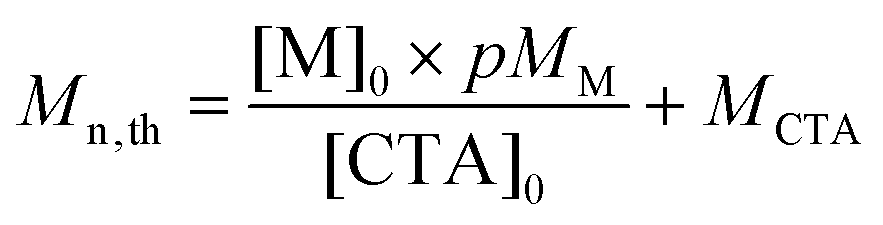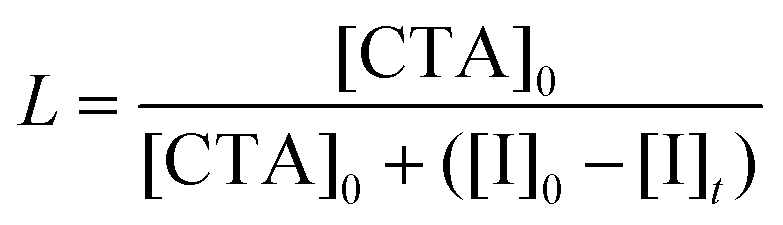DOI:
10.1039/D3PY00284E
(Paper)
Polym. Chem., 2023,
14, 2971-2978
Single-ion nano-features formed by a Li-containing block copolymer synthesized via PISA†
Received
17th March 2023
, Accepted 24th May 2023
First published on 1st June 2023
Abstract
Single-ion block copolymer nanoparticles of (poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(3-((trifluoromethane)sulfonamidosulfonyl) propyl methacrylate))-b-polystyrene, (PEGMA-co-PLiTFSI)-b-PS, were prepared using an efficient RAFT-controlled aqueous emulsion polymerization protocol. The macro-chain transfer agent (macroCTA) used, which stabilized the nanoparticles, is a lithium-ion based conductor that can be used for the formation of single-ion polymer electrolytes (SIPEs). This SIPE macroCTA was used as the stabilizer for the synthesis of PS under emulsion conditions at 70 °C, achieving near complete conversion. Kinetic studies revealed good control over the synthesis of the block copolymer (confirmed by NMR spectroscopy and GPC) and formation of spherical nanoparticles of different diameters depending on the length of the hydrophobic (PS) block (confirmed by DLS and TEM). The final high molecular weight diblock copolymer with narrow molecular weight distribution was used to prepare nanostructured films via combined solution-casting and solvent vapour annealing (SVA). Nanostructured films were characterised using AFM and GISAXS.
Introduction
Advances made in the development of controlled reversible deactivation radical polymerization (RDRP) have led to the ability to design and synthesize a variety of well-defined polymers of different structures.1–6 Among the RDRP polymerization techniques, reversible addition–fragmentation chain transfer (RAFT) polymerization is one of the most versatile and allows preparation of varied copolymers under facile and diverse conditions.5–8 The flexibility and efficiency of the RAFT method have boosted the possibility of making diverse amphiphilic block copolymers and the preparation of nanoparticles. Such block copolymer nanoparticles have often been made via post-polymerization techniques, such as solvent switch, pH switch, dialysis or thin film rehydration, typically resulting in the formation of mixed morphologies at very low concentrations (1 wt% or less).6,9 Polymerization-induced self-assembly (PISA) is a versatile method to synthesize a wide range of well-defined block copolymer nano-objects in the form of concentrated colloidal dispersions.10–13 In PISA, an insoluble block grows from one end of a soluble precursor block (macro chain transfer agent) in a suitable solvent that only dissolves the precursor. Once the insoluble block reaches a critical degree of polymerization, micellar nucleation occurs and ultimately leads to the formation of sterically stabilized diblock copolymer nano-objects. The unreacted monomer acts as a co-solvent and the high local monomer concentration within the monomer-swollen nanoparticles results in a relatively fast rate of polymerization.14,15 So far, many different combinations of shell- and core-forming blocks have been used in the PISA process and the resulting nano-objects have been used for many different types of applications from stem-cell growth, drug delivery and smart coatings to separation membranes.16–26
In recent years, solid polymer electrolytes (SPEs) have gained a great deal of attention as they offer advantages for improving the safety of existing lithium batteries.27,28 SPEs are excellent due to their mechanical strength and flexibility. SPEs can be easily prepared by solvent-casting, hot moulding or extrusion techniques.29 In this context, nanostructured block copolymer films are of interest in the design of lithium ion-containing solid copolymer electrolytes.30 These solid block copolymer electrolytes consist of ion-conducting nanochannels (e.g., lamellae, cylinders) capable of dissolving lithium salts, and a mechanically robust matrix. Current global understanding is that the lithium ion can move in the space left by the free volume of the polymer host, and the ionic conductivity (IC) is mediated by the local motion of the polymer chain segments above the glass transition temperature (Tg). Even if SPE based on semi-crystalline poly(ethylene oxide) (PEO) does not have a high IC at room temperature (RT), it is the most applied polymer host. Indeed, PEO is efficient in coordinating metal ions, due to the optimal distance and orientation of the ether oxygen atoms in its structure.31 However, to prevent the appearance of dendrites at the Li metal/electrolyte interface, the solid block copolymer electrolytes need to have a glassy domain with excellent mechanical strength, such as polystyrene (PS).30,32,33
