
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic living ROMP: block copolymers from macro-chain transfer agents†
Ankita
Mandal
 and
Andreas F. M.
Kilbinger
*
and
Andreas F. M.
Kilbinger
*
Department of chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.kilbinger@unifr.ch
First published on 18th May 2023
Abstract
Vinyl ether based macro-chain transfer agents (m-CTAs) are used to produce different di or tri-block copolymers under catalytic living ROMP conditions. Polystyrene (PS) vinyl ether m-CTA and polycaprolactone (PCL) or polylactide vinyl ether (PLA) m-CTAs are synthesized straightforwardly via ATRP and ROP respectively. Regioselectivity as well as the high metathesis activity of these m-CTAs enabled us to synthesise a range of metathesis-based A–B diblock copolymers with controlled dispersities (Đ < 1.4). In this manner, PS-ROMP (here, ROMP refers to a poly(MNI-co-DHF) block), PCL-ROMP and PLA-ROMP were synthesized using substoichiometric amounts of ruthenium complex in a living fashion. Also, a more complex PEG-PCL-ROMP tri-block terpolymer was obtained catalytically. All block copolymers were characterized by SEC and DOSY NMR spectroscopy. We believe that this methodology of using macro-chain transfer agents to prepare degradable ROMP polymers under catalytic living ROMP conditions will find applications in biomedicine.
Introduction
Over the past decades, block copolymers (BCPs) have attracted the interest of the polymer community because of their widespread applications in numerous fields such as nanotechnology, nanolithography, photonics, polymeric self-assembly, controlled drug delivery, thermoplastic elastomers, adhesives, and many others.1–6 The most common synthetic strategies are based on (i) sequential addition of monomers via a living polymerization technique, (ii) coupling reactions between different chain ends of polymers and (iii) polymerization from macroinitiators.7–14 Although many living polymerization techniques are known that allow the preparation of multiblock copolymers with controlled molecular weight and dispersity, the same polymerization technique is used to create all blocks of the copolymer in most cases.15,16 A second approach towards BCP synthesis includes the conjugation of suitably end-functionalized polymeric chains with one another. But this method heavily relies on post-polymerization modifications which are often difficult to achieve.17 An alternative strategy for the synthesis of a wide range of functionalizable BCPs is the macro-initiation technique. Macro-chain transfer agents are mostly used in RAFT and ATRP polymerization methods to yield BCPs in a straight forward approach.18,19High functional group tolerance, mild reaction conditions, precise control over molecular weight, dispersity and monomer composition have made ring opening metathesis polymerization (ROMP) one of the most efficient methods for synthesizing a diverse range of block copolymers with applications in different areas.20–25 Numerous ROMP-based block copolymers have been synthesized via sequential monomer addition using ruthenium and molybdenum-based metathesis initiators.26,27 However, in all these reports, stoichiometric amounts of metal complexes are required with respect to the number of polymer chains formed. This often results in high levels of ruthenium contamination in the synthesized block copolymers hindering potential applications in life sciences. Only few monomers can be polymerized via ROMP using symmetrical chain transfer agents (CTA) in a thermodynamically driven catalytic process.28,29 These homotelechelic polymers can further be employed to produce A–B–A type block copolymers via orthogonal polymerization techniques such as RAFT or ATRP. However, there, the ROMP part (B) shows a broad dispersity for mechanistic reasons. Furthermore, the synthesis of diblock (A–B) copolymers cannot be achieved by this method.30,31 The Boydston group has also demonstrated bidirectional polymer growth in organocatalyzed ROMP in their recent study.32,33 Moreover, our group has recently reported a catalytic living ROMP method exploiting a degenerative reversible chain transfer mechanism.34 One of the limitations of this process is that only ROMP-ROMP di or tri-block copolymers can be prepared catalytically via this methodology. A very recent report describes the synthesis of PEG/PLA-ROMP (A–B type) block copolymers using monosubstituted 1,3-diene derivatives as macro-chain transfer agents. However, as this method relies on a kinetically controlled chain transfer mechanism, the synthesized BCPs show broad dispersities.35 A catalytic living polymerization technique capable of synthesizing A–B or A–B–C type block co- and terpolymers via ROMP in combination with other polymerization methods would be a valuable addition to the polymer chemists’ synthetic toolbox.
