
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-polymerisation modification of poly(3-hydroxybutyrate) (PHB) using thiol–ene and phosphine addition†
Lucas
Al-Shok
,
James S.
Town
 ,
Despina
Coursari
,
Paul
Wilson
and
David M.
Haddleton
*
,
Despina
Coursari
,
Paul
Wilson
and
David M.
Haddleton
*
Department of Chemistry University of Warwick, Library Road, CV4 7AL, Coventry, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 5th May 2023
Abstract
As we face the issues associated with fossil resource-based polymers and their environmental impact after their useful life, polyesters become more important as “greener” alternatives due to their potential hydrolytic and enzymatic degradability in various environments. Moreover, post-modifying their structure can additionally open up access to a variety of new materials. During this work the potential to post-modifying synthetic PHB made via the organocatalysed ring-opening polymerisation of β-butyrolactone (β-BL) is shown. Modification by thiol–ene ‘click’ chemistry was succesfully conducted under UV-initiation. Surprisingly, attempting the modification under thermal conditions using dimethylphenylphosphine (DMPP) as catalyst, resulted in the attachment of the phosphine, as shown via NMR spectroscopy. Control experiments using crotonic acid, methyl crotonate and n-butyric acid indicated that the presence of a carboxylic acid group is necessary in order for the phosphine addition to occur. Further, the formation of particles shown via dynamic light scattering (DLS), zeta-potential (ZP) and transmission electron microscopy (TEM) measurements suggest an amphiphilic character of the phosphine-functionalised polymers. Finally, stability studies in the presence of salt and different pH environments revealed a high responsiveness and dependency between pH and particle size as well as surface charge.
1. Introduction
Poly(3-hydroxybutyrate) (PHB) is a high molecular weight thermoplastic polymer produced by microorganisms2,3 and has been used as a thermoplastic from a renewable route, which biodegrades under ambient and mild conditions.4–8 However, highly crystalline natural occurring PHB exhibits low thermostability and hence unfavourable properties for melt processing. Therefore, various synthetic routes of producing PHB designed to circumvent the high production costs are subject of ongoing research efforts.9–12Producing synthetic PHB paves the way for influencing the properties by changing reaction conditions and the resulting polymeric structure. Introducing functionality into a polymer is of great interest to widen the prospects for different applications either by using a functional monomer or initiator design.13–20 Another widely used approach is the modification of end groups after polymerisation.19,21 Jaffredo et al. reported the organocatalysed ring-opening polymerisation (ROP) of β-butyrolactone (β-BL) resulting in PHB exhibiting a crotonyl functionality as α-end group.1,14,22 In 2018 Coulembier expanded this work and postulated a mechanism for the ROP of β-BL catalysed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) involving an intermediate of protonated TBD and crotonate. The crotonate is acting as the polymerisation initiator selectively opening β-BL via a O-alkyl cleavage.23 These crotonate functionalities yield the potential for post-polymerisation modification. As shown by Michalak et al., successful epoxidation of crotonate functionalities can be achieved by using mCPBA followed by reacting the epoxide with alcohols and amines, respectively.24 In a follow-up study, the authors could further show the functionalisation of the crotonate end group by ozonolysis resulting in glyoxylate-end capped PHB, which could be used for designing potential biocompatible drug delivery systems.25
Moreover, thiol–ene “click chemistry” has been used to functionalise vinyl and the less reactive crotonyl groups.26 Yu and co-workers investigated the post-polymerisation modification of both allylic side chains and crotonic end groups of poly(4-allyloxymethyl-β-propiolactone).27
Further, phosphines can be regarded as potentially interesting reaction partners for crotonates as they participate in Michael-addition reactions and are commonly used as catalysts.28,29 Galkin and co-workers reported on the functionalisation of unsaturated carboxylic acids using phosphines.30–34 In particular, Bu3P has been described to form an irreversible acid-phosphine adduct.