Recently, a new class of SPEs labelled single-ion polymer electrolytes (SIPEs) has been developed where a highly delocalized anion is attached to the polymeric chain, endowing it with high mobility to the lithium counter cations. This results in the transference number of lithium being close to unity since only the lithium ions would be responsible for the ionic conductivity.34–36
The most studied SIPEs are highly delocalized anionic polyelectrolytes, including sulphonamide groups such as lithium poly(lithium 1-[3-(methacryloyloxy)-propylsulfonyl]-1-(trifluoromethylsulfonyl)imide) (PLiMTFSI) and the styrenic analogue, poly(styrene sulfonyl-(trifluoromethylsulfonyl)imide).37 However, SIPE homopolymers generally present low ionic conductivity properties because of their relatively high Tg values, limiting lithium mobility.38,39 To solve this problem Devaux et al. designed a block copolymer by combining the Li-containing block with a soft block of PEO.35 Long and co-workers optimized the relationships between chemical composition, morphology, mechanical properties and ionic conductivity by synthesizing triblock copolymers, the synthesis of which could be tedious, laborious and often costly.38 As mentioned earlier, PISA is an ideal method to prepare block copolymer particles with different morphologies at high solids content.37 PISA is also a straightforward method for the preparation of high molecular weight block copolymers due to the high monomer concentrations in the formed nanoparticles.14,40,41 This work looks into making high molecular weight SIPE-containing block copolymers via the PISA method. The first block was made through solution RAFT copolymerization of lithium 3-[(trifluoromethane)sulfonamidosulfonyl]propyl methacrylate (LiMTFSI) and polyethylene glycol methacrylate (PEGMA400). This water-soluble macro-chain transfer agent was then block extended using styrene via RAFT-controlled emulsion polymerization. The choice of a shell-forming monomer and the PISA method allowed for the synthesis of well-defined high molecular weight block copolymers in a fast manner as well as studying their self-assembly behaviour in solution in the same pot. To the best of our knowledge, this SIPE monomer and the solution self-assembly of amphiphilic block copolymer containing this monomer have never been used in a PISA formulation. Subsequently, nanostructured films were prepared using the synthesized block copolymer, P(PEGMA-co-LiTFSI)-b-PS, via a combination of solution casting and solvent vapour annealing.
Materials and methods
Materials
Polyethylene glycol monomethyl ether monomethacrylate (PEGMA400, 97.5% purity) was purchased from Polysciences. Lithium 3-[(trifluoromethane)sulfonamidosulfonyl]propyl methacrylate (LiMTFSI, 97% purity) was purchased from Specific Polymer (France). The RAFT agent 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPAD, 97% purity), 4,4′-azobis(4-cyanovaleric acid) (ACVA, 99% purity), styrene, solvents and deuterated solvents were purchased from Sigma-Aldrich. All reagents were used as received. The polystyrene-b-poly(2-vinyl pyridine)-b-polyethyleneoxide, PS-b-P2VP-b-PEO (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V:EO ≈ 65:21:13, 69.5 kg mol−1), used in this work, was purchased from Polymer Source Inc. (Canada).
:V:EO ≈ 65:21:13, 69.5 kg mol−1), used in this work, was purchased from Polymer Source Inc. (Canada).
Methods
Proton nuclear magnetic resonance spectroscopy (1H NMR).
1H NMR spectra were recorded on a 400 MHz Bruker Avance-400 spectrometer. The samples were solubilized in DMSO-d6 before analysis.
Dynamic light scattering (DLS).
The hydrodynamic radii were analysed by dynamic light scattering at 90° using an Anton Paar Litesizer TM 500. Samples were prepared at 0.1% w/w by diluting the sample with MilliQ water. The measurements were performed at 20 °C. An equilibrium step (1 minute) was set prior to each measurement.
Transmission electron microscopy (TEM).
TEM images were acquired using either a JEOL 1200 EXII (120 kV) or a JEOL 1400 P+ (120 kV). 10 μL of diluted PISA suspension (0.1% w/w) was deposited onto a carbon coated grid for 30 s and then blotted with filter paper to remove excess solution. Afterward, the sample-loaded grid was stained with 7 μL of 1% ammonium molybdate aqueous solution for 15 s before removal with filter paper.
Gel permeation chromatography (GPC).
Polymer molar mass distributions were analysed using the following two GPC systems: (1) Varian PL-50 system fitted with 2 PolarGel M 300 × 7.5 columns thermostated at 50 °C. The mobile phase was DMAc containing 0.1% w/w LiCl at a flow rate of 0.8 mL min−1. The calibration was performed using near-monodisperse poly(methyl methacrylate) (PMMA) standards ranging from 550 to 1500000 g mol−1 (EasiVial-Agilent). (2) Viscotek TDA 305 triple detector array system fitted with 2 PolarGel M 300 × 7.5 columns thermostated at 35 °C. The mobile phase was THF containing 0.3% w/w toluene at a flow rate of 1 mL min−1. The calibration was performed using near-monodisperse polystyrene (PS) standards ranging from 1200 to 400000 g mol−1 (Malvern).
