
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine-norbornene versus azide-norbornene ligation: evaluating the toolbox for polymer–polymer coupling†‡
Steffen A.
Busche
,
Stefan
Peplau
,
Luc
Zuhse
,
Désirée
Steimer
,
Dennis D.
August
and
Hans G.
Börner
 *
*
Humboldt-Universität zu Berlin, Department of Chemistry, Laboratory of Organic Synthesis of Functional Systems, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: h.boerner@hu-berlin.de
First published on 26th May 2023
Abstract
The selective ligation chemistry of the stable norbornene (Nb) with an asymmetric tetrazine (Tz), accessible in a one-pot reaction is adapted to the ligation of polymer segments yielding block copolymers. The Tz/Nb inverse electron-demand Diels–Alder reaction (IEDDA) is compared with the more classical azide (Az)-norbornene Huisgen 1,3-cycloaddition (CA) to ligate end-functionalized polymers. An endo/exo-norbornene end-functionalized polystyrene (PS1.5k-Nb) and polyethylene glycols with either tetrazine or aryl azido functionalities (PEG3k/5k-Tz, PEG3k/5k-Az) are synthesized. While the Tz/Nb IEDDA proceeds cleanly and leads to full conversion in stochiometric mixtures, the Az/Nb CA requires less effort but suffers from slower coupling kinetics and some side reactions. The coupling of PS1.5k-Nb with PEG3k/5k-Az requires 1H-NMR analysis to follow the kinetics and the products suffer from incomplete conversion as well as slight decomposition of the Az-moieties. In contrast, the ligation of PS1.5k-Nb with PEG3k/5k-Tz is robust. The reaction, which is conveniently monitored by UV/Vis spectroscopy, yields the desired PS-block-PEG in high yields and may offer potential for easy scale-up.
Introduction
Since the advent of highly specific ligation reactions,1 the synthetic toolbox for macromolecules with complex architectures has been dramatically expanded through the exploitation of effective segmental coupling strategies.2–9 Macromolecular chemistry has benefited significantly from developments in the biomolecular field providing peptide-ligation,10,11 antibody conjugation12 or DNA/RNA labeling strategies.13,14 Such ligation reactions are often bioorthogonal, stoichiometric, and ultrafast, and examples range from metal catalyzed Huisgen 1,3-dipolar cycloadditions (CAs),15–17 to thiol-ene18,19 or thiol-quinone-Michael additions,20 to Staudinger ligation,21,22 up to Diels–Alder reactions,23–25 and strain- or light promoted 1,3-cycloadditions.26,27 The use of these chemistries has enabled effective segmental coupling strategies, that combine ease of analysis and purification of the polymer blocks prior to the coupling with precise definition of the coupling sites in the polymer precursor blocks.28 Thus, if provides access routes to various block copolymers,29 macrocycles,30 graft co-polymers11,31 and molecular brushes.32,33 Molecular biology requires ligation routes to proceed highly efficient under dilute conditions, ideally tolerating all abundant biofunctionalities and often targeting submicro- or nanomolar scales. In polymer chemistry, however, the focus is on pragmatic coupling reactions where availability and stability of the coupling reactants at larger scales are important criteria. The highly efficient inverse electron-demand Diels–Alder ligation (IEDDA), in which tetrazines (Tz) react ultrafast with trans-cyclooctenes,34,35 has been adapted by Du Prez and O'Reilly et al. by replacing the latter with the readily available norbornene (Nb) derivative.29,36 The ligations of Nb-end-functional polylactide36 or polystyrene (PS)25 with 3,6-di-2-pyridyl-1,2,4,5-tetrazine-endfunctional polyethylene glycol (PEG) proceeded highly rapid in a stoichiometric manner at room temperature and could be easily monitored by the disappearance of the pink Tz color.Nb derivatives have been found to be less reactive than trans-cyclooctene, but meet the criteria of stability and, most importantly, low cost accessibility. The electron rich 3,6-di-2-pyridyl-1,2,4,5-tetrazine provides suitable coupling kinetics, but the three step synthesis is tedious and may limit scalability.29
Here we report the adaptation of an asymmetric Tz derivative for polymer segment ligation by using Tz units accessible in one-pot synthesis. Tz end-functional polymer blocks were coupled to Nb end-functional polymers and the ligation was compared to the polymer segment coupling of aryl-Az and Nb derivatives, with both reactions requiring no additive or catalyst. A series of chain-end-reactive PEG3k/5k-Tz/Az polymers were coupled to an end-functional PS1.5k-Nb block to investigate differences in coupling kinetics and to elucidate accessibility, reactivity, stability and optimal reaction conditions (Fig. 1).
