
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flame retardant phosphonate-functionalised polyethylenes†
Nichabhat
Blake
 ,
Zoë R.
Turner
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
,
Zoë R.
Turner
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk
First published on 15th June 2023
Abstract
The two-step synthesis of phosphonate-functionalised polyethylenes with enhanced thermal and flame-retardant properties has been described. Solution phase copolymerisation of ethylene and 11-bromo-1-undecene using the commercially available early transition metal catalyst, rac-ethylenebis(indenyl)zirconium dichloride, produced poly(ethylene)-co-(11-bromo-1-undecene) with comonomer incorporation levels up to 6.10 mol%. Solvent-free post-polymerisation modification of the poly(ethylene)-co-(11-bromo-1-undecene) with the phosphite esters, P(OiPr)3 and P(OPh)3 successfully converted the bromide group into the corresponding phosphonate group on polyethylene (PE–PO(OR)2). The phosphonate-functionalised polyethylene was characterised by NMR and IR spectroscopy, GPC and DSC. Higher thermal stability (higher T10%, T50%, Tmax and % residue at 700 °C) was determined by TGA with respect to pure LDPE. Microscale combustion calorimetry (MCC) tests indicate significant lower values of heat release capacity (HRC), peak heat release rate (pHRR) and total heat release (THR) compared to those of LDPE, indicating an enhanced flame retardancy. Importantly, blends of LDPE and PE–PO(OR)2 with weight ratios of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 95:5 and 99:1 demonstrate higher thermal stability compared to LDPE despite the low concentration of the phosphonate group. Additional incorporation of traditional inorganic flame retardant additives (aluminium hydroxide, ATH), LDPE:ATH:PE–PO(OR)2 with 80:10:10 wt%, presents higher thermal stability than pure LDPE or blended LDPE with PE–PO(OR)2 due to a synergistic effect from the combination of the phosphonate group and ATH.
:10, 95:5 and 99:1 demonstrate higher thermal stability compared to LDPE despite the low concentration of the phosphonate group. Additional incorporation of traditional inorganic flame retardant additives (aluminium hydroxide, ATH), LDPE:ATH:PE–PO(OR)2 with 80:10:10 wt%, presents higher thermal stability than pure LDPE or blended LDPE with PE–PO(OR)2 due to a synergistic effect from the combination of the phosphonate group and ATH.
Introduction
Despite being the most widely used plastic in the world, polyethylene has a low limiting oxygen index (LOI) and poor fire resistance which restrict its uses in many desirable applications requiring fire safety standards.1 Traditionally, flame retardancy of polyolefins was improved by using flame-retardant additives such as chlorinated or brominated compounds, often used in synergistic combination with antimony trioxide.2 Many halogenated chemicals have proven to be highly persistent, bioaccumulative and toxic in the environment and to animals and humans.3 Phosphorus flame retardants (PFRs) are considered alternatives for halogenated FRs due to their chemical versatility, multiple flame retardant mechanisms and high flame retardancy even at low loadings.4 Three different general structures of these PFRs can be recognised: the phosphite esters (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(OR)3), the phosphonates (OP(OR)2R), and the phosphinates (OP(OR)R2). Most PFRs have mechanisms of action in the solid or condensed phase by enhancing char formation.4b,c,5 In the condensed phase, PFRs act as acid precursors leading to the formation of a char layer by esterification and dehydration, which shields the material from oxygen and prevents the formation of flammable gases.6 The gas-phase mechanism generally acts in parallel with the condensed phase mechanism by releasing PO˙ radicals which react with H˙ and OH˙ radicals formed during the combustion of hydrocarbon fuels and lower their concentrations.7
P(OR)3), the phosphonates (OP(OR)2R), and the phosphinates (OP(OR)R2). Most PFRs have mechanisms of action in the solid or condensed phase by enhancing char formation.4b,c,5 In the condensed phase, PFRs act as acid precursors leading to the formation of a char layer by esterification and dehydration, which shields the material from oxygen and prevents the formation of flammable gases.6 The gas-phase mechanism generally acts in parallel with the condensed phase mechanism by releasing PO˙ radicals which react with H˙ and OH˙ radicals formed during the combustion of hydrocarbon fuels and lower their concentrations.7