Fabrication of nanostructured block copolymer films
Unblended and blended single-ion BCP films were drawn onto (2 × 2 cm) silicon substrates by using a simple tape casting technique with a 250 μm gap from an 18 wt% terpolymer solution in a di-solvent mixture of 1,4-dioxane and tetrahydrofuran (DOX/THF: 1/1 by weight). THF is a good solvent for the P(PEGMAx-co-LiMTFSIy) macroCTA while the high boiling point DOX ensures the formation of homogeneous films. The self-assembly of BCP chains was promoted by exposing films for 3 h to a continuous stream of chloroform (CHCl3) vapour produced by bubbling nitrogen gas through the liquid solvent as described previously.42 This continuous flow system was used to control the CHCl3 vapour pressure in the chamber by dilution with a separate N2 stream so that a solvent vapour consisted of 32 sccm CHCl3 vapour and 8 sccm N2 (total, 40 sccm).
AFM and GISAXS characterization studies
Atomic force microscopy (AFM Nano-Observer, CS Instruments) was used in tapping mode to characterize the surface morphology of single-ion BCP films. Silicon cantilevers (PPP-NCH, Nanosensors) with a typical tip radius of ∼5 nm were used. The resonance frequency of the cantilevers was about 235 kHz. Prior to AFM measurements, unblended and blended single-ion BCP films were treated with a fluorine-based plasma in a home-made chamber to improve the AFM topographic image contrast (plasma conditions: 45 W, 75 mTorr CF4 and 90 s). GISAXS experiments were performed on the Dutch–Belgian Beamline (DUBBLE) at the European Synchrotron Radiation Facility (ESRF) station BM26B in Grenoble.42 A monochromatic beam of 12 keV was set using a Si(111) double crystal monochromator. The sample (typical size of 150 mm2) was exposed to the X-ray beam with an angle of incidence of 0.2°, which is above the critical angle of the polymer, ensuring full penetration of the X-ray beam into the material and hence analysis of the full volume of the sample. The 2D scattering patterns were collected with a PILATUS3 S 1 M detector. The scattering vector and the sample-to-detector distance were calibrated using silver behenate as the standard, obtaining a sample-to-detector distance of 7500 mm in order to achieve a q-range from 0.01 Å to 0.068 Å.
RAFT copolymerization of poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(3-[(trifluoromethane)sulfonamidosulfonyl]propyl methacrylate) P(PEGMAx-co-LiMTFSIy).
The synthesis of the P(PEGMAx-co-LiMTFSIy) macroCTA was defined according to the following ratio: [PEGMA]:[LiMTFSI]:[CPAD]:[ACVA]; [25]:[29]:[1]:[0.2] (Scheme 1). A round bottom flask equipped with a stirrer bar was charged with PEGMA (0.81 g, 1.97 mmol), LiMTFSI (0.81 g, 2.28 mmol), CPAD (22.7 mg, 0.078 mmol), ACVA (4.5 mg, 0.016 mmol) and 3.1 g of distilled water. The flask was sealed with a rubber septum and placed in an ice bath. The reaction mixture was degassed by bubbling nitrogen for 30 min and the flask was then immersed in a pre-heated oil bath at 70 °C for 24 h under magnetic stirring. To monitor the polymerization, aliquots at different time intervals were taken using a degassed syringe under a nitrogen atmosphere. After 24 h, the monomer conversions were calculated using 1H NMR (>99% for both) before freeze drying to obtain a pink powder. The 1H NMR of the purified polymer gave a final DP of x = 27 and y = 31. The polymer was also analysed using SECDMAc (Mn = 36 kg mol−1; Đ = 1.1).
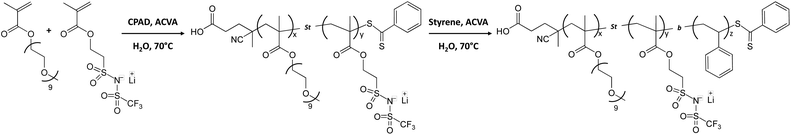 |
| | Scheme 1 Diblock copolymer synthesis of poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(lithium ((3-(methacryloyloxy)propyl)sulfonyl)((trifluoromethyl)sulfonyl)amide)-block-polystyrene, P(PEGMAx-co-LiMTFSIy)-b-PSzvia PISA at 10% (w/w) solids content in water at 70 °C. | |
Diblock synthesis of poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(lithium ((3-(methacryloyloxy)propyl)sulfonyl)((trifluoromethyl)sulfonyl)amide)-block-polystyrene P(PEGMA27-co-LiMTFSI31)-b-PSz.
The general procedure for the synthesis of P(PEGMA27-stat-LiMTFSI31)-b-PS365 was defined according to the following ratio: [Sty]:[PEGMA27-co-PLiMTFSI31]:[ACVA]; [384]:[1]:[0.2] (Scheme 1). A round bottom flask equipped with a stirrer bar was charged with styrene (0.71 g, 6.75 mmol), P(PEGMA27-co-LiMTFSI31) (0.36 g, 0.018 mmol), ACVA (1 mg, 0.004 mmol) and 9.5 g of distilled water (10% w/w). The flask was cooled down in an ice bath before degassing (N2 bubbling) for 30 min. The flask was then immersed in a pre-heated oil bath at 70 °C for 24 h under magnetic stirring. Aliquots were taken (using the procedure described earlier) at different time intervals to follow the polymerization kinetics. After 24 h of polymerization, the monomer conversions were checked using 1H NMR in a mixture of DMSO-d6 and THF. The conversion was calculated using the aromatic signals of the styrene and the polystyrene (conv. ∼ 97%). The residual styrene monomer was removed under vacuum before freeze-drying the mixture to obtain a pink powder. The final polymer, P(PEGMA27-co-LiMTFSI31)-b-PS365, was also analysed using SECDMAc (Mn = 84 kg mol−1; Đ = 1.31).