 | ||
| Fig. 1 Overview of synthetic pathway to (a) Tz-COOH and (b) Az-COOH as well as (c) idealized polymer–polymer ligation of PS1.5k-Nb with either PEG3k/5k-Tz (IEDDA) or PEG3k/5k-Az (CA). For full mechanisms of IEDDA and CA, see ESI Schemes S10 and S11.† | ||
Experimental section
See the ESI† for detailed synthesis procedure of each precursor molecule, polymer and their analysis methods. Full UV/Vis, NMR as well as SEC kinetics are also included.Results and discussion
Syntheses of polymers with Nb-, Tz- or Az-chain end functionality
To investigate the block segment ligation, a series of different polymers with the required chain end functionalities were synthesized. A polystyrene (PS) with Nb at the α-chain end was accessed by a Reversible Addition Fragmentation Transfer (RAFT) controlled polymerization of styrene, using a Nb bearing chain transfer agent (Nb-CTA).37 The PS was obtained in a controlled manner and the CTA aminolysis proceeded cleanly in the presence of hexylamine/ t-butyl acrylate to avoid the side reactions during the Az ligation step.38 The PS1.5k-Nb was isolated with Mn = 1500 g mol−1 and Đ of 1.08, as determined by size exclusion chromatography (SEC). 1H-NMR spectroscopy revealed the high degree of Nb-functionalization. An Mn,NMR of 1850 g mol−1 and a degree of polymerization of DPn = 14 was calculated by chain end group analysis comparing the integral intensities of the Nb double bond protons at 6.19–5.87 ppm with the aromatic styrene signals at 7.45–6.27 ppm (ESI Fig. S14†).A polyethylene glycol (PEG)-attached peptide synthesis resin (PAP resin) with ω-amino functional PEG-segments used to easily access the PEG-Tz and PEG-Az segments in a straightforward manner by simply coupling functional benzoic acid derivatives with PyBOP/DIPEA peptide-synthesis protocols. The required 4-(6-methyl-1,2,4,5-tetrazin-3-yl)-benzoic acid (Tz-COOH) was readily accessible in a one-pot procedure from 4-cyanobenzoic acid, hydrazine monohydrate and acetonitrile, giving the asymmetric tetrazine in 76% yields and 99% purity. The benzoic acid derivative or the 4-azidobenzoic acid (Az-COOH) was coupled to the supported PEG-NH2 and a negative Kaiser test for both coupling reactions proved the absence of free amine groups on the resins, indicating a quantitative functionalization of the PEG with Tz or Az. After liberation of the PEG-Tz and the PEG-Az from the support, the resulting polymers were isolated and 1H-NMR spectra confirmed the successful introduction of both reactive functionalities (ESI Fig. S16 and S18†). SEC measurements revealed a Mn = 3200 g mol−1 and Đ of 1.06 for the PEG3k-Tz and Mn = 3300 g mol−1 and Đ of 1.06 for the PEG3k-Az.
The strategy could be extended to solution protocols, where α-methoxy-ω-amino PEG (MeO-PEG5k-NH2) was coupled to the two different benzoic acid derivatives (cf. ESI†). After 22 h of coupling at room temperature, the product was purified by careful precipitation, yielding PEG5k-Tz with Mn = 4700 g mol−1 (Đ = 1.05) and PEG5k-Az with Mn = 4800 g mol−1 (Đ = 1.06). Both isolated products show a negative Kaiser test, demonstrating quantitative functionalization of the MeO-PEG-NH2, which is consistent with 1H-NMR analyses showing the typical resonances for either Tz or Az moieties (ESI Fig. S17 and S19†).