The prospect of this study is to contribute to the ongoing investigation of the post-polymerisation modification of crotonate-end capped synthetic PHB and to elaborate on the potential property modifications of the atactic and amorphous material. Polymers with relatively low molecular weights (>7000 g mol−1) have been synthesised to make the effect of the modified end group functionality on the polymer properties apparent. In addition to thiol–ene chemistry, the crotonate functionality could be modified by using phosphines, which unexpectedly resulted in amphiphilic properties and aggregating structures.
2. Experimental
2.1 Raw materials
β-Butyrolactone was stirred and vacuum-distilled over CaH2 and stored in a flame-dried Schlenk tube over molecular sieves under nitrogen. If not otherwise stated, all chemicals were purchased from commercial vendors, including Sigma-Aldrich, Thermo Fisher Scientific and Alfa Aesar.2.2 Synthesis of poly(3-hydroxybutyrate)
In a general procedure, 1–4 mol% TBD was added to a flame dried Schlenk tube. 1 mL of β-BL was added under stirring via a gas-tight syringe under a nitrogen atmosphere. The reaction was stirred at 60 °C and quenched by the addition of dichloromethane. The polymer solution was then stirred over ion-exchange resin Amberlite IR-120(H) to remove TBD. PHB was precipitated into cold n-pentane three times before being isolated by the removal of volatiles under vacuum at 40 °C for 24 h (Scheme 1).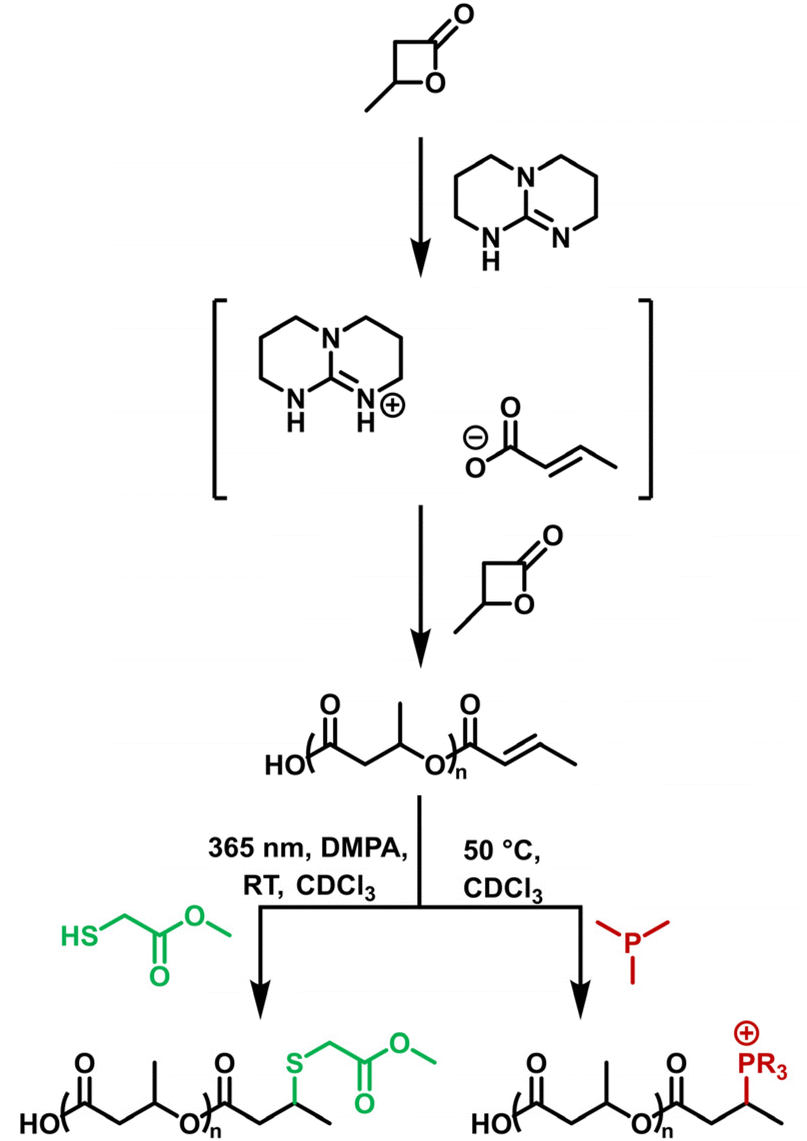 | ||
| Scheme 1 Polymerisation of β-butyrolactone using TBD as the organocatalyst results in PHB end-capped with a crotonyl functionality. The crotonyl group can be further functionalised via thiol–ene ‘click’ chemistry (green) using UV conditions and DMPA as initiator or by attaching a phosphine (red) via thermal activation. | ||
1 H NMR (400 MHz, CDCl3) δ [ppm] 6.96 (m, J = 6.5 Hz, CH, 1H), 5.83–5.79 (d, J = 15.5 Hz, CH, 1H), 5.26 (m, CH, backbone), 2.64–2.44 (m, CH2, backbone), 1.88–1.87 (d, J = 6.8 Hz, CH3, 3H), 1.28 (m, CH3, backbone).
2.3 Post-polymerisation modification: thiol–ene