Flame retardant properties of the polymer can be improved by physically mixing the polymer with flame retardant additives1b,8 or chemically binding the phosphonate group into the polymer by radical polymerisation9 or metal-catalysed copolymerisation.10 The introduction of the phosphonate group into the polymer by chemical modification not only enhances the flame retardancy efficiency with a relatively low level of comonomers but also mitigates issues related to physical blending with additives such as mobility in polymer or poor compatibility with polymer matrix.9f,11
The copolymerisation of olefins and polar comonomers appears to be the most facile approach to incorporate polar functionalities into polyolefins giving rise to higher order polymeric architectures with substantial changes in material properties.12 However, electrophilic early transition metal (groups 3 and 4) catalysis, which has been widely used in conventional olefin polymerisation, encounters severe limitations due to strong interactions between Lewis-acidic cationic metal centres and Lewis-basic polar functional groups, preventing olefin coordination and insertion and deactivating the catalyst. Also, the coordination of the Lewis basic group on the inserted comonomer at the metal centre forms a chelate structure. This effect is severe in early transition metal catalysed copolymerisation with short-chain comonomers, since a stable 5- to 7-membered chelating ring can be formed. Strategies such as functional group protection using excess Lewis acidic masking reagents such as methylaluminoxane (MAO) or trisaluminiumalkyl (AlR3) have been employed.13 Use of longer linker comonomers was reported to be able to effectively suppress polar monomer binding.14 Use of late transition metal catalysts (Ni and Pd) for copolymerisation of ethylene and polar comonomers have also been widely reported due to their lower acidity and oxophilicity than early transition metal catalysts.10,15 However, the high cost and toxicity hinder industrial upscaling.16
Herein, we report a solution for the challenges associated with the synthesis of phosphonate-functionalised polyethylene using metal catalysed copolymerisation; the two-step synthesis of phosphonate-functionalised polyethylene via solution-phase copolymerisation of ethylene and 11-bromo-1-undecene (11-Br) using commercially available rac-ethylenebis(indenyl)zirconium dichloride (Zr) as a catalyst and methylaluminoxane (MAO) as a cocatalyst (Scheme 1a) followed by post-polymerisation modification of poly(ethylene)-co-(11-bromo-1-undecene) with phosphite esters (P(OiPr)3 and P(OPh)3) (Scheme 1b). The effect of varying concentrations of catalyst, comonomer and triisobutylalminium (TIBA) on activity and comonomer incorporation level was studied. Characterisation, thermal property and flame retardancy of phosphonate functionalised polyethylene were also investigated and these polymers show remarkable enhancements in thermal stability and flame retardent properties with respect to the virgin polymer.
 | ||
| Scheme 1 Synthesis of phosphonate-functionalised polyethylene via (a) copolymerisation of ethylene and 11-bromo-1-undecene using rac-ethylenebis(indenyl)zirconium dichloride and methylaluminoxane followed by (b) post-polymerisation modification with phosphite esters. | ||
Results and discussion
Solution-phase copolymerisation of ethylene and bromoalkene
Solution-phase copolymerisation of ethylene and a series of ω-bromo-α-alkenes were carried out with Zr and MAO as a cocatalyst. The summarised copolymerisation data suggests the prominent effect of a distance between the double bond and the bromide group on the catalytic activity and comonomer incorporation level (Table 1). For the bromoalkenes with longer methylene chains, 11-bromo-1-undecene (11-Br) and 7-bromo-1-heptene (7-Br), a copolymer with higher comonomer incorporation with higher yield and activity was afforded, which is consistent with the literature.17 For the comonomers with shorter methylene chains, 5-bromo-1-pentene (5-Br) and 6-bromo-1-hexene (6-Br), the metal centre could be poisoned through backbiting of the bromide group generating chelate structures which retard the rate of copolymerisation.12 The backbiting for the long-chain comonomer to form the bigger ring, however, is not favourable due to lower entropy.17b Another potential side product is the metal–Br species from the β-heteroatom elimination resulting in inactive catalytic species.12| Entry | Comonomer | Activityb | Productivityc | Yield (g) | Incorporation leveld (mol%) | Comonomer conversion (%) | M w | M w/Mn | SCB/1000TCf | T m (°C) | Crystallinityg (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Conditions: rac-ethylenebis(indenyl)zirconium dichloride (Zr) used as a catalyst, MAO used as a cocatalyst and scavenger with [Al]0:[Zr]0 = 1000:1, [comonomer]0 = 91 mM, [TIBA]0:[comonomer]0 = 1:10, 0.5 mL toluene, 49.5 mL hexanes, ethylene 2 bar, 0.5 h, 343.15 K.