Comparison between Nb-Tz and Nb-Az ligation
It is well known that polymer-coupling strategies suffer from a reduced chain end reactivity.39 To validate the ligation chemistry and select appropriate conditions, model reactions were studied (Fig. 2a). The coupling of low molecular weight compounds Tz-COOH or Az-COOH with 5-norbornene-2-carboxylic acid (Nb-COOH) takes place in DMF under air. Pseudo-1st-order conditions were established by using excess Nb-COOH (molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 Tz/Az:Nb) to obtain quantitative conversion of the tetrazine and azido compounds. The coupling of the asymmetric tetrazine could be conveniently monitored by UV/Vis-spectroscopy at λmax = 538 nm, while 1H-NMR spectroscopy was required to follow the disappearance of the aromatic Az-COOH resonances.
:1.6 Tz/Az:Nb) to obtain quantitative conversion of the tetrazine and azido compounds. The coupling of the asymmetric tetrazine could be conveniently monitored by UV/Vis-spectroscopy at λmax = 538 nm, while 1H-NMR spectroscopy was required to follow the disappearance of the aromatic Az-COOH resonances.
 | ||
| Fig. 2 IEDDA versus CA ligation kinetics of (a) low molecular weight compounds (Tz-COOH/Az-COOH + Nb-COOH) and (b) high molecular weight polymers (PEG-Tz/PEG-Az + PS-Nb) monitored by spectroscopy to determine pseudo-1st-order rate constants k (conditions: 1.6:1 molar ratio of Nb:Tz/Az in DMF). | ||
It should be noted, that the reaction of Nb with Az produces triazolines, which can decompose partially to aziridines at elevated temperatures, as described in the literature.40,41
First, both tetrazine and azido compounds were found to be stable at reaction temperatures of 80 °C for at least 10 h, as no decomposition was observed. Remarkably, the Tz-COOH/Nb-COOH ligation reaches at 80 °C reached 90% conversion after 12 min, and quantitative coupling was achieved in less than 1 h. In comparison, the Az-COOH/Nb-COOH coupling at 80 °C proceeds much slower. Note that the reaction rates were quite similar to those of the Tz-COOH/Nb-COOH ligation at room temperature, where 90% conversion was found after 3 h and quantitative conversion after 6 h. Despite the fact that azido compounds are readily accessible on a larger scale, e.g. by halogen-azide exchange,33,42 the Az-COOH/Nb-COOH coupling proceeds very slowly at room temperature, approaching full conversion after 7 days (ESI Fig. S2†).
As expected, polymer-small molecule ligation is slower than small molecule coupling due to steric effects, despite the use of the same chemistry. It reflects the robustness of the chemistry, that the tetrazine/norbornene IEDDA proceeds at rather similar rates regardless of whether the Tz is the polymeric or the small molecule species (Fig. 2b). However, using a stochiometric ratio of the functionalities (Tz/Nb 1:1) at 80 °C, both coupling reactions (PEG3k-Tz + Nb-COOH or PS1.5k-Nb + Tz-COOH) reach 90% conversion after about 15–20 h and quantitative coupling is approached after 55 h. Increasing the tetrazine component to 10 equivalent excess accelerates the coupling considerably and quantitative conversion of PS1.5k-Nb was reached after 10 h (ESI Fig. S3†). The polymeric PEG-Tz components show superior thermal stability compared to the PEG-Az analog, which decomposes slowly and shows a 10% decay after 6.5 days (Fig. 2b). The azido coupling reaction was also much slower in this setting than the similar tetrazine ligations. Within the entire set of Nb/Az reactions, quantitative conversion was achieved only after 72 h, when 10 equivalents excess of Az-COOH was reacted with PS1.5k-Nb at 80 °C (ESI Fig. S4†). The isolated reaction product of PS1.5k-Nb with Az-COOH was analysed by MALDI-TOF-MS, suggesting the formation of triazoline- and aziridine-linkages, where the former series had higher signal intensity (ESI Fig. S37†).40 It should be noted, that the mass signal intensities in MALDI-TOF spectra are not directly quantitative results. Furthermore, no significant differences were found when the azido compound was the low or high molecular weight moiety.