In a glass vial, PHB (1300 g mol−1, 0.08 mmol) was dissolved in 3 mL CDCl3 before 2,2-dimethoxy-2-phenylacetophenone (DMPA) (1.1 eq., 0.185 mmol) were added to the solution. Subsequently, methylthioglycolate (MTG) (10 eq., 1.68 mmol) was added dropwise to the mixture. After deoxygenated by bubbling with nitrogen for 20 minutes the vial was irradiated by UV light (λ = 365 nm, 500 mJ cm−3) over night. The functionalised polymer was precipitated into n-pentane three time and all volatiles were removed under vacuum. The completion of the reaction was validated by monitoring the crotonyl-peaks (6.96 and 5.83 ppm) in the 1H-NMR.2.4 Post-polymerisation modification: phosphine
In a glass vial, PHB (1000 g mol−1, 0.1 mmol) was dissolved in 2 mL of CDCl3 and deoxygenated for 20 minutes. Afterwards, DMPP (5 eq., 0.42 mmol) was added to the solution before increasing the temperature of the reaction mixture to 50 °C. The reaction was stirred overnight and quenched by removing it from the heat source. All volatiles were removed under vacuum. The completion of the reaction was validated by monitoring the crotonyl-peaks (6.96 and 5.83 ppm) in the 1H-NMR. The functionalised polymer was purified via multiple precipitations into diethyl ether and drying under air. The reaction with Bu3P was conducted accordingly.2.5 Post-polymerisation modification: control experiments
In a glass vial, either methyl-crotonate (1 mmol), crotonic acid (0.85 mmol) or n-butyric acid (0.84 mmol) was dissolved in 2 mL of CDCl3, deoxygenated for 20 minutes, mixed with DMPP (5 eq., 5 mmol), immersed at 50 °C and stirred overnight. The reaction was quenched by removing the heat source and all volatiles were removed under vacuum. The reaction was monitored by using 1H and 31P-NMR.2.6 Stability studies
As PHB can be prone to hydrolysing in acidic and basic environments, unfunctionalised PHB and low molecular weight PHB-DMPP were suspended in 0.9 wt% acidic and basic buffer, respectively. On the next day the samples were freeze-dried and investigated via1H-NMR.2.7 Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210000–1010 Da) and polystyrene standards (12 narrow standards between 364000–160 Da (Agilent EasiVials) with Agilent GPC/SEC software.
