DOI:
10.1039/D3PY00210A
(Paper)
Polym. Chem., 2023,
14, 3897-3905
Redox-responsive nanogels for drug-delivery: thiol–maleimide and thiol–disulfide exchange chemistry as orthogonal tools for fabrication and degradation†‡
Received
25th February 2023
, Accepted 19th June 2023
First published on 18th July 2023
Abstract
Stimuli-responsive, readily functionalizable, and degradable nanogels have great potential in targeted drug delivery applications. While crosslinked polymeric nanoparticles such as nanogels modified with cell-targeting moieties can effectively deliver various hydrophobic drugs to cancer cells, they should be engineered to undergo degradation upon exposure to endogenous stimuli and release the payload once inside the cell. Herein, we report the preparation of a nanogel system crosslinked through the thiol–maleimide Michael addition reaction, which can be degraded in a reducing environment through a thiol–disulfide exchange reaction. The maleimide groups on the nanogels provided handles for conjugating cell-targeting motifs. To this end, a new monomer was designed to synthesize poly(ethylene glycol) methacrylate-based copolymers containing pendant maleimide groups linked to the polymer backbone through disulfide linkages. These copolymers were utilized to yield nanogels by crosslinking the nanoaggregates formed upon heating the copolymers in aqueous media. Nanogels were loaded with a clinically administered anticancer drug, docetaxel, and the drug release was investigated in the reductive cellular environment. Nanogels conjugated with cell-targeting peptides demonstrated preferential cellular internalization in breast cancer cells and also exhibited enhanced cytotoxicity in cells rich in glutathione. One can envision that such nanogels, employed to deliver various drugs and target motifs depending on the type of cancer, will be an attractive platform for such applications.
Introduction
The past decades have witnessed the progressive development of responsive polymeric nanoparticles.1,2 One of the fundamental reasons for the emergence of broad interest in nanoparticles stems from their ability to increase the bioavailability of hydrophobic drug molecules, many of which are insoluble in the aqueous biological milieu of our body.3,4 Additionally, the utilization of these nanosized delivery agents also addresses the pharmacokinetic limitations of conventional medication, such as short circulation time and rapid systemic clearance. As alternatives between soft polymeric versus hard inorganic nanoparticles, the polymer-based systems such as micelles,5 polymeric nanoparticles,6 and nanogels7 are more prone to deformability and degradation compared to their rigid inorganic counterparts, which assists in their overcoming of biological barriers to increase circulation time, escape immune cell uptake and reduce accumulation in the spleen.8–11 In recent years, nanogels have emerged as an attractive soft nanoparticle platform for various drug delivery applications due to their high drug-loading capacity, multi-functional nature, and high stability.12–16 Their nanoscale size offers passive tumor targeting through an enhanced permeation and retention effect17 and allows installation of appropriate targeting ligands, which enable active targeting of the diseased cells.18,19 Furthermore, employing “smart” or environmentally sensitive nanogels provides an opportunity to facilitate the efficient release of the therapeutic agent at the disease site.20
In general, the fabrication of nanogels can be achieved using one of the two common approaches: polymerization of monomers and crosslinking of polymer precursors. For example, in a seminal contribution, Matyjaszewski and coworkers utilized the inverse miniemulsion atom transfer radical polymerization method to prepare redox-responsive nanogels using direct polymerization of oligo(ethylene oxide) monomethyl ether methacrylate in the presence of a disulfide-containing dimethyl methacrylate crosslinker.21 In the second approach, a well-defined functional polymer is used as a nanogel precursor and crosslinked after assembly as a nanosized aggregate.22 In general, polymeric precursors are stabilized as nanosized aggregates using surfactants, followed by crosslinking.23–25 Thayumanavan and coworkers reported a clever approach for fabricating nanogels without using surfactants.26 They utilized the thermally promoted self-assembly of copolymers composed of oligo(ethylene glycol) and pyridyl-disulfide side chains, which undergo in situ generated thiol–disulfide exchange upon partial reduction of the disulfide groups to thiols. The monomer and polymer-based methods continue to be extensively utilized in preparing nanogels for various biomedical applications.
Stimuli-responsive nanogels change their physicochemical properties, such as size, hydrophobicity, and crosslinking density, under various environmental conditions.27 Degradable linkages in the nanogels are critical in manipulating these physicochemical properties. In this regard, stimuli-responsive linkages sensitive to stimuli such as pH, temperature, and redox have been used in designing drug delivery systems.28–38 Common degradable linkages employed are acetals,39 imines,40 hydrazones,41 dithioketals,42 and disulfide bonds.43–51 In recent years, redox-responsive disulfide bonds have been used for crosslinking nanogels because of their enhanced degradation inside cancer cells due to the high intracellular GSH concentration in the cytosol.52–55
Reactions with high efficiency are utilized to ensure the rapid crosslinking of polymeric nanogel precursors to obtain stimuli-responsive nanogels. While many ‘click’ reactions have been used to prepare stimuli-responsive nanogels,56,57 utilization of the highly efficient thiol–maleimide reaction is rare. Maleimide–thiol reaction is a highly efficient reaction used in the functionalization and crosslinking of polymers;58–64 however, maleimide–thiol conjugation is not easily cleavable, apart from the case in which specific thiols are employed.65 We envision that integrating a disulfide linkage into the maleimide–thiol conjugation system will provide advantages for rapid maleimide–thiol conjugation and facile disulfide-based degradation. We recently demonstrated that the thiol–maleimide conjugation proceeded with a high level of selectivity in the presence of disulfide linkages and utilized this selectivity to engineer fast-forming dissolvable bulk hydrogels.66 Importantly, the thiol–maleimide conjugation proceeds with high efficiency in aqueous media without releasing any byproducts, so the approach is quite benign. We envisioned that the selective thiol-based chemistry could be exploited to prepare redox-responsive nanogels that are amenable for the non-covalent and covalent functionalization for engineering targeted drug delivery systems.