The optimized reaction protocols were finally extended to conjugate polymer segments of PEG3k/5k-Tz and PS1.5k-Nb to obtain the desired PS-block-PEG copolymer as a model amphiphile.
The use of 0.005 M polymer solutions in DMF, at 80 °C and a molar ratio of functionalities of 1:1-ratio leads to the expected reduction of the reaction rates as indicated by the lower reaction constants (Fig. 3b). From the coupling kinetics, the PEG-Tz ligates very well with PS1.5k-Nb, reaching 90% conversion after 55–60 h and approaching quantitative reactions after 4 days (Fig. 3b). The molecular weight of PEG-Tz in the studied window of Mn = 3200–4700 has no dramatic effect on the coupling kinetics. However, SEC-trace analysis (Fig. 3a and ESI Fig. S33†) provided more insight into the evolution of the ligation product, where the PEG-Tz/PS1.5k-Nb coupling seems to be completed after about 57 h. Apparently, the resulting PEG5k-block-PS1.5k shows the development of a slight bimodality. However, this was already detectable in the commercial mPEG-NH2 precursor polymer. The appearance of a bimodality with the second peak located at twice the peak molecular weight most likely indicates a minor co-initiation of the ethylene oxide polymerization by water leading to H2N-PEG10k-NH2 after amination. This polymer would result in Tz-PEG10k-Tz with two PS1.5k-Nb attached. However, the overall dispersity of the resulting PEG-block-PS remained rather low at Đ = 1.15 and confirming the validity of using this route for the synthesis of well-controlled block copolymers.
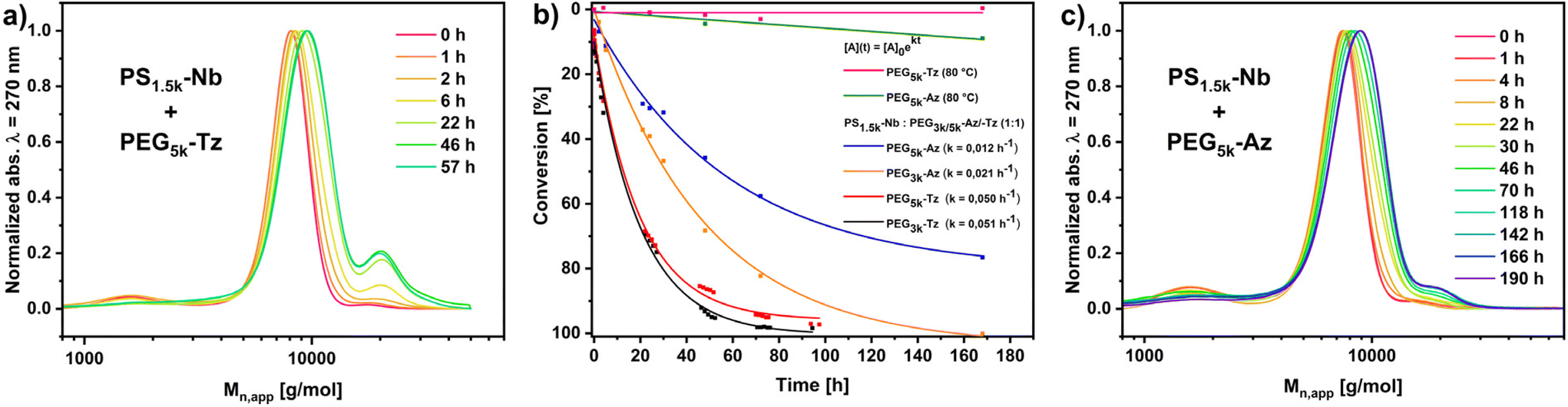 | ||
| Fig. 3 Polymer–polymer-ligation of PEG5k-Tz/Az with PS1.5k-Nb. (a) SEC-traced of the evolution of the PS1.5k-Nb PEG5k-Tz conjugation showing the disappearance of the PS1.5k-signal and shift of the PEG5k-signal; (b) time vs. conversion plot of polymer–polymer-ligation of PEG3k/5k-Tz/Az with PS1.5k-Nb monitored by UV/Vis and 1H-NMR-spectroscopy; (c) SEC-traced of the evolution of the PS1.5k-Nb PEG5k-Az conjugation showing the disappearance of the PS1.5k-signal and shift of the PEG5k-signal. [Conditions]: 1:1 ratio of PS1.5k-Nb: PEG3k/5k-Az/Tz in DMF at 80 °C, DMAc GPC, 50 °C, DMAc + LiBr. | ||
Considering the analogous ligation of PEG-Az and PS1.5k-Nb, which in any case proceeds much slower than the tetrazine variant, a pronounced effect of the PEG molecular weight on the coupling kinetics was evident. While PEG3k-Az reacted quantitatively with PS1.5k-Nb after 170 h, PEG5k-Az barely reached 80% conversion (Fig. 3b). MALDI-TOF-MS of the reaction product from PEG3k-Az and PS1.5k-Nb, confirmed the successful ligation. However, due to the complexity of two folded distributions, the resolution could only provide evidence of triazoline-linkages (ESI Fig. S38†). SEC was consistent with the kinetics, but also revealed a slightly bimodal ligation product PEG5k-block-PS1.5k (Fig. 3c). The lower degree of bimodality could be a result of the incomplete conversion, as the ABA triblock is expected to be formed slowly and preferentially at high conversion for steric reasons. This would be consistent with 1H-NMR spectroscopy, which shows that after 160 h of coupling of PS1.5k-Nb with PEG5k-Az, the resonances of Nb are still at 6.09 ppm and no complete conversion has occurred (ESI Fig. S20 and S21†).