Calculation of Mn,th.
The theoretical number-average molar masses (Mn,th) were calculated using the following equation (eqn (1)):4| | 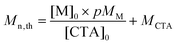 | (1) |
where [M]0 and [CTA]0 are the initial concentrations (in mol L−1) of the monomer and the chain transfer agent, p is the monomer conversion as determined by 1H NMR, and MM and MCTA are the molar masses (in g mol−1) of the monomer and the chain transfer agent, respectively.
Calculation of the theoretical number fraction of living chains (L).
The number fraction of living chains is calculated using eqn (2), neglecting all side reactions with oxygen:4| |  | (2) |
where [I]0 − [I]t is the concentration of the initiator consumed, which was calculated using the following equations when the kd value for ACVA at 70 °C was set as 2.1837 × 10−5:
| [I]0 − [I]t = [I]0 − [I]0−tkd |
Results and discussion
RAFT copolymerization of poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(lithium ((3-(methacryloyloxy)propyl)sulfonyl)((trifluoromethyl)sulfonyl)amide), P(PEGMA27-co-LiMTFSI31)
The polymerization between the poly(ethylene glycol) monomethyl ether monomethacrylate and lithium ((3-(methacryloyloxy)propyl)sulfonyl)((trifluoromethyl)sulfonyl)amide was performed by RAFT polymerization in water targeting a total molecular weight of 20 kg mol−1 with equal partition between the two monomers (10 kg mol−1 of each monomer).
After 200 minutes, 1H NMR analysis showed more than 99% conversion of both monomers (LiMTFSI and PEGMA) (Fig. S1†). Kinetic studies show a similar rate of consumption of the two monomers, indicating the formation of statistical copolymer PEGMA-co-PLiMTFSI. Furthermore, the SEC analysis of the kinetic samples showed that the copolymerization of the two monomers was well controlled, and the molecular weight increased with conversion (Fig. S2†). The final dispersity (Đ) of P(PEGMA27-co-LiMTFSI31) was 1.18. During the course of the polymerization, the DP (degree of polymerization) of each monomer was very close (DP = 27 for PEGMA; DP = 31 for PLiMTFSI) with the final Mn,th estimated to be 20.3 kg mol−1 (Table 1). The livingness of the macroCTA was estimated at 95% (≈25% of consumed initiator in 200 min) according to eqn (2), which should allow good chain extension with the second monomer (styrene).
Table 1 PEGMA and LiMTFSI conversion, theoretical and experimental number-average molar mass, degree of polymerization (DP) and dispersity of PEGMA-co-PLiMTFSI according to polymerization time
| Time (min) |
PEGMA conversion (%) and DP |
LiMTFSI conversion (%) and DP |
Polymera |
M
n,th
(kg mol−1) |
M
n,SEC
(kg mol−1) |
Đ
|
|
DPs calculated from conversion.
Calculated from NMR conversions.
Obtained from SEC analysis.
|
| 20 |
19 |
14 |
PEGMA5-co-PLiMTFSI4 |
3.7 |
10.1 |
1.12 |
| 5 |
4 |
| 30 |
21 |
24 |
PEGMA6-co-PLiMTFSI7 |
5.1 |
17.2 |
1.13 |
| 6 |
8 |
| 60 |
63 |
67 |
PEGMA16-co-PLiMTFSI19 |
13.2 |
27.0 |
1.17 |
| 16 |
21 |
| 120 |
96 |
95 |
PEGMA24-co-PLiMTFSI29 |
17.1 |
29.7 |
1.19 |
| 24 |
29 |
| 200 |
>99 |
>99 |
PEGMA27-co-PLiMTFSI31 |
20.3 |
34.7 |
1.18 |
| 25 |
29 |
Synthesis of poly(ethylene glycol) monomethyl ether monomethacrylate-co-poly(lithium ((3-(methacryloyloxy)propyl)sulfonyl)((trifluoromethyl)sulfonyl)amide)-block-polystyrene, P(PEGMA27-co-LiMTFSI31)-b-PS365via PISA
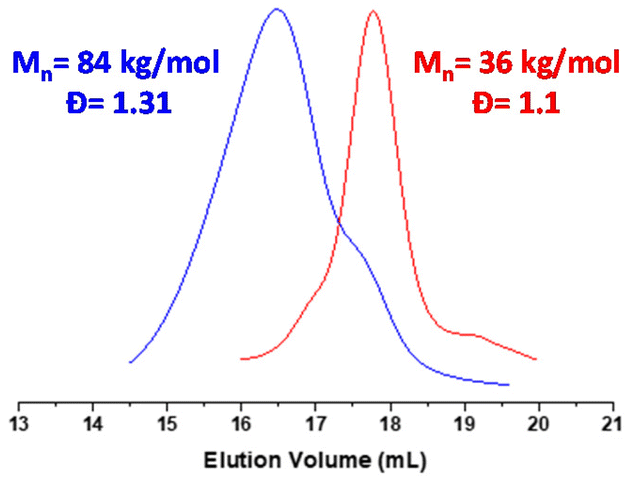
The macroCTA synthesized previously was used to control the polymerization of styrene under aqueous emulsion conditions. The polymerization was successfully conducted at 10% w/w with a target DP of 375. The polymerization was monitored by 1H NMR and by SEC. The conversions calculated using the 1H NMR spectra show more than 85% within the first 2 hours and near complete conversion (circa 97%) at 4 hours (Fig. S3 and S4†), yielding a final composition of P(PEGMA27-co-LiMTFSI31)-b-PS365. As seen in other PISA formulations, the conversion follows two different rates. The first regime marks the polymerization of styrene (between T zero and 100 minutes, 40% styrene conversion was achieved) while the faster second regime indicates the formation of nanoparticles and higher local concentration of styrene.43 The SEC analysis of the kinetic samples shows a linear increase of the molecular weight with conversion. The polymer dispersity increases from 1.17 to 1.31, indicating good control over the polymerization of styrene (Fig. 1). However, the SEC traces (Fig. 2) show a clear shoulder at lower molecular weight (higher elution volume), indicating partial chain extension. Deconvolution of this peak revealed that the final block copolymer contains 4–5% of unreacted macroCTA chains (Fig. S5†).