Herein, we report the synthesis and evaluation of redox-responsive nanogels obtained using the thiol–maleimide conjugation. For this purpose, a novel disulfide-containing maleimide side chain polymer was synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization. An oligo(ethylene glycol)-based comonomer was used to provide aqueous solubility and undergo thermally promoted self-assembly to yield nanoaggregates. The otherwise unstable nanoaggregates were crosslinked using a water-soluble bis-thiol crosslinker to produce stable nanogels. These nanogels could be loaded with hydrophobic dyes and drugs and conjugated with peptide-based cancer cell targeting motifs (Scheme 1). Nanogels were prone to degradation under reducing environments, releasing their encapsulated cargo. Finally, a clinically used anticancer drug, docetaxel, was loaded into the targeting group appended nanogels, and their cellular internalization and toxicity were investigated.
 |
| | Scheme 1 Schematic illustration of the preparation of drug-loaded targeted redox-responsive nanogels. | |
Experimental section
Reagents and materials
Diethyl ether was purchased from Merck. 1,4-Dithio-DL-threitol (DTT) (98%) was purchased from Alfa Aesar. Furan (>99%), maleic anhydride (99%), 3-amino-1-propanol (99%), 2-hydroxymethyl methacrylate (HEMA, 98%), 4-(dimethylamino)pyridine (DMAP, >99%), 4,4′-dithiodibutyric acid (95%), N,N′-dicyclohexylcarbodiimide (DCC), poly(ethylene glycol) methyl ether methacrylate (PEGMEMA, Mn = 300), 2,2′-azobis(2-methylpropionitrile) (AIBN), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CTA), 2,2′-(ethylenedioxy)-diethanethiol, Nile red (NR), docetaxel, and glutathione were purchased from Sigma-Aldrich. All solvents were purchased from Merck and used as received. The furan-protected maleimide–disulfide acid was prepared according to a literature procedure.66
Instrumentation
Instrumentation details are provided in the ESI.‡
Cell lines
MDA-MB-231 was purchased from ATCC (LGC Standards, Germany). Please see the ESI for technical details.‡
Synthesis of a masked maleimide–disulfide-containing monomer (FMDSMA)
Masked maleimide–disulfide acid (0.854 g, 0.002 mol), HEMA (1.00 g, 0.007 mol), and DMAP (0.565 g, 0.004 mol) were added into a round bottom flask and dissolved in anhydrous CH2Cl2 (1.5 mL). DCC (0.397 g, 0.002 mol) was dissolved in another flask in anhydrous CH2Cl2 (1 mL). The DCC solution was slowly added to the first flask at 0 °C. The reaction mixture was stirred at room temperature for 24 hours. Cold CH2Cl2 (25 mL) was then added to the reaction mixture and precipitated dicyclohexylurea was removed by filtration. After evaporation, the residue was dissolved in cold EtOAc (25 mL) and precipitated dicyclohexylurea was filtered, and the filtrate was concentrated. The concentrated solution was purified by column chromatography on SiO2, using EtOAc and CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluents, affording 0.9 g of product (82% yield). 1H NMR (CDCl3, δ, ppm), 6.51 (s, 2H, CH
:1) as eluents, affording 0.9 g of product (82% yield). 1H NMR (CDCl3, δ, ppm), 6.51 (s, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.12 (s, 1H, CH2C), 5.60 (s, 1H CH2C), 5.26 (s, 2H, bridgehead protons), 4.34 (s, 4H, C(O)OCH2CH2O(O)C), 4.04 (t, 2H, C(O)OCH2CH2), 3.57 (t, 2H, NCH2CH2), 2.84 (s, 2H, bridge protons), 2.72 (t, 4H, SCH2CH2), 2.44 (t, 4H, C(O)CH2CH2), 2.03 (m, 4H, SCH2CH2CH2), 1.94 (m, 2H, NCH2CH2CH2), 1.53 (s, 3H, CCH3). 13C NMR (CDCl3, δ, ppm), 176.20, 172.80, 172.71, 167.12, 136.54, 135.92, 126.13, 80.96, 62.38, 62.16, 61.37, 47.42, 37.77, 37.68, 35.75, 32.50, 32.42, 26.67, 24.14, 24.10, 18.30.
CH), 6.12 (s, 1H, CH2C), 5.60 (s, 1H CH2C), 5.26 (s, 2H, bridgehead protons), 4.34 (s, 4H, C(O)OCH2CH2O(O)C), 4.04 (t, 2H, C(O)OCH2CH2), 3.57 (t, 2H, NCH2CH2), 2.84 (s, 2H, bridge protons), 2.72 (t, 4H, SCH2CH2), 2.44 (t, 4H, C(O)CH2CH2), 2.03 (m, 4H, SCH2CH2CH2), 1.94 (m, 2H, NCH2CH2CH2), 1.53 (s, 3H, CCH3). 13C NMR (CDCl3, δ, ppm), 176.20, 172.80, 172.71, 167.12, 136.54, 135.92, 126.13, 80.96, 62.38, 62.16, 61.37, 47.42, 37.77, 37.68, 35.75, 32.50, 32.42, 26.67, 24.14, 24.10, 18.30.