In contrast to that, the 1H-NMR of the final coupling product PEG5k-block-PS1.5k from the Tz/Nb ligation shows the absence of the typical Tz- and Nb-resonances at 8.66 and 6.09 ppm, respectively (ESI Fig. S22 and S23†). This demonstrates the efficiency of the Nb/Tz IEDDA compared to the analogous Nb/Az CA chemistry.
Finally, to confirm the ligation of both PEG and PS blocks a diffusion-ordered spectroscopy (DOSY43) NMR analysis was performed, comparing the starting polymers with each of the resulting ligation products (ESI Fig. S24–S32†). Significant shifts in the diffusion coefficients were observed for both ligation types and no signals from unreacted precursors were observed within the error of the method (ESI Fig. S29 and S32†). Due to the increase in molecular weight from the PS1.5k-Nb to the PS-block-PEG product the diffusion coefficient increases from logD = −9.6 m2 s−1 (PS1.5k-Nb) to −9.9 m2 s−1 (PS1.5k-b-PEG3k) and −10.0 m2 s−1 (PS1.5k-b-PEG5k), which clearly underlines the successful ligation reactions.
Conclusion
A polymer ligation chemistry for segment-segment coupling was adapted, taking advantage of the easy accessibility of asymmetric tetrazine (Tz) moieties and demonstrating the ability to effectively ligate to a stable and commercially available norbornene (Nb) derivative. The strategy was compared to the alternative route of reacting azido- (Az) with norbornene derivatives. The Nb/Tz chemistry followed an inverse electron-demand Diels–Alder (IEDDA) mechanism, which shows advantages over the Nb/Az cycloaddition (CA). Stochiometric IEDDA ligation of PEG-Tz and PS-Nb allowed for quantitative solution coupling, resulting in the expected PEG-block-PS copolymer. In contrast, the analogous Nb/Az CA route suffered from significantly slower coupling kinetics of the PEG-Az and PS-Nb, which plateaued at incomplete conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. Busche and S. Peplau contributed equally to this work. The authors thank Dr A. Dallmann (HU Berlin) for the setup of the DOSY-NMR experiments and Carolin M. Schröter (HU Berlin) for the MALDI-TOF-MS measurements. This work was supported by the German Research Foundation (DFG; grant: Copy Mussel Poly, BO 1762/12-1, AOBJ 668918).References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed. Engl., 2011, 50, 60–62 CrossRef CAS PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burked, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- O. S. Taskin, B. A. Temel, M. A. Tasdelen and Y. Yagci, Eur. Polym. J., 2015, 62, 304–311 CrossRef CAS.