Herein, we report the synthesis of di or tri – block copolymers such as polystyrene (PS)-ROMP, polycaprolactone (PCL)-ROMP, polylactide (PLA)-ROMP and polyethylene glycol (PEG)-polycaprolactone (PCL)-ROMP using either PS, PCL, PLA or PEG-PCL vinyl ether-terminated macro chain transfer agents (m-CTAs) under catalytic living ROMP conditions. As the ROMP block of these copolymers is prepared via copolymerizing 2,3-dihydrofuran (DHF) with norbornene derivatives, it can be easily degraded by addition of dilute HCl (due to the presence of acid-labile vinyl ethers) making them more environmentally sustainable.36
Recently our group has reported a strategy for the one-pot synthesis of living and degradable ROMP polymers by copolymerizing 2,3-dihydrofuran (DHF) with several norbornene derivatives using vinyl ethers as chain transfer agents under catalytic living ROMP conditions.37 In that report, the ultrafast and regioselective metathesis activity of vinyl ethers38,39 was also exploited to synthesize a PEG-ROMP diblock copolymer using a PEG-based macro chain transfer agent. Herein, we have expanded this strategy to synthesize different A–B and A–B–C type block co- and terpolymers with good control over molecular weight and dispersity using catalytic amounts of Grubbs’ ruthenium complexes.
Results and discussion
To begin with, a polystyrene (PS) vinyl ether macro-chain transfer agent m-CTA1 (Mn, SEC (CHCl3) = 3.12 kDa, Đ = 1.16, ESI, Fig. S1† and Fig. 1) was synthesized via ATRP using the CuBr/PMDETA catalyst system.40 Next, a one pot copolymerization of M1 and DHF was performed using m-CTA1 and G2 in a ratio of G2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m-CTA1:M1:DHF = 1:10:600:1200.37 (ESI, Table S1,† entry 1) A SEC measurement of the precipitated copolymer (P1) indicated good control over molecular weight in agreement with a degree of polymerization determined by the M1:m-CTA1 ratio as well as dispersity (Mn,SEC (CHCl3) = 16.89 kDa, Đ = 1.33, Mn (theo.) = 16.43 kDa, Fig. 3A). The absence of any m-CTA1 signal in the SEC elugram as well as a single diffusing species in DOSY NMR spectroscopy (Fig. 3B) revealed the successful synthesis of the di-block copolymer. As discussed in the previous report,37 norborneneimide derivatives are slow to be ring-opened by the propagating Ru Fischer-carbene. This allows for a rapid regioselective exchange between the propagating Fischer-carbene and the vinyl ether of m-CTA1 under these conditions. Once all m-CTA1 has been transferred to the polymer chain ends, the degenerative, reversible and regioselective Fischer-carbene exchange between the end groups occurs faster than monomer propagation giving the macroscopic impression of all chains growing at the same time. This quasi-simultaneous growth of all chains leads to a straightforward synthesis of block copolymers with controlled molecular weight (determined by the monomer/m-CTA ratio) and narrow molecular weight distributions in one pot37 (Fig. 2A and B).
