube were added compound 2 (1 g, 4.3 mmol, 1 eq.) and benzylamine (1.38 g, 12.9 mmol, 3 eq.). The mixture was stirred for 48 h at 100 °C. The crude product was purified by column chromatography on silica gel (from 95/5 pentane up to 70/30 pentane/AcOEt) to yield a colourless oil (0.93 g, 74%).

1H NMR (400 MHz, CDCl3): δ (ppm) 7.41–7.30 (m, 5H, aromatics), 4.52 (d, 3J = 5.9 Hz, 2H, CO–NH–C2–Ar), 3.30 (s, 2H, S–C2–CO–NH), 2.53 (t, 3J = 7.4 Hz, 2H, CH3–(CH2)5–CH2–C2–S), 1.61–1.51 (m, 2H, CH3–(CH2)5–C2–CH2–S), 1.44–1.19 (m, 10H, CH3–(C2)5–CH2–CH2–S), 0.91 (t, 3J = 7.0 Hz, 3H, C3–(CH2)5–CH2–CH2–S).
13C NMR (101 MHz, CDCl3): δ (ppm) 168.8 (s, S–CH2–O–NH), 138.0 (s, aromatic carbon), 128.8 (s, 2 aromatic carbons), 127.7 (s, 2 aromatic carbons), 127.6 (s, aromatic carbon), 43.8 (s, CO–NH–H2–Ar), 36.3 (s, S–H2–CO–NH), 29.1, 29.2, 29.2, 31.8 and 33.3 (s, S–CH2–H2–H2–H2–H2–H2–CH2–CH3), 28.8 (s, CH3–CH2–CH2–CH2–CH2–CH2–CH2–H2–S), 22.6 (s, CH3–H2–CH2–CH2–CH2–CH2–CH2–CH2–S), 14.1 (s, H3–CH2–CH2–CH2–CH2–CH2–CH2–CH2–S).
HRMS (ESI+): calc. m/z for [M + H]+: 294.1886, measured: 294.1893.
Diethyl 2,2′-(hexane-1,6-diylbis(sulfanediyl))bis(2,2-difluoroacetate) 10
In a 250 mL round-bottom flask were placed hexane-1,6-dithiol (10 g, 66.5 mmol, 1 eq.) and ethyl 2-bromo-2,2-difluoroacetate (40.5 g, 200 mmol, 3 eq.), dissolved in 100 mL of DMF. Then, potassium carbonate (18.4 g, 133 mmol, 2 eq.) was added slowly at 0 °C. The mixture was then stirred and allowed to warm to room temperature. After 2 h, the reaction was quenched with water (100 mL). The mixture was extracted with diethyl ether (3 × 100 mL) and the organic layers were washed with a saturated sodium bicarbonate solution (3 × 100 mL), then with water (3 × 100 mL) and finally dried over MgSO4. The solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (from 99/1 up to 80/20 pentane/AcOEt) to yield a colourless oil (15.4 g, 58%).

1H NMR (400 MHz, CDCl3): δ (ppm) 4.38 (q, 3J = 7.1 Hz, 4H, CH3–C2–O–CO), 2.89 (t, 3J = 7.4 Hz, 4H, S–C2–CH2–CH2), 1.76–1.66 (m, 4H, S–CH2–C2–CH2), 1.49–1.43 (m, 4H, S–CH2–CH2–C2), 1.40 (t, 3J = 7.1 Hz, 6H, C3–CH2–O–CO).
19F NMR (377 MHz, CDCl3): δ (ppm) −82.8 (s).
13C NMR (101 MHz, CDCl3): δ (ppm) 161.9 (t, 2J = 32.9 Hz, CH3–CH2–O–O), 120.7 (t, 1J = 286 Hz, O–CO–F2–S), 63.6 (s, CH3–H2–O–CO), 29.4 (s, S–CH2–H2–CH2), 28.6 (t, 3J = 3.1 Hz, S–H2–CH2–CH2), 28.0 (s, S–CH2–CH2–H2), 13.9 (s, H3–CH2–O–CO).
HRMS (ESI+): calc. m/z for [M + NH4]+: 412.1234, measured: 412.1229.