 |
| | Fig. 1 Evolution of number-average molecular weight and dispersity of the target P(PEGMA27-co-LiMTFSI31)-b-PS365. Mn,th (blue dashed line) represents the theoretical number-average molecular weight calculated according to eqn (1). | |
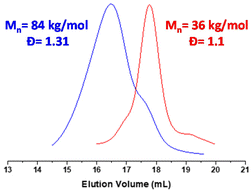 |
| | Fig. 2 SEC traces of P(PEGMA27-co-LiMTFSI31) (in red) and (PEGMA27-co-PLiMTFSI31)-b-PS365 (in blue) obtained using DMAc as eluent and RI detector. | |
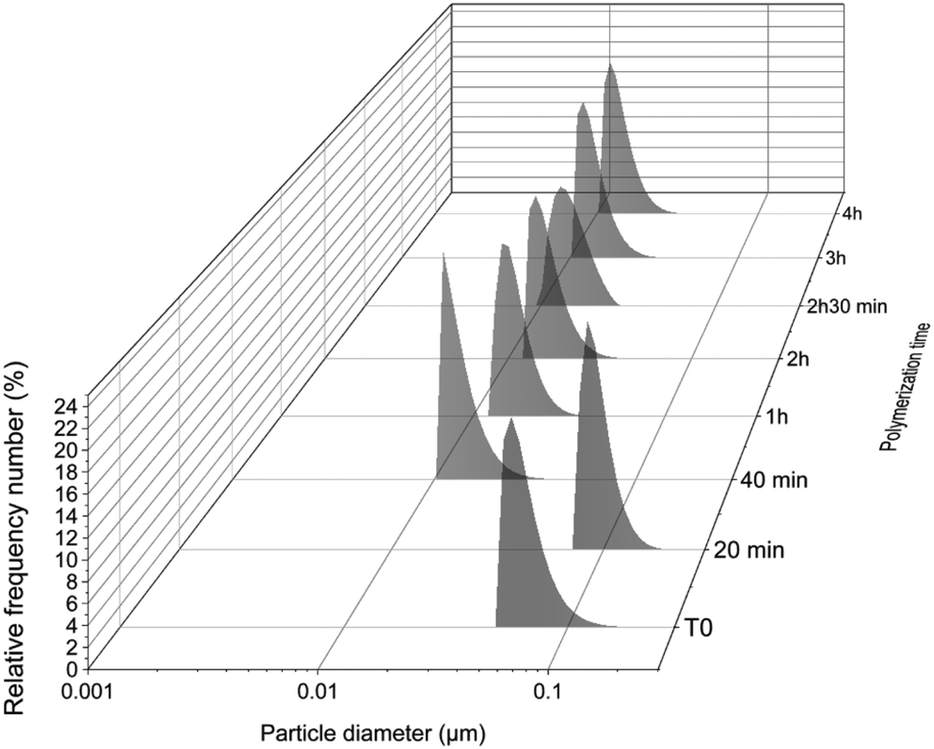
In order to verify the self-assembly of the block copolymer, the kinetic samples were analysed using DLS (Fig. 3). In the first 20 minutes, particles of around 100 nm were detected, which were probably styrene droplets as the polymerization was under an emulsion regime. As the polymerization carried on and PS chains were formed, the size of the particles was reduced by a factor of 10, reaching an average size of around 10–15 nm with polydispersities of 0.23 to 0.28 (Fig. S6†). The final particle size at the end of polymerization (almost full conversion) was 15 nm (PDI = 0.23). TEM studies of the kinetic samples (Fig. 4) revealed that, as is often the case with aqueous emulsion polymerization formulations, only kinetically trapped spheres were observed. The first sample at T = 20 (DPPS = 19) (Fig. 4A) shows ill-defined particles as the sample contains a lot of unreacted monomer (94.79%). However, as the length of PS increases, well-defined spherical nanoparticles are formed. At full conversion (DPPS = 365; Fig. 4G and H), spherical nanoparticles of 30–70 nm, with a distinct core–shell structure, are formed.