:1 with a 40 mg mL−1 solution of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) in THF. PHB samples were mixed 1:1 with a 40 mg mL−1 solution of super-dihydroxybenzoic acid (sDHB, 9:1 (w/w) mixture of 2,5-DHB and 2-hydroxy-5-methoxybenzoic acid). The samples, 0.5 μL of each, were then spotted on an MTP 384 ground steel target plate and analysed using a Bruker AutoFlex speed ToF/ToF analyser, equipped with a 337 nm nitrogen laser.
210000–1010 Da) and polystyrene standards (12 narrow standards between 364000–160 Da (Agilent EasiVials) with Agilent GPC/SEC software.
:1 with a 40 mg mL−1 solution of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) in THF. PHB samples were mixed 1:1 with a 40 mg mL−1 solution of super-dihydroxybenzoic acid (sDHB, 9:1 (w/w) mixture of 2,5-DHB and 2-hydroxy-5-methoxybenzoic acid). The samples, 0.5 μL of each, were then spotted on an MTP 384 ground steel target plate and analysed using a Bruker AutoFlex speed ToF/ToF analyser, equipped with a 337 nm nitrogen laser.
3. Results and discussion
3.1 Synthesis of poly(3-hydroxybutyrate)
PHB with molecular weights between 1000 and 5300 g mol−1 were synthesised by the reaction of β-BL with TBD in a flame-dried Schlenk tube overnight at 60 °C, according to the procedure described by Jaffredo.1 Molecular weight was determined using 1H-NMR with the chemical shifts at 5.7 ppm and 6.9 ppm as an indication of the crotonate end group (Fig. S5–S8†). MALDI-ToF MS confirmed the presence of both the crotonate and acid α, ω terminal groups of PHB (Fig. S10–S13†).3.2 Post-polymerisation modification
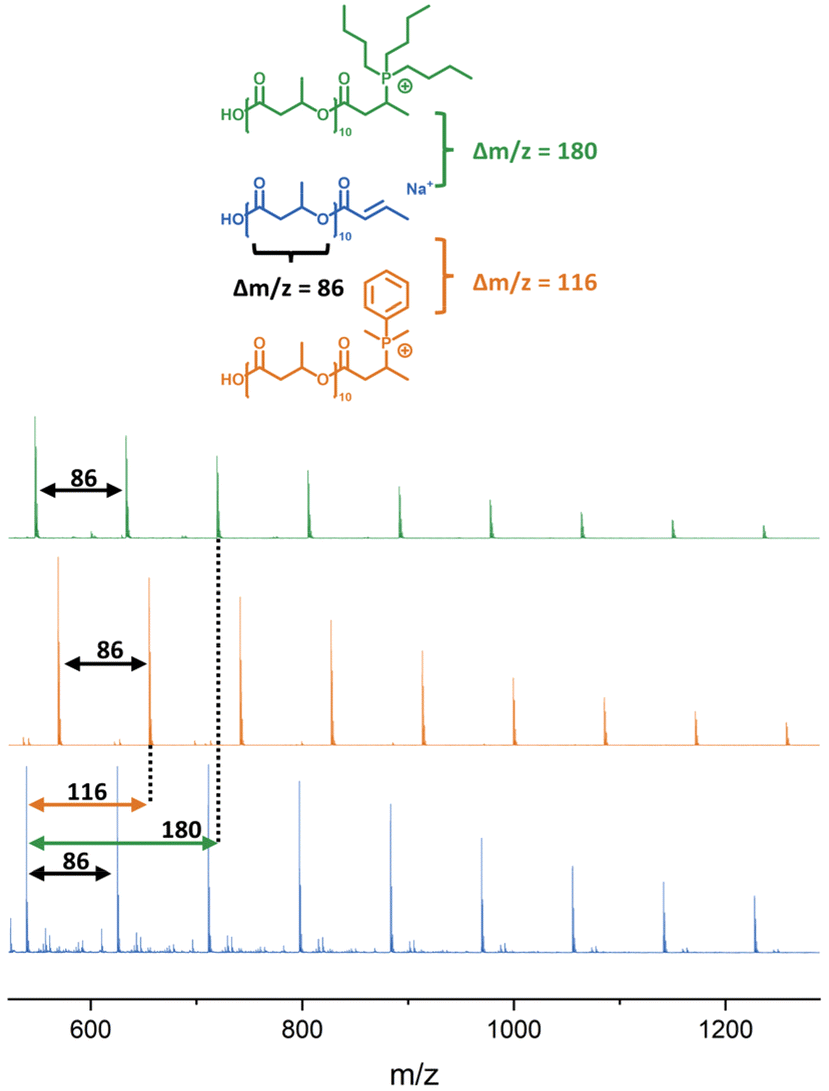 | ||
| Fig. 1 (a) 1H-NMR after thiol–ene functionalisation of low molecular weight PHB with MTG. (b) MALDI-ToF MS of functionalised PHB-MTG showing sodium adducts. | ||
 | ||
| Fig. 2 MALDI-ToF spectra of unfunctionalised (uf) PHB compared to PHB functionalised with DMPP and Bu3P, respectively. | ||
 | ||
| Fig. 3 (a) DOSY-NMR of DMPP-functionalised PHB. (b) 1H–31P correlation NMR of DMPP-functionalised PHB showing phosphine oxide (blue) and functionalised polymer (red). | ||
 | ||
| Fig. 4 1H (f2)–31P (f1) NMR correlation spectrum of functionalised crotonic acid using DMPP (a) and Bu3P (b), respectively. Signals labelled with a red background correspond to oxidised phosphine, a green background refers to functionalised crotonic acid. The insert shows the resolved splitting pattern of the methyl group showing a characteristic doublet–doublet splitting and therefore suggesting coupling to an adjacent phosphorous. | ||