Synthesis of a masked maleimide–disulfide-containing copolymer
PEGMEMA (0.500 g, 1.60 mmol), FMDSMA (0.185 g, 0.3 mmol), AIBN (1.02 mg, 0.006 mmol), and CTA (0.015 g, 0.050 mmol) were added to a round bottom flask and dissolved in DMF (1.5 mL). The solution was purged with N2 for 45 minutes. The reaction vessel was immersed in a preheated oil bath at 70 °C. After 24 hours, the solution was concentrated using a rotary evaporator, and the residue was precipitated in cold ether. The precipitated polymer was then collected and dried under high vacuum, affording 0.700 g of product (73% yield). Mn,theo = 12800 g mol−1, Mn,GPC = 10800 g mol−1, and Mw/Mn = 1.20. 1H NMR (CDCl3, δ, ppm), 6.51 (s, CHCH), 5.25 (s, CH bridgehead protons), 4.26 (s, C(O)OCH2CH2OC(O)), 4.08 (t, (O)COCH2CH2), 3.60 (m, OCH2CH2O), 3.38 (s, 3H, CCH3), 2.85 (s, bridge protons), 2.73 (t, SCH2CH2), 2.47 (t, 4H, C(O)CH2CH2). The retro Diels–Alder reaction was carried out by dissolving the product (0.7 g) in anhydrous toluene (25 mL), and refluxing the solution for 8 hours. The solution was then concentrated, and the residue precipitated in cold ether and dried under high vacuum to yield 0.650 g of product (92% yield). 1H NMR (CDCl3, δ, ppm), 6.72 (s, CHCH), 4.26 (s, C(O)OCH2CH2O(O)C), 4.08 (t, C(O)OCH2CH2), 3.60 (m, OCH2CH2O), 3.38 (s, CH3C), 2.73 (t, SCH2CH2), 2.47 (t, C(O)CH2CH2).
Orthogonality of the thiol–disulfide exchange and thiol–maleimide reaction
For an equivalent amount of thiol and maleimide reaction, a copolymer (10 mg, 0.001 mmol) and 2-mercapto-ethanol (0.38 mg, 0.005 mmol) were reacted in PBS (0.10 mL) in a thermal shaker with constant shaking (100 rpm) for 30 minutes at 37 °C. The solution was freeze-dried, and the polymer was redissolved in CH2Cl2 (0.2 μL) and precipitated in diethyl ether (1.5 mL). The solution was centrifuged at 13000 rpm for five minutes, and the supernatant was separated. The polymer part was analyzed using 1H NMR spectroscopy. The supernatant was redissolved in CH2Cl2 (0.50 mL) and filtered through a short silica oxide plug using methanol as the eluent. The filtered solution was then evaporated, redissolved in acetonitrile, and analyzed using LCMS. The same procedure was applied to the control experiment, where four equivalents of thiol were used.
Determination of the LCST of the copolymer
The LCST of a copolymer solution (0.1 M PBS) was determined by measuring the transmittance values of a polymer solution between 20 and 80 °C via a UV-vis spectrophotometer at a wavelength of 600 nm. The size of polymeric particles at room temperature (25 °C) and above the LCST (60 °C) was measured using dynamic light scattering (DLS).
Preparation of nanogels
For nanogel preparation, nanoaggregates were first prepared by dissolving the copolymer (5 mg) in PBS (1 mL, 0.1 M) at 60 °C. After 10 minutes at 60 °C, 2,2′-(ethylenedioxy)-diethanethiol as a crosslinker (0.082 mg, 0.0005 mmol) was added to the solution, and the reaction mixture was stirred for 30 minutes at 60 °C, affording 4.80 mg of nanogel (96% yield).
Evaluation of thiol content in nanogels
The unreacted thiol concentration in the NG was measured using Ellman's reagent (4.0 mg mL−1). A nanogel solution (30 μL) was diluted with PBS (0.970 mL), and Ellman's reagent (0.250 mL) was then added. After 15 minutes of incubation at room temperature, the absorbance of the solution at 412 nm was measured using a UV-vis spectrophotometer.
Loading and release of NR
For NR loading, NR was dissolved in acetone (37 μg mL−1), and this solution (1 mL) was added to the NG solution (1 mL, 5 mg mL−1). After mixing, the acetone was allowed to evaporate slowly at 25 °C for 24 hours. The solution was filtered through filters (0.45 μm pore size) to remove excess insoluble NR. The amount of loaded NR was calculated as follows: the nanogels were first incubated in a DTT solution (0.20 M) for 1 hour. The solution was then freeze-dried and redissolved in DMSO. The fluorescence signal at 623 nm was measured using a spectrometer, and the content was quantified employing an NR calibration curve. To visualize the degradation of the NGs, DTT (0.20 M) was added to an NR-loaded NG solution. Digital photographs of this solution under UV light were taken before and 10 minutes after DTT addition. NR release from the NGs was also monitored by incubating them in reducing (0.01 M GSH) and non-reducing environments. The released amount was quantified at different time points using a fluorescence spectrophotometer.
Conjugation of cRGDfC to nanogels
To the nanogel solution (1 mL, 5 mg/mL) in PBS, cRGDfC (250 μg, 0.430 μmol) was added, and the mixture was stirred for 24 hours at room temperature. Unreacted peptides were removed using a dialysis bag (cutoff: 12 kDa). After purification of the nanogels, 100 μL of the nanogel solution was used against the BCA working reagent (2 mL) for the determination of conjugated peptides. The blue mixture was incubated for 30 minutes at 60 °C. After 30 minutes, the blue color mixture turned purple, and its absorbance was measured at 562 nm using a UV-vis spectrophotometer.