Diethyl 2,2′-(hexane-1,6-diylbis(sulfanediyl))diacetate 11
In a 100 mL round-bottom flask were placed hexane-1,6-dithiol (4 g, 26.6 mmol, 1 eq.) and ethyl 2-bromoacetate (13.3 g, 79.8 mmol, 3 eq.), dissolved in 25 mL of DMF. Then, potassium carbonate (7.4 g, 53.2 mmol, 2 eq.) was added slowly at 0 °C. The mixture was then stirred and allowed to warm to room temperature. After 2 h, the reaction was quenched with water (50 mL). The mixture was extracted with diethyl ether (3 × 50 mL) and the organic layers were washed with a saturated sodium bicarbonate solution (3 × 50 mL), then with water (3 × 50 mL) and finally dried over MgSO4. The solvent was removed under reduced pressure. The resulting product (8.16 g, 95%) was used without further purification.
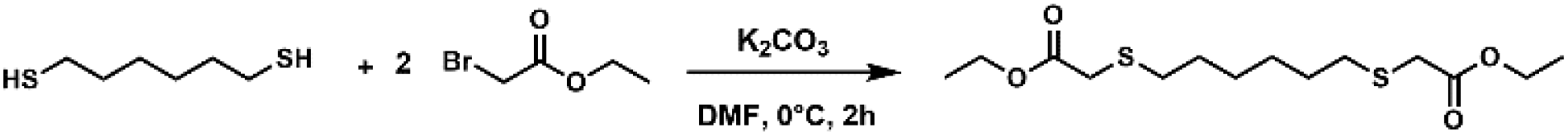
1H NMR (400 MHz, CDCl3) : δ (ppm) 4.18 (q, 3J = 7.1 Hz, 4H, CH3–C2–O–CO), 3.19 (s, 4H, CO–C2–S), 2.62 (t, 3J = 7.6 Hz, 4H, S–C2–CH2–CH2), 1.66–1.53 (m, 4H, S–CH2–C2–CH2), 1.45–1.35 (m, 4H, S–CH2–CH2–C2), 1.28 (t, 3J = 7.7 Hz, 6H, C3–CH2–O–CO).
13C NMR (101 MHz, CDCl3): δ (ppm) 170.5 (s, O–O–CH2–S), 61.2 (s, CH3–H2–O–CO), 33.7 (s, O–CO–H2–S), 32.5 (s, S–CH2–H2–CH2), 28.8 (s, S–H2–CH2–CH2), 28.2 (s, S–CH2–CH2–H2), 14.2 (s, H3–CH2–O–CO).
HRMS (ESI+): calc. m/z for [M + H]+: 323.1345, measured: 323.1344.
Synthesis of fluorinated and non-fluorinated polyamides (FPA and NFPA)
For the synthesis of the polyamide networks, TREN and compounds 10 or 11 were mixed in a SpeedMixer for 3 minutes and then cured at 100 °C for 2 hours for the fluorinated materials (FPA) and 30 hours for the non-fluorinated material (NFPA). The resulting samples were cut into small pieces (ca. 1 cm2) and then shaped under a hot press for 1 h under 3 tonnes of pressure at different temperatures, depending on the initial material formulation.Computational details
The computational work was carried out using the Gaussian09 suite of programs.96 The geometry optimizations were performed without any symmetry constraint using the BP86 functional and the 6-311G(d,p) basis functions for all atoms. The effect of dispersion forces (Grimme's D3 empirical method97) and solvation (SMD,98 using an arbitrary ε value of 10) were included during the optimization. The ZPVE, PV, and TS corrections at 298 K were obtained with Gaussian09 from the solution of the nuclear equation using the standard ideal gas and harmonic approximations at T = 298.15 K, which also verified the nature of all optimized geometries as local minima or first-order saddle points. A correction of 1.95 kcal mol−1 was applied to all G values to change the standard state from the gas phase (1 atm) to solution (1 M).99Author contributions
D. B. and G. T. performed all the experimental work and collected the data. R. P. performed the DFT calculations. S. C., V. L. and E. L. supervised the project. D. B. and R. P. wrote the first draft of the manuscript which was reviewed and edited by R. P., S. C., V. L. and E. L. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded by the French National Research Agency ANR (AFCAN project: ANR-19-CE06-0014). R. P. is grateful to the CALMIP mesocenter of the University of Toulouse for the allocation of computational resources.References
- B. L. Deopura, R. Alagirusamy, M. Joshi and B. Gupta, Polyesters and Polyamides, 2008 Search PubMed.
- X. Cui and D. Yan, Eur. Polym. J., 2005, 41, 863–870 CrossRef CAS.
- P. Sikorski, N. A. Jones, E. D. T. Atkins and M. J. Hill, Macromolecules, 2001, 34, 1673–1676 CrossRef CAS.
- R. J. Palmer and Updated by Staff, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2005 Search PubMed.