Reaction with a mixture of methyl-crotonate and n-butyric acid in the presence of DMPP or Bu3P gave a similar functionalisation to the crotonic acid and PHB phosphine additions (Fig. S54–S57†), suggesting the successful functionalisation of methyl-crotonate. Thus, in order for the double bond functionalisation to happen, an additional acid group is required. Interestingly, for crotonic acid the signal of the carboxylic acid shifts from 12 ppm to 6 ppm, indicating a less acidic proton or an interaction between the carbonyl-oxygen and the neighbouring phosphorous. A similar shift was also observed for the mixture of n-butyric acid and methyl-crotonate, suggesting a similar O–P interaction. Alternatively, the residual peak at 6 ppm could corresponds to complexed water.
Repeating the NMR measurement in DMSO resulted in a significant shift toward higher field (3.4 ppm) agreeing with a residual water signal. Galkin et al. described a mechanism of their ‘phosphabetaine’ formation, involving the deprotonation of the carboxylic acid and the identification of water incorporated in the crystal lattice which supports this explanation.32
3.3 Self-assembly of functionalised PHB
The phosphine-modification of PHB using DMPP and Bu3P resulted in an amphiphilic molecule, which became evident by observing the formation of dispersions in aqueous media after functionalisation. In contrast, unfunctionalised PHB precipitates in aqueous media and forms visible sedimentation.PHB with molecular weights between 1000 and 5300 g mol−1 were functionalised using both Bu3P and DMPP. Following the determination of the critical aggregation concentration (CAC), the particles were characterised via zeta-potential (ZP) measurements, DLS and TEM (Fig. 5). Functionalised PHB-DMPP with a molecular weight of 1000 g mol−1 gave a CAC of 0.12 mg mL−1 observed via fluorescence of pyrene in the presence of different concentrations of functionalised polymer (Fig. S18, S27, S36 and S45†).38 In comparison, sodium dodecyl sulphate (SDS), which is commonly used as surfactant additive typically shows a CAC of 0.008 mol L−1 (2.3 mg mL−1) in aqueous media.39 This would be several magnitudes higher than the measured CAC for any phosphine-functionalised PHB, which showcases the effectiveness as potential surfactants.