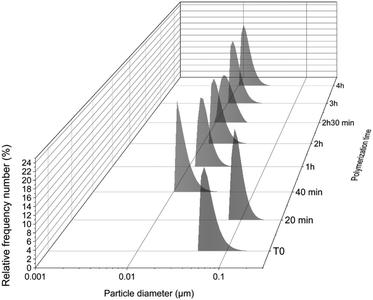 |
| | Fig. 3 Average particle diameter according to polymerization time of P(PEGMA27-co-LiMTFSI31)-b-PSz, where at T = 20 min, z = 19; T = 40 min, z = 50; T = 1 h, z = 97; T = 2 h, z = 328; T = 2 h 30 min, z = 336; T = 3 h, z = 359; and T = 4 h, z = 365. | |
 |
| | Fig. 4 TEM images of P(PEGMA27-co-LiMTFSI31)-b-PSz synthesized at 10% w/w solids in water at 70 °C; (A) n = 19, (B) n = 50, (C) n = 97, (D) n = 328, (E) n = 336, (F) n = 359 and (G and H) n = 365. TEM samples were prepared in MilliQ water at 0.1% w/w at room temperature. | |
As demonstrated in the past decade by results of various different studies,11,13,44 the versatility of RAFT-mediated PISA enables the facile synthesis of a wide range of block copolymers with variable block lengths and morphologies in solution.45–47 Here we also looked into how these PISA-prepared block copolymers self-assemble in thick film configurations. For the preparation of block copolymer films, the final kinetic sample, P(PEGMA27-co-LiMTFSI31)-b-PS365, was isolated and dissolved in a di-solvent mixture (DOX/THF: 1/1 by weight, 18 wt%). The solution was then cast on a silicon substrate using tape casting. An atomic force microscopy (AFM) topographic investigation of the prepared film (∼9 μm) revealed short-range ordered P(PEGMA27-co-LiMTFSI31) nanodomains, with a period (p) of circa 44 nm, after exposing the BCP layer to chloroform vapour for 3 h (Fig. 5). The nearest-neighbour distance distribution (NNDD) associated with P(PEGMA27-co-LiMTFSI31) nanodomains indicates a mean period, p, of 44 nm (full width at half maximum (FWHM) = 19 nm) (see Fig. S7†).48
 |
| | Fig. 5 A 3 × 3 μm AFM topographic image of a solvent-annealed (3 h, CHCl3) P(PEGMA27-co-LiMTFSI31)-b-PS365 film, treated with CF4 plasma, showing the formation of poorly ordered (black) P(PEGMA27-co-LiMTFSI31) domains drowned in (bright) PS matrix. Inset: a magnified AFM topographic view of the P(PEGMA27-co-LiMTFSI31)-b-PS365 film top surface that clearly shows short-range ordered structure with a period of ∼44 nm. Scale bars: 250 nm. | |
To improve the long-range order of the single-ion conducting nanochannels, a blending strategy was employed. A PEO-containing triblock terpolymer able to self-assemble into highly ordered lamellae with an out-of-plane (OP) orientation was used as the structural matrix. Three different films were prepared from a blend of PS-b-poly(2-vinyl pyridine)-b-poly(ethylene oxide) (PS-b-P2VP-b-PEO) with different amounts (0–50% w/w) of the P(PEGMA27-co-LiMTFSI31)-b-PS365. After the solvent-annealing step, the neat PS-b-P2VP-b-PEO film showed long-range ordered OP lamellae with a period of around 43 nm as measured from the diffused halo formed on the 2D-fast Fourier transform (2D-FFT, not shown here) (Fig. 6A).
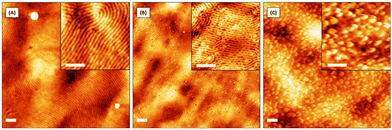 |
| | Fig. 6 3 × 3 μm AFM topographic images of solvent-annealed (3 h, CHCl3) PS-b-P2VP-b-PEO films doped with different amounts of P(PEGMA27-co-LiMTFSI31)-b-PS365: (A) 0 wt%, (B) 30 wt% and (C) 50 wt%. Insets: magnified views revealing the different morphologies obtained by varying the single-ion BCP amount within the blended PS-b-P2VP-b-PEO films treated with CF4 plasma. Scale bars: 250 nm. | |
To confirm the formation of OP lamellae within the neat PS-b-P2VP-b-PEO films over large areas, grazing incidence small angle X-ray scattering (GISAXS) measurements were also performed. The 2D GISAXS pattern of the neat film and its associated horizontal 1D line-cut profile at the Yoneda peak position are shown in Fig. 7. The horizontal 1D line-cut profile exhibits diffraction peaks with a positional ratio of 1:2:3 that matches well with the formation of vertically aligned lamellae. From the first-order peak, q*, located at 0.0143 Å−1, an inter-lamellae distance, d, of 43.9 nm was estimated (d = 2π/q*), matching the value extracted from the AFM topographic image. Addition of 30 wt% of single-ion BCP induced the formation of a morphology consisting of discrete perforations within OP lamellae with p ∼ 40 nm as extracted from the 2D-FFT (Fig. 6B). In general, the perforation formed within a perforated lamellar (PL) phase follows either a conventional hexagonal or tetragonal symmetry.49 In the AFM topographic image, a rather patchy surface is observed, which is due to the formation of perforations with ill-defined morphology. The GISAXS results presented in Fig. 8 also suggest that the perforations within the PL structure have short-range ordering since the horizontal 1D line-cut profile (along the Yoneda peak of the BCP film) mainly consists of the diffraction peaks having a positional ratio of 1:2:3:4, which corresponds to an OP lamellar stacking (p ∼ 40.9 nm, q* = 0.0153 Å−1). The small shoulder located at ∼1.15q* probably corresponds to a local hexagonal arrangement of perforations with a period (phex = (2/√3) 2π/qhex ∼ 40.4 nm, qhex ∼ 1.15q*) fitting well with the lamellar periodicity. Conversely, doping the PS-b-P2VP-b-PEO films with 50 wt% of the same single-ion BCP resulted in the loss of the nanostructure as attested both by the AFM topographic image presented in Fig. 6C and its associated featureless GISAXS profile at the Yoneda peak position (Fig. S8†). These results indicate that long-range ordered nanochannels could only be achieved at low single-ion BCP loading, potentially affecting the overall ionic conductivity of the blended films.