Loading and release of docetaxel (DTX)
To calculate the DTX loading, a NG solution (0.1 mL, 5 mg mL−1) was diluted with a PBS solution (0.9 mL, 0.1 M). DTX (0.25 mg) was then dissolved in acetone (1.0 mL). The DTX solution (1.0 mL, 0.25 mg mL−1) was then added to the NG solution. Acetone in the final solution was allowed to evaporate slowly at 25 °C for 24 hours. Unloaded DTX was removed by the filtration method (0.45-μm pore size filter). The DTX content was calculated after treating the NG solution with DTT (0.20 M) for one hour. The final solution was diluted with CH3CN (1:1) and analyzed using LCMSMS, and the DTX content was calculated as 28 (±2) wt%. For DTX release studies, a DTX-loaded NG solution (1 mL, 5 mg mL−1) was put in a dialysis bag (cutoff: 3.5 kDa), and release was started in a 40 mL PBS and 40 mL GSH solution (0.010 M) in a thermal shaker with constant shaking (100 rpm) at 37 °C. Samples (1 mL) were taken, and the new solution was added at every time point; the samples were analyzed using LCMSMS.
In vitro experiments
The cytotoxicity of the nanogels was investigated via the CCK-8 viability assay on MDA-MB-231 cancer cells. The cells (6000 cells per well) were seeded on a 96-well plate in quadruplicate with a culture medium (100 μL) and incubated for 24 hours at 37 °C for cell growth and adhesion. After incubation, one part of the cells was treated with GSH solution (5 mM) for one hour at 37 °C. After one hour, the GSH solution was removed and washed three times with a cell medium. The cells were then treated with cellular media containing different concentrations of free drug, empty NG, drug-loaded NG, and drug-loaded and cRGDfC-conjugated nanogels. For free drug treatment, DTX (10−4 M) was dissolved in culture media containing DMSO (2.5%), and then serial dilutions were made from a stock solution (10−4 M) using cell media; in that way, the DMSO concentration in each well was kept below 0.5% (v/v). For the control, an empty NG solution in cell media (1 mL, 0.4 mg mL−1) was prepared and diluted with fresh cell media to add to the wells. For the drug-loaded nanogels, nanogels with different DTX concentrations (10−6 − 10−11 M) were prepared. A cRGDfC-conjugated nanogel solution (0.4 mg mL−1) with varying concentrations of DTX was prepared in cell media and added to cells treated with GSH and those without GSH. The plate was kept with the corresponding solutions for 48 hours. For the pulse-chase experiment, the same sample preparation and procedure were used; however, cells containing drug solutions were replaced with fresh media after four hours. After 20 h, CCK-8 solution was added to each well to culture for four hours. After incubation with CCK-8, the absorbance values at 450 nm were measured via a microplate reader. Results were obtained using GraphPad prism software in nonlinear regression mode.
Internalization experiments
For the cellular internalization experiment, MDA-MB-231 adenocarcinoma cells (150000 cells per well) were seeded in a 12-well plate as duplicates in culture media (1 mL). The cells were incubated at 37 °C for 24 hours. The cells were then incubated with non-functionalized nanogels, cRGDfC-conjugated nanogels, and free cRGDfV (10 μg mL−1) containing cRGDfC-conjugated NGs, all containing the same amount of Nile red dye, at 37 °C for a predetermined time. After six hours, cellular media were removed and washed two times with PBS (500 μL), and 4% of formaldehyde solution (300 μL) was added to the wells at 37 °C for 10 minutes. After removing the solution and completing the washing process, DAPI (300 μL) containing PBS was added to the wells for 15 minutes at room temperature for nucleus staining. After washing the cells with PBS, cell images were obtained using a Zeiss Observer Z1 fluorescence microscope. For the flow cytometry experiment, MDA-MB-231 adenocarcinoma cells (200000 cells per well) were seeded into a 12-well plate as duplicates in culture media (1 mL) and incubated at 37 °C for 24 hours. The properties of the samples were the same as those used in the cellular internalization experiment. After 24 hours, cell media were removed, and the cells were trypsinized with a 0.05% trypsin solution for 2 minutes at 37 °C. After 2 minutes, culture media (700 μL) were added to the wells and analyzed using flow cytometry (Guava easyCyte).
Results and discussion
Synthesis of a masked maleimide–disulfide-containing monomer
Due to our interest in designing redox-responsive nanogels that are prepared using the efficient thiol–maleimide reaction, a furan-protected maleimide and disulfide-bearing methacrylate (FMDSMA) monomer was synthesized. As a monomer precursor, masked maleimide–disulfide acid was prepared according to a previously reported protocol.66 Esterification reaction was used for coupling the masked maleimide–disulfide acid with 2-hydroxyethyl methacrylate to produce a FMDSMA monomer (Fig. 1a). The composition of the synthesized monomer was deduced from the 1H and 13C NMR spectra (Fig. 1b, Fig. S1a and b‡). In the 1H NMR spectrum, the proton resonances at 5.60 and 6.12 ppm belonging to the methacrylic double bond and at 6.51 and 5.26 ppm belonging to protons on the bicyclic moiety confirmed the structure (Fig. 1b). Likewise, the carbon resonance at 172.80 ppm and 172.71 ppm belonging to carbonyl carbons of the ester groups and at 167.12 ppm for the carbonyl in the methacrylate group corroborated the structure. As expected, the carbonyl bond stretching around 1700 cm−1 was present in the FTIR spectrum (Fig. S2‡).