 | ||
| Fig. 5 (a) Table summarising observed CAC, Dh, ZP and particle diameters (PD) for PHB functionalised with DMPP or Bu3P, respectively. Molecular weights were determined by 1H-NMR. (b) TEM pictures taken from the low molecular weight PHB functionalised with DMPP or Bu3P. | ||
ZP measurements (Fig. S19, S28, S37 and S46†) of unfunctionalised and functionalised polymer showed a significant difference of the precursor PHB with ZPs between −67 and −20 mV, PHB-DMPP with a positive ZP between 49–67 mV and PHB-Bu3P with 37–68 mV.
DLS measurements (Fig. S20, S29, S38 and S47†) provided further evidence that the functionalised polymers formed particles with aggregates of 130–269 nm (PHB-DMPP) and 187–272 nm (PHB-Bu3P). Dry-state transmission electron microscopy (TEM) showed particles with comparable diameters between 93 and 279 nm (Fig. 5b and S30, S39, S48†). In general, the large size of particles possibly suggests a form of self-assembly or ordered stacking of multiple chains, possibly facilitated by a positively charged phosphine and a negatively charged carboxylate end group.
The particle size distributions (PDI) for the functionalised particles lie between 0.12 and 0.27, showing stable particles with moderately broad dispersity. In particular, low molecular weight conjugates show narrower distributions, which suggests higher stability at low molecular weight.
3.4 Self-assembly as a function of salt and pH
In order to elaborate on the presumably charged surface of the dispersed particles, further stability studies were conducted by investigating the aggregation behaviour of low molecular weight PHB-DMPP and PHB-Bu3P in different salt and buffer solutions testing the influence of salt concentration and pH, respectively (Table 1).

|
In comparison to aggregates formed in deionised water, NaCl solutions (Fig. S57–S59†) with 0.9 wt% (physiological concentration) showed immediate sedimentation suggesting a disruption of particles by blocking the charged sites with counter ions. Decreasing the NaCl-concentration 10-fold resulted in relatively large particles for PHB-DMPP (281 nm) and PHB-Bu3P (212 nm) with PDI's of 0.22 and 0.18, respectively. A decrease in ZP compared to deionised water suggests a decreased surface charge due to interactions with counter ions. Notably, PHB-DMPP seemed to form particles at 0.09 wt% NaCl with a positive ZP of 14 mV in contrast to the negative −6 mV of PHB-Bu3P, possibly due to less prominent salt interactions of PHB-DMPP. Interestingly, sedimentation was observed for 0.09 wt% NaCl suspensions of PHB-DMPP and PHB-Bu3P after 1 h, which would be indicative for decreased stability over time.
Testing the aggregation in dependence of pH 4 (Fig. S61–S63†), pH 7 (Fig. S64–S67†) and pH 9 (Fig. S68–S71†) revealed no aggregation in pH 4 irrespective of concentration and phosphine functionality. Taking in to account the pKa values of both cotonic acid (4.6),40 DMPP (6.5)41 and Bu3P (8.4)42 provides an estimation that both functionalised polymers could bear protonated end groups at pH 4 and are therefore not able to form suspended particles.
For pH 7 the most distinct differences between PHB-DMPP and PHB-Bu3P can be observed. At 0.9 wt%, PHB-Bu3P forms aggregates with an increased Dh compared to deionised water, whereas PHB-DMPP shows immediate sedimentation. However, upon decreasing the salt concentration to 0.09 wt% the Dh for PHB-Bu3P is comparable to the prior, while PHB-DMPP forms measurable particles with similar diameters. This different behaviour toward salt concentration could highlight the sensitive aggregation of PHB-DMPP close to the pKa of the phosphine.
At basic pH both PHB-DMPP and PHB-Bu3P show similar stable aggregation and particle sizes compared to deionised water. The ZP shows an even strong shift toward negative values (−35 to −60 mV) compared to neutral pH which could be caused by an increased number of anions associated with the particle surface.