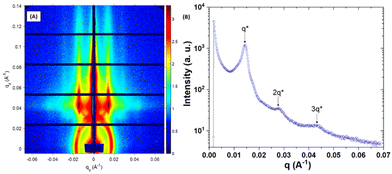 |
| | Fig. 7 (A) Synchrotron 2D-GISAXS pattern obtained at room temperature and (B) its corresponding intensity profile of a neat PS-b-P2VP-b-PEO film exposed to CHCl3 vapour for 3 h. The GISAXS pattern line-cut along qy integrated between qz values 0.037–0.043 Å−1 revealed an OP lamellar structure with a period of ∼43.9 nm (q* = 0.0143 Å−1). | |
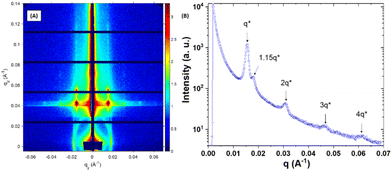 |
| | Fig. 8 (A) Synchrotron 2D-GISAXS pattern obtained at room temperature and (B) its corresponding intensity profile of a solvent-annealed (3 h, CHCl3) PS-b-P2VP-b-PEO film blended with 30 wt% of P(PEGMA27-co-LiMTFSI31)-b-PS365. The GISAXS pattern line-cut along qy integrated between qz values 0.037–0.043 Å−1 revealed an OP lamellar structure with a period of ∼40.9 nm (q* = 0.0153 Å−1) as well as the presence of an additional peak located at 1.15q* that would correspond to the first-order (10) reflection of a hexagonal lattice formed by the perforations. | |
Conclusions
The synthesis of well-defined P(PEGMA-co-LiMTFSI) and P(PEGMA-co-LiMTFSI)-b-PS copolymers were performed in two steps. First, RAFT solution polymerization was used to synthesize the water-soluble SIPE containing the first block. Then, a surfactant-free RAFT emulsion polymerization of styrene was performed to obtain the high molecular weight diblock copolymer. The kinetic studies of both blocks confirmed good control over the polymerization as indicated by low dispersity indexes for both blocks. The resulting amphiphilic block copolymer formed spherical nanoparticles in solution during the polymerization. The size of the spherical nanoparticles increased with the increasing length of the growing hydrophobic PS block. Only spherical particles were formed. This is not surprising considering the charged nature of the stabilizing block. The diblock copolymer with the highest molecular weight was then used to prepare self-assembled films. The films were prepared via a combination of drop-casting and SVA. Unblended block copolymer films microphase-separated into structures with poorly ordered morphology. Upon blending 30 wt% of the synthesized block copolymer with PS-b-P2VP-b-PEO, a film with perforated lamellar structure with an out-of-plane orientation was formed. Such an ordered structure would be highly desired to manufacture solid state polymer electrolytes for battery applications.
Author contributions
We, the undersigned, declare that this manuscript is original, has not been published before and is not currently being considered for publication elsewhere. We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us. We understand that the corresponding author is the sole contact for the editorial process. She is responsible for communicating with the other authors about progress, submissions of revisions and final approval of proofs.
Conceptualization: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, Karim Aissou, and Mona Semsarilar; Methodology: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, and Mona Semsarilar; Validation: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, Karim Aissou, and Mona Semsarilar; Formal analysis: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, Karim Aissou, and Mona Semsarilar; Investigation: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, and Arnaud Chaix; Resources: Karim Aissou and Mona Semsarilar; Writing of the original draft: Hamza Chouirfa and Belkacem Tarek Benkhaled; Writing – review and editing: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, Karim Aissou, and Mona Semsarilar; Visualization: Hamza Chouirfa, Chaimaa Gomri, Belkacem Tarek Benkhaled, Arnaud Chaix, Karim Aissou, and Mona Semsarilar; Supervision: Mona Semsarilar; Project administration: Mona Semsarilar; Funding acquisition: Karim Aissou and Mona Semsarilar.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge the French national research agency (ANR) for funding this work under the contract references of PEPPISA-19-CE06-0011-01 and AFM-Ring-18-CE09-00xx. The authors would also like to thank MUSE (University of Montpellier) for providing financial help under reference ERC-IMOPS.