- C. Loth, L. Charles, J.-F. Lutz and M. Nerantzaki, ACS Macro Lett., 2021, 10, 481–485 CrossRef CAS PubMed.
- S. Vandewalle, S. Billiet, F. Driessen and F. E. Du Prez, ACS Macro Lett., 2016, 5, 766–771 CrossRef CAS PubMed.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- Z. Ni, L. Zhou, X. Li, J. Zhang and S. Dong, PLoS One, 2015, 10, e0141918 CrossRef PubMed.
- P. Wilke, T. Kunde, S. Chattopadhyay, F. E. Du Prez and H. G. Börner, Chem. Commun., 2017, 53, 593–596 RSC.
- A. Maggi, E. Ruivo, J. Fissers, C. Vangestel, S. Chatterjee, J. Joossens, F. Sobott, S. Staelens, S. Stroobants, P. Van Der Veken, L. Wyffels and K. Augustyns, Org. Biomol. Chem., 2016, 14, 7544–7551 RSC.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
- E. Paredes and S. R. Das, ChemBioChem, 2011, 12, 125–131 CrossRef CAS PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- A. Verch, H. Hahn, E. Krause, H. Cölfen and H. G. Börner, Chem. Commun., 2010, 46, 8938–8940 RSC.
- J.-F. Lutz, H. G. Börner and K. Weichenhan, Aust. J. Chem., 2007, 60, 410–413 CrossRef CAS.
- B. D. Fairbanks, D. M. Love and C. N. Bowman, Macromol. Chem. Phys., 2017, 218, 1700073 CrossRef.
- M. M. Stamenović, P. Espeel, W. V. Camp and F. E. Du Prez, Macromolecules, 2011, 44, 5619–5630 CrossRef.
- J. Horsch, P. Wilke, M. Pretzler, S. Seuss, I. Melnyk, A. Fery, A. Rompel and H. G. Börner, Angew. Chem., Int. Ed., 2018, 57, 15728–15732 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Brase, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- H. Akat, B. Gacal, D. K. Balta, N. Arsu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2109–2114 CrossRef CAS.
- A. Beloqui, S. R. Mane, M. Langer, M. Glassner, D. M. Bauer, L. Fruk, C. Barner-Kowollik and G. Delaittre, Angew. Chem., Int. Ed. Engl., 2020, 59, 19951–19955 CrossRef CAS PubMed.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Börner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071–1074 CrossRef CAS PubMed.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chemistry, 2021, 27, 5057–5073 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- A. Kocaarslan, Z. Eroglu, Ö. Metin and Y. Yagci, Beilstein J. Org. Chem., 2021, 17, 2477–2487 CrossRef CAS PubMed.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- J. D. Megiatto Jr. and D. I. Schuster, J. Am. Chem. Soc., 2008, 130, 12872–12873 CrossRef PubMed.
- H. Durmaz, A. Dag, N. Cerit, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5982–5991 CrossRef CAS.
- Y. Shi, X. Wang, R. W. Graff, W. A. Phillip and H. Gao, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 239–248 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O’Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362–1366 CrossRef CAS PubMed.
- S. Cho, J. Son, I. Kim, H. Ahn, H.-S. Jang, S. H. Joo, K. H. Park, E. Lee, Y. Kim and S.-K. Ahn, Polymer, 2019, 175, 49–56 CrossRef CAS.
- P. Walker and W. A. Waters, J. Chem. Soc., 1962, 1632–1638 RSC.
- D. J. Lunn, E. H. Discekici, J. Read de Alaniz, W. R. Gutekunst and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2903–2914 CrossRef CAS.
- A. C. Oehlschlager, P. Tillman and L. H. Zalkow, Chem. Commun., 1965, 596–598 RSC.
- F. Sebest, L. Casarrubios, H. S. Rzepa, A. J. P. White and S. Díez-González, Green Chem., 2018, 20, 4023–4035 RSC.
- J.-F. Lutz, H. G. Börner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00320e |
| ‡ Dedicated to the creative polymer chemist Professor Yusuf Yagci, who brought together scientists as well as polymers. |
| This journal is © The Royal Society of Chemistry 2023 |