b kgPE molM−1 h−1 bar−1.
c kgPE gcat−1 h.
d Determined by 1H NMR spectroscopy in C2D2Cl4 at 130 °C (see ESI†).
e g mol−1.
f Determined by GPC-IR.
g Determined by DSC.
h [comonomer]0 = 182 mM.
|
|||||||||||
| 1 | 5-Bromo-1-pentene | 2634 | 12.6 | 3.2 | 1.7 | 38 | 53600 |
4.5 | 2.4 | 120 | 68 |
| 2 | 6-Bromo-1-hexene | 552 | 2.6 | 0.7 | 1.1 | 7 | 30100 |
5.3 | 6.2 | 117 | 60 |
| 3 | 7-Bromo-1-heptene | 3167 | 15.2 | 3.8 | 2.9 | 75 | 52100 |
6.0 | 5.9 | 119 | 47 |
| 4 | 11-Bromo-1-undecene | 3056 | 14.6 | 3.7 | 3.8 | 90 | 36900 |
4.4 | n/a | 121 | 23 |
| 5h | 11-Bromo-1-undecene | 2963 | 14.1 | 5.3 | 4.9 | 74 | 27400 |
4.3 | 8.9 | 116 | 24 |
| 6h | 1-Dodecene | 2394 | 11.4 | 4.3 | 6.8 | 88 | 21600 |
3.9 | 16.4 | 125 | 44 |
Copolymerisation of ethylene and 1-dodecene was also carried out to study the impact of polar comonomer (11-Br) vs. non-polar olefin (1-dodecene) on copolymerisation activity and incorporation level. Under analogous conditions (Table 1, entries 5 and 6), comparable activity and comonomer incorporation level were observed suggesting a trivial effect of the functional bromide group on copolymerisation with ethylene. A monomodal molecular weight distribution was displayed from their GPC traces (Fig. S59†). The short chain branching (SCB) profile of poly(ethylene)-co-(1-dodecene) shows a higher Mw fraction containing higher short chain branches (SCB)/1000TC suggesting more 1-dodecene was incorporated at the higher molar mass fractions (Fig. S59†). The SCB profile from using 11-Br suggests that the comonomer was more evenly distributed across the range of molecular weights. The 13C{1H} NMR spectrum of poly(ethylene)-co-(1-dodecene) shows signals at 120.55 and 99.91 ppm corresponding to the olefinic end group (Fig. S12†). These signals were diminished from those of poly(ethylene)-co-(11-bromo-1-undecene) (Fig. S10†).