:m-CTA1:M1:DHF = 1:10:600:1200.37 (ESI, Table S1,† entry 1) A SEC measurement of the precipitated copolymer (P1) indicated good control over molecular weight in agreement with a degree of polymerization determined by the M1:m-CTA1 ratio as well as dispersity (Mn,SEC (CHCl3) = 16.89 kDa, Đ = 1.33, Mn (theo.) = 16.43 kDa, Fig. 3A). The absence of any m-CTA1 signal in the SEC elugram as well as a single diffusing species in DOSY NMR spectroscopy (Fig. 3B) revealed the successful synthesis of the di-block copolymer. As discussed in the previous report,37 norborneneimide derivatives are slow to be ring-opened by the propagating Ru Fischer-carbene. This allows for a rapid regioselective exchange between the propagating Fischer-carbene and the vinyl ether of m-CTA1 under these conditions. Once all m-CTA1 has been transferred to the polymer chain ends, the degenerative, reversible and regioselective Fischer-carbene exchange between the end groups occurs faster than monomer propagation giving the macroscopic impression of all chains growing at the same time. This quasi-simultaneous growth of all chains leads to a straightforward synthesis of block copolymers with controlled molecular weight (determined by the monomer/m-CTA ratio) and narrow molecular weight distributions in one pot37 (Fig. 2A and B).
 | ||
| Fig. 1 Structures of the catalyst, monomers, and macro-chain transfer agents used in this report. | ||
 | ||
| Fig. 2 (A) Proposed mechanism of catalytic living ROMP for the syntheses of different block copolymers using vinyl ethers as macro-chain transfer agents. (B) Mechanism of catalytic living ROMP illustrating reversible degenerative exchange between Fischer-carbene end groups. | ||
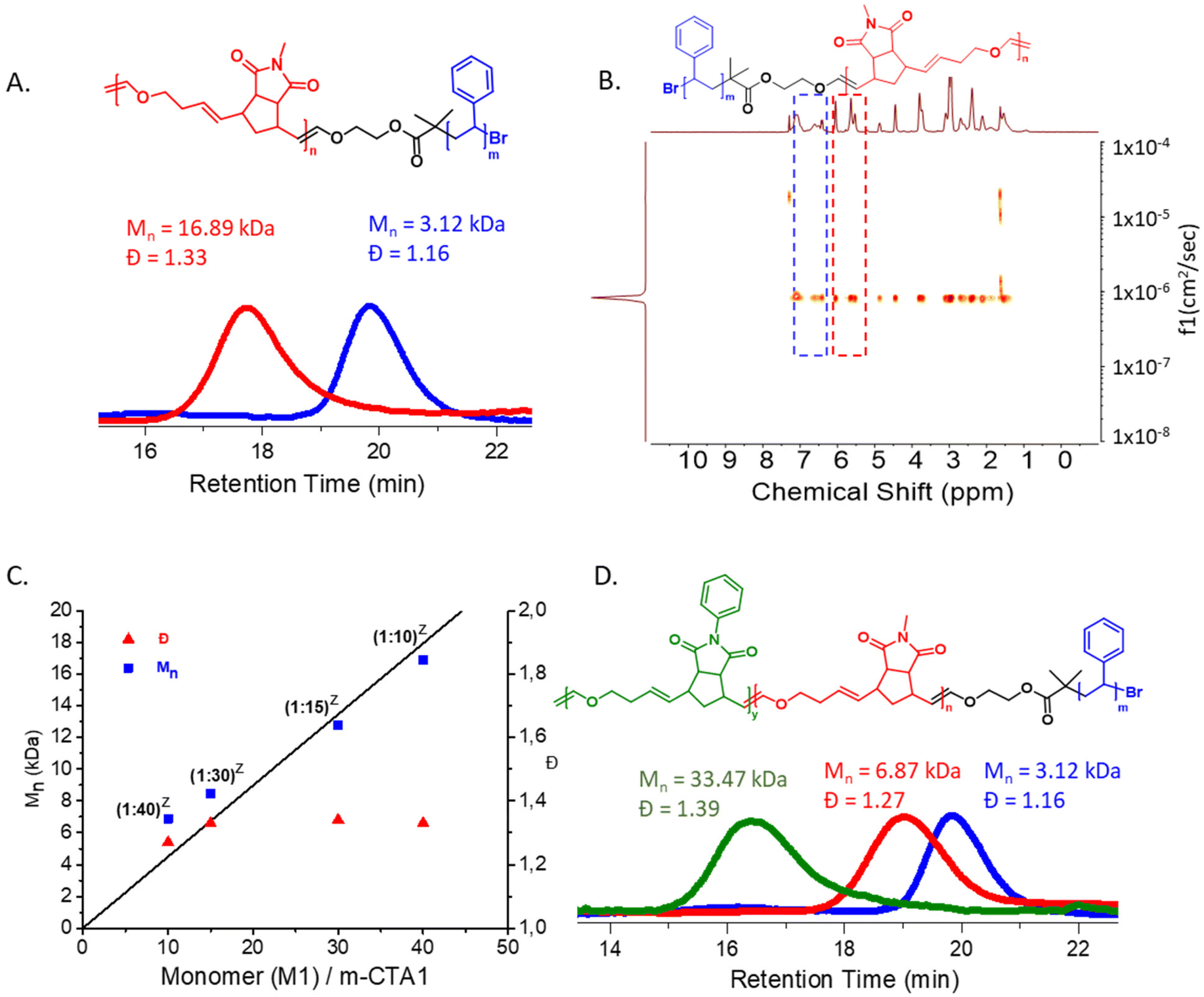 | ||
| Fig. 3 (A) SEC (CHCl3) traces of the m-CTA1 (blue) and the diblock copolymer P1 (red) (B) DOSY NMR spectra (400 MHz, CDCl3) of P1 (C) Plot of the number average molecular weight (Mn,SEC (CHCl3) and molecular weight dispersity (Đ) versus the [M1]/[m-CTA1] ratio showing a linear correlation. The ratios reported in brackets (z) denote the [G2]/[m-CTA1] ratio. (D) SEC (CHCl3) traces of the m-CTA1 (blue), the diblock copolymer P4 (red) and the triblock terpolymer P7 (green). | ||