References
- N. Ballard and J. M. Asua, ACS Macro Lett., 2020, 190–196 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- G. Moad and E. Rizzardo, Polym. Int., 2019, 69, 19–22 Search PubMed.
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811–820 CrossRef CAS PubMed.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- M. Semsarilar, E. R. Jones and S. P. Armes, Polym. Chem., 2014, 5, 195–203 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- L. Upadhyaya, M. Semsarilar, R. Fernández-Pacheco, G. Martinez, R. Mallada, A. Deratani and D. Quemener, Polym. Chem., 2016, 7, 1899–1906 RSC.
- L. Upadhyaya, B. Oliveira, V. J. Pereira, M. T. Barreto Crespo, J. G. Crespo, D. Quemener and M. Semsarilar, Sep. Purif. Technol., 2020, 251, 117375 CrossRef CAS.
- H. Phan, R. Cavanagh, D. Destouches, F. Vacherot, B. Brissault, V. Taresco, J. Penelle and B. Couturaud, ACS Appl. Polym. Mater., 2022, 4, 7778–7789 CrossRef CAS.
- L. Luppi, T. Babut, E. Petit, M. Rolland, D. Quemener, L. Soussan, M. A. A. Moradi and M. Semsarilar, Polym. Chem., 2019, 10, 336–344 RSC.
- U. Farooq, L. Upadhyaya, A. Shakeel, G. Martinez and M. Semsarilar, J. Membr. Sci., 2020, 611, 118181 CrossRef CAS.
- U. Farooq, L. Upadhyaya, A. Shakeel, G. Martinez and M. Semsarilar, J. Membr. Sci., 2020, 611, 118181 CrossRef CAS.
- J. Ma, H. M. H. M. Andriambololona, D. Quemener and M. Semsarilar, J. Membr. Sci., 2018, 548, 42–49 CrossRef CAS.
- L. Upadhyaya, M. Semsarilar, S. Nehache, D. Cot, R. Fernández-Pacheco, G. Martinez, R. Mallada, A. Deratani and D. Quemener, Macromolecules, 2016, 49, 7908–7916 CrossRef CAS.
- C. György, P. M. Kirkman, T. J. Neal, D. H. H. Chan, M. Williams, T. Smith, D. J. Growney and S. P. Armes, Angew. Chem., Int. Ed., 2023, 62, e202218397 CrossRef.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed.
- H. Phan, M. Cossutta, C. Houppe, C. Le Cœur, S. Prevost, I. Cascone, J. Courty, J. Penelle and B. Couturaud, J. Colloid Interface Sci., 2022, 618, 173–184 CrossRef CAS PubMed.
- Y. Zhang, C. A. Lim, W. Cai, R. Rohan, G. Xu, Y. Sun and H. Cheng, RSC Adv., 2014, 4, 43857–43864 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- K. Aissou, M. Mumtaz, Ö. Usluer, G. Pécastaings, G. Portale, G. Fleury, E. Cloutet and G. Hadziioannou, Macromol. Rapid Commun., 2016, 37, 221–226 CrossRef CAS PubMed.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- J.-C. Daigle, A. Vijh, P. Hovington, C. Gagnon, J. Hamel-Pâquet, S. Verreault, N. Turcotte, D. Clément, A. Guerfi and K. Zaghib, J. Power Sources, 2015, 279, 372–383 CrossRef CAS.
- R. Khurana, J. L. Schaefer, L. A. Archer and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7395–7402 CrossRef CAS PubMed.
- C. Cao, Y. Li, Y. Feng, C. Peng, Z. Li and W. Feng, Energy Storage Mater., 2019, 19, 401–407 CrossRef.
- D. Devaux, L. Liénafa, E. Beaudoin, S. Maria, T. N. T. Phan, D. Gigmes, E. Giroud, P. Davidson and R. Bouchet, Electrochim. Acta, 2018, 269, 250–261 CrossRef CAS.
- L. Porcarelli, M. A. Aboudzadeh, L. Rubatat, J. R. Nair, A. S. Shaplov, C. Gerbaldi and D. Mecerreyes, J. Power Sources, 2017, 364, 191–199 CrossRef CAS.
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS.
- C. Jangu, A. M. Savage, Z. Zhang, A. R. Schultz, L. A. Madsen, F. L. Beyer and T. E. Long, Macromolecules, 2015, 48, 4520–4528 CrossRef CAS.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- K. Nieswandt, P. Georgopanos, C. Abetz, V. Filiz and V. Abetz, Materials, 2019, 12, 3–6 CrossRef PubMed.
- K. Nieswandt, P. Georgopanos and V. Abetz, Polym. Chem., 2021, 12, 2210–2221 RSC.
- G. Portale, D. Hermida-Merino and W. Bras, Eur. Polym. J., 2016, 81, 415–432 CrossRef CAS.
- S. J. Hunter, J. R. Lovett, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Polym. Chem., 2021, 12, 3629–3639 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- K. Aissou, A. Nunns, I. Manners and C. A. Ross, Small, 2013, 9, 4077–4084 CrossRef CAS PubMed.
- L. Zhu, P. Huang, S. Z. D. Cheng, Q. Ge, R. P. Quirk, E. L. Thomas, B. Lotz, J.-C. Wittmann, B. S. Hsiao, F. Yeh and L. Liu, Phys. Rev. Lett., 2001, 86, 6030–6033 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*