Compared with other polar comonomers, Zr exhibits higher catalytic activity and better tolerability toward the bromide group which could be explained by the activation and reactivation route.17a,18 During the copolymerisation, the bromide group of the free comonomer or the end group of growing polymer chains can compete with ethylene to coordinate at the metal centre and form the dormant species. Unlike other heteroatoms, the dormant species can be reactivated due to much lower metal–halogen bond energy and the active site can therefore be regenerated. This mechanism also implies that the more electronegative fluoride and chloride groups will have higher binding energies to the metal centre compared to the bromide and iodide groups; hence, will be difficult to liberate during the reactivation step to regenerate the active cationic species.17a
As the highest comonomer incorporation and activity was obtained using 11-Br, an in-depth study of ethylene/11-Br copolymerisation was carried out to study the effect of varying concentrations of catalyst, comonomer and TIBA on the activity and comonomer incorporation level. The copolymerisation data are summarised in Table 2. Under analogous conditions, a significant reduction in activity was observed from copolymerisation of ethylene and 11-Br (entries 2–9) compared to ethylene homopolymerisation (entry 1). The decreased activity could be explained by the so-called “Polar Monomer Problem”.12 The copolymerisation without TIBA as a protecting agent for 11-Br showed that only 34% of 11-Br was copolymerised, resulting in copolymer containing only 0.50 mol% incorporation level (entry 2). The molar ratio of 2:1 of TIBA:comonomer was used for the copolymerisation of ethylene and 11-Br (entry 3). Excess TIBA was widely employed to mitigate the Lewis basic functional group effects.19 Copolymer with 1.12 mol% incorporation level was obtained which 93% of 11-Br. However, its comonomer incorporation cannot be accurately determined from its 1H NMR spectrum due to the presence of signals tentatively corresponding to the residual oxides (Fig. S4†). A similar issue with the 1H NMR spectrum was also encountered from the copolymerisation with an absence of TIBA (Fig. S4†). To reduce the undesired effect from the excess TIBA, a molar ratio of 1:10 TIBA:11-Br was employed leading to higher activity, productivity and incorporation level (Table 2, entries 3–9).
| Entry | Zr (mM) | Ratiob | 11-Br (mM) | Activityc | Productivityd | Yield (g) | Incorporatione (mol%) | Comonomer conv. (%) | M w | M w/Mn | T m (°C) | Crystallinityf (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Conditions: rac-ethylenebis(indenyl)zirconium dichloride (Zr), MAO used as a cocatalyst and scavenger with [Al]0:[Zr]0 = 1000:1, 0.5 mL toluene, 49.5 mL hexanes, ethylene 2 bar, 0.5 h, 343.15 K.
b [TIBA]0:[11-Br]0.
c kgPE molM−1 h−1 bar−1.
d kgPE gcat−1 h.
e Determined by 1H NMR in C2D2Cl4 at 130 °C.
f Determined by DSC.
g g mol−1.
h Time = 1, 45 and 60 minutes for entries 1, 8 and 9.
|
||||||||||||
| 1h | 0.024 | 0 | 0 | 12880 |
1.0 | 0.5 | — | — | 292300 |
4.4 | 133 | 78 |
| 2 | 0.2 | 0 | 50 | 556 | 1.33 | 5.0 | 0.50 | 34 | 29400 |
3.4 | 131 | 85 |
| 3 | 0.2 | 2 | 50 | 604 | 1.44 | 6.0 | 1.12 | 93 | 5800 | 4.2 | 120 | 95 |
| 4 | 0.1 | 0.1 | 91 | 793 | 1.89 | 3.8 | 2.44 | 61 | 42400 |
7.6 | 127 | 49 |
| 5 | 0.05 | 0.1 | 91 | 1246 | 2.98 | 3.9 | 3.70 | 88 | 61000 |
8.2 | 125 | 37 |
| 6 | 0.025 | 0.1 | 91 | 3125 | 7.47 | 3.7 | 4.07 | 92 | 36900 |
4.4 | 122 | 20 |
| 7 | 0.025 | 0.1 | 182 | 4322 | 10.33 | 5.3 | 5.36 | 78 | 32900 |
3.6 | 125 | 23 |
| 8h | 0.025 | 0.1 | 182 | 2995 | 10.73 | 5.4 | 5.80 | 85 | 39800 |
4.5 | 124 | 22 |
| 9h | 0.025 | 0.1 | 274 | 2460 | 11.75 | 5.9 | 6.10 | 65 | 27000 |
3.4 | 123 | 24 |
Higher yield, productivity, activity and comonomer incorporation level were observed with decreasing catalyst concentration (entries 4–6). 92% of 11-Br was copolymerised when using 0.025 mM of Zr, followed by 88 and 61% when using 0.05 and 0.1 mM of Zr, respectively. The comonomer incorporation level determined from 1H NMR spectroscopy is in the range of 2.44–6.10 mol% which is higher than those reported in the literature.17a,18,20 The signal at 3.49 ppm corresponds to the methylene protons adjacent to the bromide group indicating the successful incorporation of 11-Br (Fig. S5†). A reduction in melting temperature (Tm) and crystallinity from the copolymer containing bromide group compared to those of homopolyethylene was observed from DSC (Table 2).