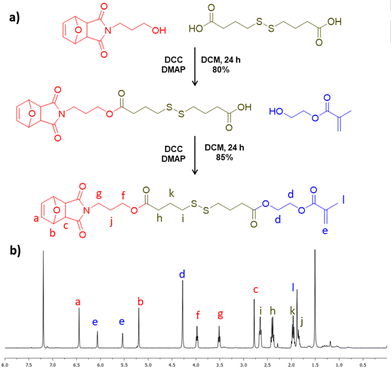 |
| | Fig. 1 (a) Synthetic pathway and (b) 1H NMR spectrum of the masked maleimide–disulfide-containing monomer. | |
Synthesis of the redox-responsive copolymer
A copolymer of FMDSMA and PEGMEMA as a building block of nanogels was synthesized using RAFT polymerization to yield well-defined polymers (Fig. 2a). A polymer that would contain enough functionality for crosslinking and functionalization and yet be water-soluble was targeted using an FMDSMA:PEGMEMA ratio of 1:5. The experimental monomer ratio in the copolymer was calculated to be 1:6 from its 1H NMR spectrum (Fig. S3a‡), and the number average molecular weight was deduced to be 10800 gmol−1, with 1.2 PDI using size exclusion chromatography (SEC) (Fig. S4a‡). The furan-protected maleimide moieties were unmasked via the retro Diels–Alder reaction by refluxing polymer solution in toluene. The number average molecular weight was deduced to be 10100 gmol−1 with a molecular weight distribution of 1.3 by SEC analysis (Fig. S4b‡). The unmasking of the maleimide groups was confirmed using 1H NMR spectroscopy (Fig. 2b and Fig. S3b‡); the appearance of vinylic proton resonance at 6.7 ppm and the disappearance of proton resonances at 6.5 and 5.2 ppm confirmed the presence of the thiol-reactive maleimide unit.
 |
| | Fig. 2 (a) Synthesis of the maleimide-containing copolymer and (b) 1H NMR spectrum of the copolymer. | |
The selectivity of thiol–maleimide reaction over thiol–disulfide exchange reaction
For well-defined crosslinking, it is crucial to explore the reactivity of free thiol moieties of the crosslinker against maleimide moieties and disulfide bonds on the polymer. Our previous study showed that the thiol functional group selectively undergoes a Michael addition reaction with maleimide groups instead of a thiol-exchange reaction with disulfide groups.65 In this study, the preferred reaction with the maleimide group was confirmed. The maleimide and disulfide-group containing copolymers were reacted with a stoichiometric equivalent of 2-mercaptoethanol. 1H NMR analysis of the final product shows that the vinylic proton resonance of the maleimide ring at 6.72 ppm disappeared and new peaks appeared at 3.87 ppm, which indicates successful thiol–maleimide addition (Fig. S5‡). Furthermore, the supernatant of the above reaction was analyzed using LCMS for possible side products that may be released through any thiol–disulfide exchange reaction. The reaction between the copolymer and the stoichiometric equivalent of 2-mercaptoethanol did not yield any side product, which may be formed due to the thiol–disulfide exchange reaction (Fig. S6a‡). On the other hand, the reaction between the copolymer and four equivalents of 2-mercaptoethanol led to the formation of side products, proving that excess thiols not only react with the maleimide group but also result in thiol–disulfide exchange reactions (Fig. S6b‡).
Preparation of redox-responsive NGs
It is well-established that polymers containing oligo ethylene side chains demonstrate a thermo-responsive behavior and can self-assemble to yield nanoaggregates in an aqueous solution above their lower critical solution temperature (LCST).67 Due to our interest in the fabrication of nanoaggregates using the thermo-responsive behavior of the obtained copolymers, the LCST behavior of copolymers was studied (Fig. 3a). A clear polymer solution at 25 °C and a cloudy polymer solution above 60 °C indicated that the copolymer reversibly undergoes self-assembly to form nanoaggregates. DLS measurements also confirmed that polymeric nanoaggregates were formed with an average diameter of 86 nm at 60 °C (Fig. 3b). The nanoaggregates could be crosslinked at 60 °C upon adding a small bis-thiol, namely, 2,2′-(ethylenedioxy)-diethanethiol, into the solution. The optimum crosslinking ratio for maleimide to thiol was determined as 37%, which afforded stable hydrogels which could be degraded in a reducing environment. The hydrodynamic diameter of crosslinked particles was determined as 18 nm using TEM (Fig. 3c) and 67 nm diameter using DLS (Fig. 3d). The smaller size obtained using TEM, as compared to DLS measurements, was presumably due to the shrinkage in the dehydrated state of the nanogels during the TEM measurements. Ellman's analysis of the obtained nanogels demonstrated that a negligible amount of free thiol groups remained in the NGs (ca. 2%). The temperature-responsive behavior of the nanogels was evaluated using DLS over a range of 25–70 °C with 5 °C increments, and it was observed that the NGs were able to maintain their integrity (Fig. S7a‡). However, the size of the NGs decreased slightly near the LCST, presumably due to the thermoresponsive nature of the PEG units (Fig. S7b‡).
 |
| | Fig. 3 (a) Change in % transmittance of the copolymer solution with temperature changes, (b) hydrodynamic size as determined by DLS of the copolymer solution at 25 and 60 °C, (c) TEM image, and (d) DLS size distribution histogram of crosslinked NGs. | |
NR loading and redox-responsive release from nanogels
Due to our interest in hydrophobic drug delivery, nanogels were loaded with a hydrophobic dye molecule Nile red (NR). Loading of the water-insoluble dye NR was carried out by the co-solvent (acetone) evaporation method. Fig. S8a‡ demonstrates the successful loading of NR into the nanogels. In contrast, when NR was mixed with a non-crosslinked polymer solution through co-solvent evaporation, no significant loading was observed (Fig. S8b‡). The degradability of nanogels in a reducing environment was evaluated with NR-loaded nanogels (NR@NG). Under UV light, NR@NG gave a nice pink color, but the color disappeared when the nanogels were incubated in DTT solution (200 mM) for 10 minutes due to the release of the water-insoluble molecule NR upon NG degradation under a reducing environment (Fig. 4a). To evaluate NR release under milder conditions for a period of time, the fluorescence intensity of NR-loaded nanogels was monitored for five days. While there was no significant fluorescence intensity change in the PBS solution (Fig. 4b), it dramatically decreased in a GSH solution (10 mM), indicating that nanogels were degrading and releasing their water-insoluble cargo (Fig. 4c). DLS measurements also indicated that NGs started to disintegrate at pH 5.5 (Fig. S9‡), and in the GSH solution (10 mM) after four hours at 37 °C (Fig. 4e) while remaining stable in the PBS solution after the same period of time (Fig. 4d). Given that the NGs appeared to disintegrate under acidic conditions, we investigated the release of NR from the NGs in a pH 5.5 buffer. As shown in Fig. S10,‡ the intensity of NR incubated at pH 5.5 was found to be significantly lower in comparison with NGs that were incubated in a pH 7.4 buffer.