1H-NMR were recorded for PHB before and after being kept in acidic and basic buffer, respectively. Neither unfunctionalised nor functionalised PHB showed detectable degradation products after the treatment (Fig. S72 and S73†), suggesting minimal hydrolysation in 0.9 wt% acidic and basic buffer.
4 Conclusion
In this work we have demonstrated the successful post-polymerisation modification of synthetic PHB from TBD-catalysed ROP of β-BL. The resulting crotonate end group could be functionalised under conventional UV-mediated thiol–ene conditions. However, when conducted under thermal conditions, the double bond was functionalised with the used phosphine catalyst (DMPP). The structure of the product was determined via1H-, 31P-, DOSY- and 1H–31P-NMR correlation experiments. A carboxylic acid is required for this reaction to occur, as has been shown with control experiments where methyl-crotonate could only be functionalised in the presence of n-butyric acid. In order to investigate the self-assembly behaviour, polymers with molecular weights between 1000 and 5300 g mol−1 were synthesised and subsequently functionalised with DMPP and Bu3P, respectively. DLS and TEM measurements showed stable aggregates with diameters between 100 and 300 nm, suggesting an intermolecular aggregation process resulting in self-assembled structures. However, at higher molecular weights, hydrophobic interactions seem to dominate and cause random aggregation. Further, stability studies in different salt concentrations and pH environments revealed high stability in alkaline pH, good to low stability in pH 7 and low stability in pH 4 as well as physiological concentrations of NaCl. These results highlight the importance of the role of the charged end groups of the functionalised polymers and how the stability can be influenced by the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Al-Shok thanks the University of Warwick for a PhD studentship through the Warwick Centre for Doctoral Training in Analytical Science. We thank the Research Technology Platforms (RTP) of the University of Warwick and Dr Daniel Lester for providing training and equipment and EPSRC for equipment funded in part by EPSRC EP/V036211/1 and EP/V007688/1.References
- C. G. Jaffredo, J. F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2012, 33, 1938 CrossRef CAS PubMed.
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1 CrossRef CAS PubMed.
- Y. Poirier, D. Dennis, K. Klomparens, C. Nawrath and C. Somerville, FEMS Microbiol. Lett., 1992, 103, 237 CrossRef CAS.
- W. J. Orts, G. A. R. Nobes, J. Kawada, S. Nguyen, G.-E. Yu and F. Ravenelle, Can. J. Chem., 2008, 86, 628 CrossRef CAS.
- J. Mergaert, A. Webb, C. Anderson, A. Wouters and J. Swings, Appl. Environ. Microbiol., 1993, 59, 3233 CrossRef CAS PubMed.
- L. Savenkova, Z. Gercberga, O. Muter, V. Nikolaeva, A. Dzene and V. Tupureina, Process Biochem., 2002, 37, 719 CrossRef CAS.
- L. Savenkova, Z. Gercberga, V. Nikolaeva, A. Dzene, I. Bibers and M. Kalnin, Process Biochem., 2000, 35, 573 CrossRef CAS.
- M. P. Arrieta, J. López, E. Rayón and A. Jiménez, Polym. Degrad. Stab., 2014, 108, 307 CrossRef CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC.
- R. M. Shakaroun, P. Jéhan, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2020, 11, 2640 RSC.
- J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696 CrossRef CAS PubMed.
- Z. Jedlinski, P. Kurcok and R. W. Lenz, Macromolecules, 1998, 31, 6718 CrossRef CAS.
- C. G. Jaffredo and S. M. Guillaume, Polym. Chem., 2014, 5, 4168 RSC.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2013, 4, 3837 RSC.
- M. Helou, G. Moriceau, Z. W. Huang, S. Cammas-Marion and S. M. Guillaume, Polym. Chem., 2011, 2, 840 RSC.