Next, a series of PS-ROMP diblock copolymers (P2–P5) were synthesized by varying the M1:m-CTA1 ratio. All polymers were characterized by SEC and DOSY NMR spectroscopy. (ESI, Table S1,† entries 2–5, Fig. S2–S5 and S14–S17†). A linear correlation between molecular weight and the M1:m-CTA1 ratio was observed as expected for a living copolymerization (Fig. 3C).
After that, monomer M2 and DHF were also copolymerized in an analogous manner using m-CTA1 to yield polymer P6 (Mn,SEC (CHCl3) = 9.31 kDa, Đ = 1.31, ESI, Table S1,† entry 6, Fig. S6 and S18†).
Since these di-block copolymers contain an acid labile backbone functionality,37,41 treatment of P5 with dilute HCl resulted in the degradation of the ROMP block whereas the polystyrene block did not degrade under these conditions (ESI, Fig. S12†).
Next, a tri-block terpolymer was synthesized via reinitiating polymer P4 with a catalytic amount of G2 and adding a mixture of monomer M2 and DHF. The synthesis of this PS-ROMP-ROMP tri-block terpolymer (P7) (Mn,SEC (CHCl3) = 33.47 kDa, Đ = 1.39) is a significant proof of the proposed mechanism (Fig. 3D).
After that, to show the versatility of this method, different macro-chain transfer agents were synthesized. Ring opening polymerization (ROP) was employed to synthesize biodegradable polycaprolactone (PCL) m-CTA2 (Mn,SEC (CHCl3) = 2.21 kDa, Đ = 1.22) and polylactide (PLA) m-CTA3 (Mn,SEC (DMF) = 1.93 kDa, Đ = 1.18) based macro-chain transfer agents.42,43 A one pot copolymerization of M1 and DHF using m-CTA2 yielded the polymers P8 (Mn,SEC (CHCl3) = 14.81 kDa, Đ = 1.34) and P9 (Mn,SEC (CHCl3) = 11.36 kDa, Đ = 1.30) respectively (ESI, Table S2†). A single diffusing species in the DOSY NMR spectrum as well as a clear shift in the SEC elugram compared to the PCL macro-chain transfer agent (m-CTA2) confirmed the successful PCL-ROMP di-block copolymer formation (Fig. 4A and B). In a similar way, M1 and DHF were copolymerized using the PLA-based macro-CTA m-CTA3 and G2 with the ratio of G2:m-CTA3:M1:DHF = 1:50:2000:4000 to obtain a low dispersity di-block copolymer P10 (Mn,SEC (DMF) = 21.86 kDa, Đ = 1.32, (Fig. 4C and D).