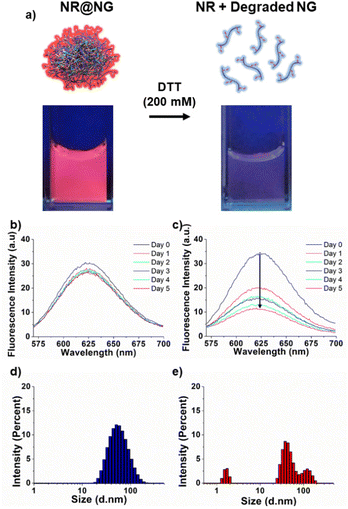 |
| | Fig. 4 (a) Schematic representation of degradation and visual images of redox-responsive degradation of hydrophobic dye NR-containing nanogels under UV light. Fluorescence intensity of NR-containing nanogel solutions incubated in (b) PBS and (c) GSH solutions (0.010 M) for five days. (d) DLS analysis of NG after 4 hours in PBS and (e) after 4 hours in GSH solution (0.010 M). | |
In vitro DTX release, cytotoxicity, and cellular uptake
DTX-loaded NGs were prepared by the co-solvent (acetone) evaporation method. The DTX loading was calculated after degrading NGs in DTT solution (200 mM) and determined from the weight percentage ratio of the amount of encapsulated drug to polymer and was found to be 36 (±2) wt%. Next, we monitored the DTX release profile from the drug-loaded NGs in neutral (pH 7.4, PBS), acidic (pH 5.5), reducing (10 mM GSH), and acidic–reducing environments (pH 5.5, 10 mM GSH). As shown in Fig. 5a, there is a significant difference in drug release between neutral and reducing environments. It was hypothesized that NGs lose their structural integrity upon exposure to GSH and start to release their cargo. As a comparison, DTX release from NGs at pH 7.4 plateaus at 11% after 2 hours. Additionally, the pH of the environment plays a critical role in the release of DTX; an acidic environment (pH 5.5) boosts the release compared to the release in 10 mM GSH alone. According to the DLS measurements shown in Fig. S9,‡ it was observed that the NGs were unstable under acidic conditions and expanded. We hypothesize that this loss of nanogel stability is due to the presence of pH-sensitive, hydrolyzable ester bonds between the polymer backbone and crosslinking junctions, which results in the release of cargo, albeit less than that observed in the presence of both acidic and reducing environments.
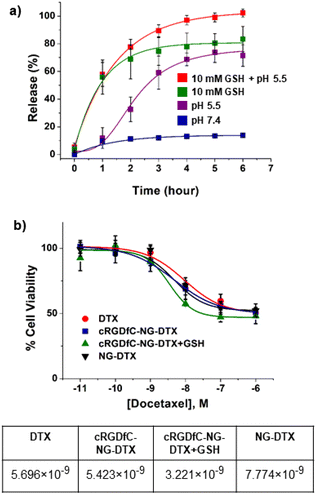 |
| | Fig. 5 (a) DTX release in a reducing, non-reducing, acidic and neutral environments for 24 hours at 37 °C and (b) cell viabilities of MDA-MB-231 tumor cells treated with the DTX, cRGDfC-NG-DTX, cRGDfC-NG-DTX+GSH, and NG-DTX. | |
The cytotoxic effect of DTX-loaded NGs was assessed using human breast carcinoma MDA-MB-231 cells. Due to our interest in active targeting and redox-responsive degradation of NGs, cell viability tests were performed with free DTX, NG-DTX, cRGDfC-NG-DTX, and cRGDfC-NG-DTX+GSH. For the latter, MDB-MB-231 cells were previously treated with GSH to reach an efficient intracellular GSH concentration. The results showed that the EC50 value of cRGDfC-NG-DTX and NG-DTX are somewhat similar. Additionally, cRGDfC-NG-DTX+GSH showed slightly improved cytotoxic efficacy on MDB-MB-231 cells when GSH was present in the intracellular environment (Fig. 5b). Although these differences in cytotoxicity are marginal, a clear effect of the targeting group is observed in the internalization studies. The in vitro cellular internalization of cRGDfC-NG loaded with the fluorescent NR dye was assessed with MDA-MB-231 cells. The cells were treated with an equivalent amount of NR-containing NG, cRGDfC-NG, and cRGDfV+cRGDfC-NG for 3 hours, where the cRGDfV peptide, which has a higher affinity for the integrin receptors, was introduced as a competitive inhibitor to establish that the enhanced uptake was due to receptor-mediated internalization. The NR-loaded NGs were gradually internalized by the MDA-MB-231 cells as suggested by the increase in red fluorescence (Fig. 6a). The presence of cRGDfC on the surface of the NG (cRGDfC-NG) increased the NR internalization compared to the NG without the cRGDfC (NG) motif. The addition of the competitive integrin-targeting peptide cRGDfV resulted in lower NR internalization, as indicated by fluorescence microscopy. The uptake of cRGDfC-NG was also investigated and quantified using flow cytometry. The mean fluorescence intensity values of the MDA-MB-231 cells treated with NR-loaded cRGDfC-NG compared to NG (non-targeted) showed that the targeting group bearing nanogels was preferentially internalized (Fig. 6b).