The molecular weights of the copolymers are much lower than those of polyethylene and considerably decline at higher concentrations of 11-Br (Table 2). The reduction in molecular weight for the copolymers could be attributed to the chain transfer reactions induced by the spatial active site of Zr. In addition to the chain transfer to aluminium alkyl, β-H transfers to metal and monomer are also significant in the polymerisation using Zr.18,21 The competing coordination to the metal centre between the 11-Br and ethylene could hinder the incorporation of ethylene and accelerate the chain transfer reactions resulting in a reduction in molecular weights of the resultant copolymers. Broad molecular weight distribution (Mw/Mn = 7.6–8.2) was observed at copolymerisation using 0.1 and 0.05 mM of Zr. Better control on molecular weight distribution was observed when the Zr concentration was reduced to 0.025 mM, displaying narrower Mw/Mn values between 3.4–4.5. The FT-IR spectrum of the copolymer shows the adsorption band assigned to C–Br stretching at 656 cm−1 (Fig. S40†). The bands at 719 and 1463 cm−1 correspond to the bending vibrations and the rocking vibration of methylene protons of polyethylene. Additionally, the bands at 2916 and 2848 cm−1 are attributed to the asymmetric and symmetrical stretching vibrations of CH2 of polyethylene.
Post-polymerisation modification of brominated functionalised polyethylene
Attempted copolymerisation of ethylene and phosphonate comonomer, CH2CH(CH2)nPO(OiPr)2 (n = 2–6), using Zr/MAO was carried out. However, NMR and IR spectroscopy, and DSC analyses indicate an absence of phosphonate group on the polyethylene. An alternative route for the synthesis of phosphonate-functionalised polyethylene was therefore investigated. Reactions of poly(ethylene)-co-(11-bromo-1-undecene) and phosphite esters (P(OR)3, R = iPr and Ph) were used to synthesise phosphonate-functionalised polyethylene using the Michaelis–Arbuzov reaction.22,23
Post-polymerisation modification of bromo-functionalised polyethylene using triisopropyl phosphite
Poly(ethylene)-co-(11-bromo-1-undecene) with 5.80 mol% comonomer incorporation level was reacted with an excess of triisopropyl phosphite (P(OiPr)3, 50 equiv.) in a neat condition at 130 °C for 48 h. Aliquots were collected at 3, 6, 24 and 48 h and analysed by 1H NMR spectroscopy (Fig. S32†). The 1H NMR spectrum of the aliquot taken at 48 h shows that 96% of the bromide group was converted to the diisopropyl phosphonate group (Fig. S32†). The reaction temperature was increased from 130 to 180 °C to reduce the reaction time. Under analogous conditions. aliquots were taken at 3, 6, 24 and 48 h. The 1H NMR spectra of the aliquot collected after 3 and 6 h shows 97 and 100% of the bromide group converted to the phosphonate group, respectively (Fig. S33†).The signal of the methylene protons adjacent to the bromide group (CH2Br, δ = ca. 3.50 ppm) was absent while the one from the phosphonate moiety, PO(OCHMe2)2, was observed at 4.74 ppm. The COSY NMR spectrum shows a through-bond correlation between the signals corresponding to the methine protons and the methyl protons from the phosphonate group (4.74 ppm vs. 1.40 ppm) (Fig. S16†). The 31P{1H} NMR spectrum shows a sharp signal at 29.5 ppm indicating the presence of the phosphonate group into the polyethylene (Fig. S15†). It is noted that the signal of triisopropyl phosphite at ca. 140 ppm was not observed. The 1H–31P HMBC NMR spectrum shows cross-peaks at 4.7 ppm (1H)-29.5 ppm (31P{1H}) and 1.7 ppm (1H)-29.5 ppm (31P{1H}) (Fig. S19†). This provides support for the assignment of the resonances corresponding to the iPr group of the phosphonate moiety and its neighbouring methylene protons. The DSC analysis of the phosphonate copolymer shows a melting temperature at 123 °C and a degree of crystallinity of 40% (Fig. S67†). The presence of the phosphonate moiety on the polyethylene backbone is also confirmed by FT-IR spectroscopy (Fig. S41†). The peaks corresponding to PO at 1243 cm−1 and P–O–C at 1007–982 cm−1 were observed together with the transmission bands at 2916, 2848, 1463 and 719 cm−1 assigned to the (CH2)n of the copolymer.