 | ||
| Fig. 4 (A) SEC (CHCl3) traces of the m-CTA2 (blue) and the diblock copolymer P8 (red) (B) DOSY NMR spectra (400 MHz, CDCl3) of P8 (C) SEC (DMF) traces of the m-CTA3 (blue) and the diblock copolymer P10 (red) (D) DOSY NMR spectra (400 MHz, CDCl3) of P10. | ||
Next, we focused our attention on synthesizing a more complex A–B–C type triblock terpolymer. To achieve this, the PEG-PCL based macro-CTA m-CTA4 was synthesized via ROP using the Sn(Oct)2 catalyst system (Mn,SEC (DMF) = 4.16 kDa, Đ = 1.23).44m-CTA4 was employed for the catalytic living copolymerization of M1 and DHF to obtain P11 (Mn,SEC (DMF) = 27.68 kDa, Đ = 1.35). SEC analysis showed a complete disappearance of the m-CTA4 signal along with a significant increase in the molecular weight of the final tri-block terpolymer. Additionally, DOSY NMR spectroscopy supported the successful synthesis of the PEG-PCL-ROMP tri-block terpolymer (Fig. 5A and B).
 | ||
| Fig. 5 (A) SEC (DMF) traces of polyethylene glycol monomethyl ether 2000 (green), m-CTA4 (blue) and the triblock terpolymer P11 (red) (B) DOSY NMR spectra (400 MHz, CDCl3) of P11. | ||
Conclusions
In conclusion, we have successfully developed a straightforward strategy for the synthesis of A–B or A–B–C type block co- and terpolymers catalytically (using up to 80 times less Ru complex than in a classical metathesis polymerization) exploiting the regioselectivity and high metathesis activity of vinyl ethers as macro-chain transfer agents. Polystyrene, polycaprolactone and polylactide based macro –CTAs (m-CTA1–3) were employed in the syntheses of PS-ROMP, PCL-ROMP, PLA-ROMP di-block copolymers under catalytic living ROMP conditions. A poly ethylene glycol-polycaprolactone based diblock macro-CTA (m-CTA4) was also used to grow a ROMP polymer block yielding a PEG-PCL-ROMP triblock terpolymer using substoichiometric amounts of ruthenium complex for the very first time. We believe that this versatile method will provide a sustainable pathway towards the catalytic synthesis of different metathesis-based degradable block copolymers finding widespread applications in numerous fields.Author contributions
A. M. and A. F. M. K. designed the experiments. A. M. carried out all the syntheses, experiments, and analyses. A. M. wrote the first draft of the manuscript. A. F. M. K. edited the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swiss National Science Foundation (SNSF) and the Fribourg Center for Nanomaterials (FriMat) for financial support.References
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- H. Feng, X. Lu, W. Wang, N. G. Kang and J. W. Mays, Polymers, 2017, 9(10), 494 CrossRef PubMed.
- N. Kumar, M. N. V. Ravikumar and A. J. Domb, Adv. Drug Delivery Rev., 2001, 53, 23–44 CrossRef CAS PubMed.
- M. Karayianni and S. Pispas, J. Polym. Sci., 2021, 59, 1874–1898 CrossRef CAS.
- V. Agrahari and V. Agrahari, Drug Discovery Today, 2018, 23, 1139–1151 CrossRef CAS PubMed.
- T. H. Epps and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- S. Pal, A. Mandal, L. Hong, R. D. Ortuso, A. Petri-Fink, S. Salentinig and A. F. M. Kilbinger, Macromolecules, 2022, 55, 2854–2860 CrossRef CAS.
- M. A. Hillmyer and F. S. Bates, Macromolecules, 1996, 29, 6994–7002 CrossRef CAS.