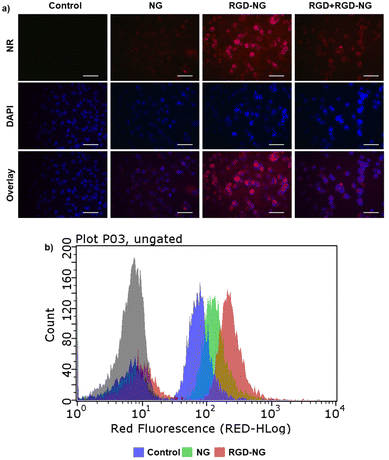 |
| | Fig. 6 (a) NR-internalization of NG, RGD-NG, and RGD+RGD-NG into MDA-MB-231 cells after 3 hours. NR provides a red fluorescence signal, whereas the cell nuclei are blue due to DAPI staining (scale bar: 50 μm), and (b) flow cytometry histograms of the control, and upon NG and cRGDfC-NG treatment. | |
Conclusions
In summary, a redox-responsive nanogel system for application in targeted drug delivery was prepared using the reactivity differences between the thiol–maleimide and disulfide-thiol exchange chemistry. A new monomer containing a disulfide linkage and a masked maleimide group was designed for this purpose. Copolymers of this unique monomer with oligo(ethylene glycol)-based monomers furnish a thermo-responsive polymer that yields nano-sized aggregates upon heating in aqueous media. The difference in the reactivity of the disulfide and maleimide groups towards thiols enables crosslinking of aggregates using bis-thiol-based crosslinkers. Some of the residual maleimide groups in the nanogels are functionalized by the peptide-targeting ligand cRGDfC through a thiol–maleimide addition reaction. Nanogels could be loaded with hydrophobic dyes and drugs, and a redox-responsive release was observed under a reducing environment. Preferential cellular internalization and enhanced cytotoxicity against MDA-MB-231 were observed for the targeting group constructs. Overall, the robust fabrication of these redox-responsive and functionalizable nanogels offers a reliable targeted drug delivery platform that is versatile enough to be evaluated for the treatment of various cancers by loading and conjugating appropriate drugs and targeting moieties.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge The Presidency of Republic of Turkey Directorate of Strategy and Budget for Infrastructure Grant No. 2009K120520.
References
- K. K. Hershberger, A. J. Gauger and L. M. Bronstein, ACS Appl. Bio Mater., 2021, 4, 4720–4736 CrossRef CAS PubMed.
- N. R. Ko and J. K. Oh, Biomacromolecules, 2014, 15, 3180–3189 CrossRef CAS PubMed.
- S. Kalepu and V. Nekkanti, Drug Delivery Transl. Res., 2016, 6, 319–332 CrossRef CAS PubMed.
- A. P. Griset, J. Walpole, R. Liu, A. Gaffey, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2009, 131, 2469–2471 CrossRef CAS PubMed.
- G. S. Kwon and T. Okano, Adv. Drug Delivery Rev., 1996, 21, 107–116 CrossRef CAS.
- A. Z. Wilczewska, K. Niemirowicz, K. H. Markiewicz and H. Car, Pharmacol. Rep., 2012, 64, 1020–1037 CrossRef CAS PubMed.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS PubMed.
- Y. Hui, X. Yi, F. Hou, D. Wibowo, F. Zhang, D. Zhao, H. Gao and C. X. Zhao, ACS Nano, 2019, 13, 7410–7424 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- P.-L. Latreille, V. Adibnia, A. Nour, J.-M. Rabanel, A. Lalloz, J. Arlt, W. C. K. Poon, P. Hildgen, V. A. Martinez and X. Banquy, Nat. Commun., 2019, 10, 4294 CrossRef PubMed.
- A. C. Anselmo, M. Zhang, S. Kumar, D. R. Vogus, S. Menegatti, M. E. Helgeson and S. Mitragotri, ACS Nano, 2015, 9, 3169–3177 CrossRef CAS PubMed.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- H. Zhang, Y. Zhai, J. Wang and G. Zhai, Mater. Sci. Eng., C, 2016, 60, 560–568 CrossRef CAS PubMed.
- F. Pinelli, Ó. F. Ortolà, P. Makvandi, G. Perale and F. Rossi, Nanomedicine, 2020, 15, 2707–2727 CrossRef CAS PubMed.
- N. M. Matsumoto, D. C. González-Toro, R. T. Chacko, H. D. Maynard and S. Thayumanavan, Polym. Chem., 2013, 4, 2464–2469 RSC.
- J. H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS PubMed.
- H. Maeda, J. Drug Targeting, 2017, 25, 781–785 CrossRef CAS PubMed.
- J. Chen, J. Ding, W. Xu, T. Sun, H. Xiao, X. Zhuang and X. Chen, Nano Lett., 2017, 17, 4526–4533 CrossRef CAS PubMed.
- H. He, A. W. Cattran, T. Nguyen, A. L. Nieminen and P. Xu, Biomaterials, 2014, 35, 9546–9553 CrossRef CAS PubMed.
- S. Maya, B. Sarmento, A. Nair, N. Rejinold, S. Nair and R. Jayakumar, Curr. Pharm. Des., 2013, 19, 7203–7218 CrossRef CAS PubMed.
- K. O. Jung, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef PubMed.
- L. Chambre, W. S. Saw, G. Ekineker, L. V. Kiew, W. Y. Chong, H. B. Lee, L. Y. Chung, Y. Bretonnière, F. Dumoulin and A. Sanyal, Bioconjugate Chem., 2018, 29, 4149–4159 CrossRef CAS PubMed.
- S. Mitra, U. Gaur, P. C. Ghosh and A. N. Maitra, J. Controlled Release, 2001, 74, 317–323 CrossRef CAS PubMed.