The bromine concentration in the copolymer samples was analysed using the oxygen combustion flask technique with the analytical uncertainty of 0.3% absolute. 9.65 wt% of bromine was detected from poly(ethylene)-co-(11-bromo-1-undecene) with a 5.8 mol% incorporation level. After the post-polymerisation modification with P(OiPr)3, 0.23 wt% of bromine was reported despite the absence of resonances assigned to bromide moiety from the 1H and 13C{1H} NMR spectra of PE–PO(OiPr)2 (Fig. S13 and S21†).
Post-polymerisation modification of bromo functionalised polyethylene using triphenyl phosphite
Poly(ethylene)-co-(11-bromo-1-undecene) with 5.11 mol% comonomer incorporation level was reacted with an excess of triphenyl phosphite (P(OPh)3, 50 equiv.) in neat conditions at 180 °C for 24, 48 and 72 h. The 1H NMR spectra of the resulting copolymers, after purification, suggest the incomplete conversion of the bromide group to the phosphonate group, indicated by a signal at 3.49 ppm corresponding to the methylene protons of the bromide group (CH2Br) (Fig. S35†). Signals in the region of 7.15–7.50 ppm are attributed to the aromatic protons on the phenyl ring of the phosphonate moiety. Based on the integral numbers, the mole ratios of the bromide group to the phosphonate group is 50:50, 30:70 and 20:80 determined from the terpolymers obtained from the reactions carried out for 24, 48 and 72 h (PE–Br–PO(OPh)2_50%, PE–Br–PO(OPh)2_70% and PE–Br–PO(OPh)2_80%), respectively.
Similar to the post-polymerisation modification using triisopropyl phosphite, higher conversion was achieved by increasing the reaction temperature from 180 to 200 °C. After 72 h at 200 °C, the bromide group was fully converted to the phosphonate group evidenced by its 1H NMR spectrum (Fig. S22†). The 31P{1H} NMR spectrum shows a singlet at 25.14 ppm confirming the presence of the phosphonate group on the polyethylene backbone (Fig. S23†). The 1H–31P HMBC NMR spectrum shows cross-peaks at 7.39–7.28, 2.16, 1.88 ppm (1H) with 25.2 ppm (31P{1H}) supporting the NMR spectroscopic assignment of the signals corresponding to the phenyl rings and the methylene groups adjacent to the phosphonate group (Fig. S24†). Melting temperatures (Tm) in the range of 122–124 °C and degree of crystallinity of 20–32% were determined from DSC analysis. The FTIR spectrum of PE–Br–PO(OPh)2 shows the absorption bands at 1270 cm−1 (PO) and 1190 and 930 cm−1 (P–O–Ph) indicating the presence of the phosphonate moiety (Fig. S42†). The bands between 1594–1463 cm−1 were assigned to C–C stretching vibration of the aromatic ring.