- M. A. Dyson, A. M. Sanchez, J. P. Patterson, R. K. O'Reilly, J. Sloan and N. R. Wilson, Soft Matter, 2013, 9, 3741–3749 RSC.
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2166–2170 CrossRef CAS.
- D. Benoit, E. Harth, P. Fox, R. M. Waymouth and C. J. Hawker, Macromolecules, 2000, 33, 363–370 CrossRef CAS.
- J. A. Opsteen and J. C. M. Van Hest, Chem. Commun., 2005, 57–59 RSC.
- X. Wang, Y. Luo, B. Li and S. Zhu, Macromolecules, 2009, 42, 6414–6421 CrossRef CAS.
- L. A. L. Fliervoet, M. Najafi, M. Hembury and T. Vermonden, Macromolecules, 2017, 50, 8390–8397 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- T. L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- K. Nomura and M. M. Abdellatif, Polymer, 2010, 51, 1861–1881 CrossRef CAS.
- L. M. Pitet, J. Zhang and M. A. Hillmyer, Dalton Trans., 2013, 42, 9079–9088 RSC.
- M. A. Sowers, J. R. Mccombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- E. M. Kolonko, J. K. Pontrello, S. L. Mangold and L. L. Kiessling, J. Am. Chem. Soc., 2009, 131, 7327–7333 CrossRef CAS PubMed.
- V. Komanduri, Y. Janpatompong, R. Marcial-Hernandez, D. J. Tate and M. L. Turner, Polym. Chem., 2021, 12, 6731–6736 RSC.
- A. K. Diallo, L. Annunziata, S. Fouquay, G. Michaud, F. Simon, J. M. Brusson, S. M. Guillaume and J. F. Carpentier, Polym. Chem., 2014, 5, 2583–2591 RSC.
- R. M. Thomas and R. H. Grubbs, Macromolecules, 2010, 43, 3705–3709 CrossRef CAS.
- M. K. Mahanthappa, F. S. Bates and M. A. Hillmyer, Macromolecules, 2005, 38, 7890–7894 CrossRef CAS.
- C. W. Bielawski, T. Morita and R. H. Grubbs, Macromolecules, 2000, 33, 678–680 CrossRef CAS.
- P. Lu, N. M. Alrashdi and A. J. Boydston, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2977–2982 CrossRef CAS.
- P. Lu and A. J. Boydston, Polym. Chem., 2019, 10, 2975–2979 RSC.
- M. Yasir, P. Liu, I. K. Tennie and A. F. M. Kilbinger, Nat. Chem., 2019, 11, 488–494 CrossRef CAS PubMed.
- I. Mandal, A. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2022, 11, 1384–1389 CrossRef CAS PubMed.
- J. D. Feist, D. C. Lee and Y. Xia, Nat. Chem., 2022, 14, 53–58 CrossRef CAS PubMed.
- A. Mandal, I. Mandal and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2023, 62(4), e202211842 CrossRef CAS PubMed.
- Y. Minenkov, G. Occhipinti and V. R. Jensen, Organometallics, 2013, 32, 2099–2111 CrossRef CAS.
- Z. Liu and J. D. Rainier, Org. Lett., 2005, 7, 131–133 CrossRef CAS PubMed.
- Q. Li, L. Zhang, L. Bai, J. Miao, Z. Cheng and X. Zhu, Prog. Chem., 2010, 22, 2079–2088 CAS.
- A. Mandal, I. Mandal and A. F. M. Kilbinger, Macromolecules, 2022, 55, 7827–7833 CrossRef CAS.
- S. S. Liow, V. T. Lipik, L. K. Widjaja and M. J. M. Abadie, J. Polym. Res., 2012, 19, 9748 CrossRef.
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. D. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Polymers, 2021, 13, 1822 CrossRef CAS PubMed.
- W. Xiong, L. Peng, H. Chen and Q. Li, Int. J. Nanomed., 2015, 10, 2985–2996 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00387f |
| This journal is © The Royal Society of Chemistry 2023 |