- A. L. Daniel-da-Silva, L. Ferreira, A. M. Gil and T. Trindade, J. Colloid Interface Sci., 2011, 355, 512–517 CrossRef CAS PubMed.
- L. Messager, N. Portecop, E. Hachet, V. Lapeyre, I. Pignot-Paintrand, B. Catargi, R. Auzély-Velty and V. Ravaine, J. Mater. Chem. B, 2013, 1, 3369–3379 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908–5916 RSC.
- R. Zhang, T. Nie, Y. Fang, H. Huang and J. Wu, Biomacromolecules, 2022, 23, 1–19 CrossRef PubMed.
- I. Altinbasak, M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7603–7624 RSC.
- H. Wang, L. Gao, T. Fan, C. Zhang, B. Zhang, O. A. Al-Hartomy, A. Al-Ghamdi, S. Wageh, M. Qiu and H. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 54621–54647 CrossRef CAS PubMed.
- A. S. Hoffman, P. S. Stayton, M. E. H. El-Sayed, N. Murthy, V. Bulmus, C. Lackey and C. Cheung, J. Biomed. Nanotechnol., 2007, 3, 213–217 CrossRef CAS.
- K. Bauri, M. Nandi and P. De, Polym. Chem., 2018, 9, 1257 RSC.
- R. Bej, K. Achazi, R. Haag and S. Ghosh, Biomacromolecules, 2020, 21, 3353–3363 CrossRef CAS PubMed.
- N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2018, 19, 3572–3585 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
- A. E. Ekkelenkamp, M. R. Elzes, J. F. J. Engbersen and J. M. J. Paulusse, J. Mater. Chem. B, 2018, 6, 210–235 RSC.
- S. Kumar and P. De, Polymer, 2014, 55, 824–832 CrossRef CAS.
- A. P. Griset, J. Walpole, R. Liu, A. Gaffey, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2009, 131, 2469–2471 CrossRef CAS PubMed.
- Y. Li, Q. N. Bui, L. T. M. Duy, H. Y. Yang and D. S. Lee, Biomacromolecules, 2018, 19, 2062–2071 CrossRef CAS PubMed.
- M. S. Alkanawati, M. Machtakova, K. Landfester and H. Thérien-Aubin, Biomacromolecules, 2021, 22, 2976–2984 CrossRef CAS PubMed.
- J. Huang, C. Zheng, H. Xiao, H. Huang, Y. Wang, M. Lin, J. Pang, Y. Wang, Y. Yuan and X. Shuai, J. Controlled Release, 2021, 340, 259–270 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, J. T. Wang, X. Zhang and Y. Yang, Polym. Chem., 2021, 12, 554–563 RSC.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47, 1449–1451 RSC.
- N. Boehnke, J. K. Kammeyer, R. Damoiseaux, H. D. Maynard, N. Boehnke, J. K. Kammeyer, D. Maynard and R. Damoiseaux, Adv. Funct. Mater., 2018, 28, 1705475 CrossRef.
- A. Degirmenci, R. Sanyal, M. Arslan and A. Sanyal, Polym. Chem., 2022, 13, 2595–2607 RSC.
- R. Kilic Boz, D. Aydin, S. Kocak, B. Golba, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2022, 33, 847 CrossRef PubMed.
- O. Gok, P. Erturk, B. Sumer Bolu, T. N. Gevrek, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 2463–2477 CrossRef CAS PubMed.
- I. Altinbasak, R. Sanyal and A. Sanyal, RSC Adv., 2016, 6, 74757–74764 RSC.
- S. Santra, S. Kolay, S. Sk, D. Ghosh, A. Mishra, L. Roy, K. Sarkar and M. R. Molla, Polym. Chem., 2022, 13, 3294–3303 RSC.
- M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 1763–1773 RSC.
- Z. Jiang and S. Thayumanavan, Isr. J. Chem., 2020, 60, 132–139 CrossRef CAS PubMed.
- D. J. Phillips and M. I. Gibson, Antioxid. Redox Signal., 2014, 21, 786–803 CrossRef CAS PubMed.
- B. Saha, S. Bhattacharyya, S. Mete, A. Mukherjee and P. De, ACS Appl. Polym. Mater., 2019, 1, 2503–2515 CrossRef CAS.
- R. Bej, J. Sarkar and S. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 194–202 CrossRef CAS.
- L. Chambre, A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2018, 29, 1885–1896 CrossRef CAS PubMed.
- B. Aktan, L. Chambre, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 490–497 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- Y. Oz and A. Sanyal, Chem. Rec., 2018, 18, 570–586 CrossRef CAS PubMed.
- M. Arslan, B. S. Bolu, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7137–7146 RSC.
- L. Chambre, H. Maouati, Y. Oz, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2020, 31, 2116–2124 CrossRef CAS PubMed.
- N. Cengiz, T. N. Gevrek, R. Sanyal, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2020, 31, 1382–1391 CrossRef CAS PubMed.
- Y. Oz, M. Arslan, T. N. Gevrek, R. Sanyal and A. Sanyal, ACS Appl. Mater. Interfaces, 2016, 8, 19813–19826 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- I. Altinbasak, S. Kocak, R. Sanyal and A. Sanyal, Biomacromolecules, 2022, 23, 3525–3534 CrossRef CAS PubMed.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
Footnotes |
| † Dedicated to the warm memory of Prof. Yusuf Yagci. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR spectrum of the polymer, 13C NMR spectrum of the monomer, LCMS spectrum monitoring reactions, UV-vis spectrum of nanogels, FTIR spectrum of the monomer, and SEC plots of polymers. See DOI: https://doi.org/10.1039/d3py00210a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Salli
Kocak
a,
Rana
Sanyal
ab and
Amitav
Sanyal
a,
Salli
Kocak
a,
Rana
Sanyal
ab and
Amitav
Sanyal