- B. Brehmer, Bio-Based Plastics, John Wiley & Sons Ltd, Chichester, UK, 2013, pp. 275–293 Search PubMed.
- S. Russo and E. Casazza, Polymer Science: A Comprehensive Reference, Elsevier, 2012, vol. 4, pp. 331–396 Search PubMed.
- W. E. Hanford and R. M. Joyce, J. Polym. Sci., 1948, 3, 167–172 CrossRef CAS.
- R. S. Davé, R. L. Kruse, L. R. Stebbins and K. Udipi, Polymer, 1997, 38, 939–947 CrossRef.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef.
- H. Mutlu and M. A. R. Meier, Macromol. Chem. Phys., 2009, 210, 1019–1025 CrossRef CAS.
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1800524 CrossRef PubMed.
- M. Winnacker and B. Rieger, Macromol. Rapid Commun., 2016, 37, 1391–1413 CrossRef CAS PubMed.
- M. A. Ali, S. Tateyama, Y. Oka, D. Kaneko, M. K. Okajima and T. Kaneko, Macromolecules, 2013, 46, 3719–3725 CrossRef CAS.
- M. Li and T. J. Dingemans, Polymer, 2017, 108, 372–382 CrossRef CAS.
- M. Li, J. Bijleveld and T. J. Dingemans, Eur. Polym. J., 2018, 98, 273–284 CrossRef CAS.
- E. Tarkin-Tas and L. J. Mathias, Macromolecules, 2010, 43, 968–974 CrossRef CAS.
- T. Agag, C. R. Arza, F. H. J. Maurer and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4335–4342 CrossRef CAS.
- H. Bouchékif, D. Tunc, C. Le Coz, A. Deffieux, P. Desbois and S. Carlotti, Polymer, 2014, 55, 5991–5997 CrossRef.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- W. He, Y. Tao and X. Wang, Macromolecules, 2018, 51, 8248–8257 CrossRef CAS.
- A. Sathyan, R. C. Hayward and T. Emrick, Macromolecules, 2019, 52, 167–175 CrossRef CAS.
- C. Yi, J. Zhao, Z. Zhang and J. Zhang, Ind. Eng. Chem. Res., 2017, 56, 13743–13750 CrossRef CAS.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- D. Boucher, J. Madsen, L. Yu, Q. Huang, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Macromolecules, 2021, 54, 6772–6779 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Macromol. Chem. Phys., 2016, 217, 2541–2550 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- A. V. Tobolsky, I. B. Prettyman and J. H. Dillon, Rubber Chem. Technol., 1944, 17, 551–575 CrossRef CAS.
- M. D. Stern and A. V. Tobolsky, Rubber Chem. Technol., 1946, 19, 1178–1192 CrossRef.
- C. Pronoitis, M. Hakkarainen and K. Odelius, Polym. Chem., 2021, 12, 5668–5678 RSC.
- K. Liang, G. Zhang, J. Zhao, L. Shi, J. Cheng and J. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 5673–5683 CrossRef CAS.
- Y. Chen, H. Zhang, S. Majumdar, R. A. T. M. van Benthem, J. P. A. Heuts and R. P. Sijbesma, Macromolecules, 2021, 54, 9703–9711 CrossRef CAS PubMed.
- M. E. Smith and H. Adkins, J. Am. Chem. Soc., 1938, 60, 657–663 CrossRef.
- N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS PubMed.
- V. Gotor, R. Brieva, C. González and F. Rebolledo, Tetrahedron, 1991, 47, 9207–9214 CrossRef CAS.
- I. K. Miller, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 1403–1417 CrossRef CAS.
- E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 1–5 Search PubMed.
- J. E. Dander, E. L. Baker and N. K. Garg, Chem. Sci., 2017, 8, 6433–6438 RSC.
- L. Becerra-Figueroa, A. Ojeda-Porras and D. Gamba-Sánchez, J. Org. Chem., 2014, 79, 4544–4552 CrossRef CAS PubMed.
- M. Shi and S. C. Cui, Synth. Commun., 2005, 35, 2847–2858 CrossRef CAS.
- E. Bon, D. C. H. Bigg and G. Bertrand, J. Org. Chem., 1994, 59, 4035–4036 CrossRef CAS.
- J. M. Hoerter, K. M. Otte, S. H. Gellman, Q. Cui and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 647–654 CrossRef CAS PubMed.
- S. E. Eldred, D. A. Stone, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2003, 125, 3422–3423 CrossRef CAS PubMed.