- C. Guillaume, N. Ajellal, J.-F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 907 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260 RSC.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573 RSC.
- S. M. Guillaume, Eur. Polym. J., 2013, 49, 768 CrossRef CAS.
- D. Kai, K. Zhang, S. S. Liow and X. J. Loh, ACS Appl. Bio Mater., 2019, 2, 127 CrossRef CAS PubMed.
- T. Ouchi, T. Uchida, H. Arimura and Y. Ohya, Biomacromolecules, 2003, 4, 477 CrossRef CAS PubMed.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2013, 46, 6765 CrossRef CAS.
- S. Moins, C. Henoumont, J. De Winter, A. Khalil, S. Laurent, S. Cammas-Marion and O. Coulembier, Polym. Chem., 2018, 9, 1840 RSC.
- M. Michalak, M. Kawalec and P. Kurcok, Polym. Degrad. Stab., 2012, 97, 1861 CrossRef CAS.
- M. Michalak, A. A. Marek, J. Zawadiak, M. Kawalec and P. Kurcok, Eur. Polym. J., 2013, 49, 4149 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381 CrossRef CAS.
- Y. Yu, M. Kim, G. S. Lee, H. W. Lee, J. G. Kim and B.-S. Kim, Macromolecules, 2021, 54, 10903 CrossRef CAS.
- D. H. Valentine Jr. and J. H. Hillhouse, Synthesis, 2003, 317 Search PubMed.
- C. Gimbert, M. Lumbierres, C. Marchi, M. Moreno-Mañas, R. M. Sebastián and A. Vallribera, Tetrahedron, 2005, 61, 8598 CrossRef CAS.
- Y. V. Bakhtiyarova, R. R. Minnullin, I. V. Galkina, R. A. Cherkasov and V. I. Galkin, Russ. J. Gen. Chem., 2015, 85, 2037 CrossRef CAS.
- V. I. Galkin, A. V. Salin, Y. V. Bakhtiyarova and A. A. Sobanov, Russ. J. Gen. Chem., 2009, 79, 919 CrossRef CAS.
- V. I. Galkin, Y. V. Bakhtiyarova, R. I. Sagdieva, I. V. Galkina and R. A. Cherkasov, Heteroat. Chem., 2006, 17, 557 CrossRef CAS.
- V. I. Galkin, Y. V. Bakhtiyarova, N. A. Polezhaeva, I. V. Galkina, R. A. Cherkasov, D. B. Krivolapov, A. T. Gubaidullin and I. A. Litvinov, Russ. J. Gen. Chem., 2002, 72, 376 CrossRef CAS.
- V. I. Galkin, Y. V. Bakhtiyarova, N. A. Polezhaeva, I. V. Galkina, R. A. Cherkasov, D. B. Krivolapov, A. T. Gubaidullin and I. A. Litvinov, Russ. J. Gen. Chem., 2002, 72, 384 CrossRef CAS.
- T. A. Albright, W. J. Freeman and E. E. Schweizer, J. Org. Chem., 1975, 40, 3437 CrossRef CAS.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189 CrossRef CAS PubMed.
- J. Jover and J. Cirera, Dalton Trans., 2019, 48, 15036 RSC.
- A. P. Narrainen, S. Pascual and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 439 CrossRef CAS.
- B. Hammouda, J. Res. Natl. Inst. Stand. Technol., 2013, 118, 151 CrossRef CAS.
- C. M. Palmeira, M. I. Rana, C. B. Frederick and K. B. Wallace, Biochem. Biophys. Res. Commun., 2000, 272, 431 CrossRef CAS PubMed.
- R. C. Bush and R. J. Angelici, Inorg. Chem., 1988, 27, 681 CrossRef CAS.
- C. A. Streuli, Anal. Chem., 1960, 32, 985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00272a |
| This journal is © The Royal Society of Chemistry 2023 |