- N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS PubMed.
- D. A. Kissounko, I. A. Guzei, S. H. Gellman and S. S. Stahl, Organometallics, 2005, 24, 5208–5210 CrossRef CAS.
- J. M. Hoerter, K. M. Otte, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 5177–5183 CrossRef CAS PubMed.
- R. Vanjari, B. K. Allam and K. N. Singh, Tetrahedron Lett., 2013, 54, 2553–2555 CrossRef CAS.
- J. W. Wu, Y. D. Wu, J. J. Dai and H. J. Xu, Adv. Synth. Catal., 2014, 356, 2429–2436 CrossRef CAS.
- J. Liang, J. Lv and Z. C. Shang, Tetrahedron, 2011, 67, 8532–8535 CrossRef CAS.
- G. W. Wang, T. T. Yuan and D. D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380–1383 CrossRef CAS PubMed.
- K. Eersels and G. Groeninckx, Polymer, 1996, 37, 983–989 CrossRef CAS.
- L. Wang, X. Dong, Y. Gao, M. Huang, C. C. Han, S. Zhu and D. Wang, Polymer, 2015, 59, 16–25 CrossRef CAS.
- S. Fakirov and Z. Denchev, Transreactions in Condensation Polymers, John Wiley & Sons, Ltd, 2007, pp. 319–389 Search PubMed.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- T. Liu, S. Zhang, C. Hao, C. Verdi, W. Liu, H. Liu and J. Zhang, Macromol. Rapid Commun., 2019, 40, 1800889 CrossRef PubMed.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360 RSC.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- H. Zhang, S. Majumdar, R. A. T. M. Van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 12, 272–277 CrossRef PubMed.
- M. Podgórski, N. Spurgin, S. Mavila and C. N. Bowman, Polym. Chem., 2020, 11, 5365–5376 RSC.
- M. Delahaye, F. Tanini, J. O. Holloway, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5207–5215 RSC.
- M. Podgórski, S. Mavila, S. Huang, N. Spurgin, J. Sinha, C. N. Bowman, M. Podgórski, S. Mavila, S. Huang, N. Spurgin, J. Sinha and C. N. Bowman, Angew. Chem., Int. Ed., 2020, 59, 9345–9349 CrossRef PubMed.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Macromolecules, 2022, 55, 1669–1679 CrossRef CAS.
- F. Cuminet, D. Berne, S. Lemouzy, E. Dantras, C. Joly-Duhamel, S. Caillol, E. Leclerc and V. Ladmiral, Polym. Chem., 2022, 8, 5255–5446 Search PubMed.
- S. Lemouzy, F. Cuminet, D. Berne, S. Caillol, V. Ladmiral, R. Poli and E. Leclerc, Chem. – Eur. J., 2022, 28, e202201135 CrossRef CAS PubMed.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- L. Pettazzoni, F. Leonelli, A. Martinelli, L. M. Migneco, S. Alfano, D. Di Luca, L. Celio and V. Di Lisio, J. Appl. Polym. Sci., 2022, 52408 Search PubMed.
- C. Wakselman, J. Fluor. Chem., 1992, 59, 367–378 CrossRef CAS.
- M. A. Ali, A. Nath, M. M. Islam, S. B. Shaheed and I. N. Dibbo, RSC Adv., 2022, 12, 11255–11261 RSC.
- G. Li, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2020, 10, 710–716 RSC.
- W. Wu, Bull. Korean Chem. Soc., 2014, 35, 2673–2678 CrossRef CAS.
- X. Yang, L. Fan and Y. Xue, RSC Adv., 2014, 4, 30108–30117 RSC.
- Y. Yang, J. Liu, F. S. Kamounah, G. Ciancaleoni and J.-W. Lee, J. Org. Chem., 2021, 86, 16867–16881 CrossRef CAS PubMed.
- D. Berne, B. Quienne, S. Caillol, E. Leclerc and V. Ladmiral, J. Mater. Chem. A, 2022, 10, 25085–25097 RSC.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Polymers, 2018, 10, 43 CrossRef PubMed.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- A. Gablier, M. O. Saed and E. M. Terentjev, Soft Matter, 2020, 16, 5195–5202 RSC.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047–2056 RSC.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 RSC.
- A. M. Hubbard, Y. Ren, A. Sarvestani, D. Konkolewicz, C. R. Picu, A. K. Roy, V. Varshney and D. Nepal, ACS Omega, 2022, 7, 29125–29134 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00577a |
| This journal is © The Royal Society of Chemistry 2023 |