Thermal stability and flammability studies
Thermogravimetric analysis (TGA) is one of the widely used techniques for the study of the thermal stability of materials and also indicates the decomposition of the polymers at various temperatures.24 The thermal stability of the investigated polymers was evaluated by the temperatures at 10 and 50% weight loss (T10% and T50%) and the temperature of maximum rates of weight loss (Tmax) as well as the solid residual char yields at 700 °C. TGA was conducted under nitrogen and air atmosphere with a heating rate of 20 °C min−1. The TGA and DTG plots of the pyrolysis and thermal-oxidative of the commercial LDPE and phosphonate-functionalised polyethylene are displayed in Fig. 1. The summarised results are shown in Table 3. | ||
| Fig. 1 TGA curves of reference polymers under nitrogen (a) and air (c) atmospheres and DTG curves of reference polymers under nitrogen (b) and air (d) atmospheres. | ||
| Entry | Sample | T m (°C) | Crystallinity (%) | Nitrogen atmosphere | Air atmosphere | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T 10% (°C) | T 50% (°C) | T max (°C) | Residueb (%) | T 10% (°C) | T 50% (°C) | T max (°C) | Residueb (%) | ||||
| a TGA tests were performed between 50–800 °C under nitrogen and air atmospheres with a heating rate of 20 °C min−1. b Residue at 700 °C. | |||||||||||
| 1 | LDPE | 110 | 47 | 471 | 503 | 503 | 0.16 | 300 | 355 | 335 | 0.22 |
| 2 | PE–PO(OiPr)2 | 123 | 19 | 307 | 509 | 293, 509 | 1.1 | 295 | 492 | 284, 487 | 4.01 |
| 3 | PE–Br–PO(OPh)2_50% | 124 | 23 | 462 | 505 | 508 | 0.95 | 377 | 481 | 393, 483 | 2.83 |
| 4 | PE–Br–PO(OPh)2_70% | 122 | 32 | 423 | 503 | 506 | 1.16 | 360 | 496 | 342, 496 | 3.24 |
| 5 | PE–Br–PO(OPh)2_80% | 124 | 20 | 457 | 503 | 505 | 1.66 | 362 | 505 | 344, 503 | 3.35 |
| 6 | PE–PO(OPh)2 | 117 | 18 | 475 | 510 | 510 | 1.22 | 455 | 506 | 333, 507 | 3.51 |
Under nitrogen atmosphere, TGA and DTG curves of PE–PO(OiPr)2, PE–PO(OPh)2, and LDPE demonstrate comparable T50% and Tmax (500–510 °C) (Table 3, Fig. 1a and b). However, T10%, T50% and Tmax obtained from LDPE were lower when performed under an air atmosphere than nitrogen atmosphere. A single-decomposition step was observed from testing LDPE under both atmospheres evidenced by a single DTG peak (Fig. 1b and d). The residue of the LDPE at 700 °C was at 0.16–0.22%. The presence of the phosphonate group on polyethylene leads to higher thermal stability; thermal degradation of PE–PO(OiPr)2 and PE–PO(OPh)2 gives higher T50% and Tmax and residual percentage at 700 °C than those of LDPE. The T10% of PE–PO(OiPr)2 conducted under an air atmosphere (295 °C) is lower than LDPE (300 °C) which is attributed to the lower bond dissociation energy of OP–O and P–O–C bonds than C–C bonds.25 The phosphonate group decomposes during the first stage of heating between ca. 200–350 °C resulting in the formation of a carbonaceous char layer, shielding the material from oxygen and preventing the formation of flammable gases.4b Therefore, the degradation of polyethylene occurred at higher temperatures indicated by higher T50% and Tmax and an increased residual char yield (3.51–4.01%) compared to LDPE.26 Higher T10% from PE–PO(OPh)2 than PE–PO(OiPr)2 (455 vs. 270 °C) is attributed to the aromatic rings incorporated into PE backbone resulting in the delayed first stage of decomposition (Fig. 1c and d).27 The highest char yield (4.01%) was gained from thermal-oxidative degradation of PE–PO(OiPr)2 (Table 3, entry 2), whilst the highest Tmax and T50% were observed from the TGA test of PE–PO(OPh)2 under an air atmosphere (Table 3, entry 6).
The effect of the incorporation level of the phosphonate group on thermal stability was observed from the TGA curves of PE–Br–PO(OPh)2_x% (x = 50, 70 and 80) and PE–PO(OPh)2 conducted under nitrogen and air atmosphere (Fig. 2). During the first thermal-oxidative degradation stage, a higher phosphonate incorporation level results in lower T10%. During the second decomposition stage, higher T50% and Tmax were observed as well as higher char residual percentage correlated with a higher phosphonate incorporation level.
 | ||
| Fig. 2 TGA curves of PE–PO(OPh)2 and PE–Br–PO(OPh)2_x% (x = 50, 70 and 80) under nitrogen (a) and air (c) atmospheres and DTG curves of reference polymers under nitrogen (b) and air (d) atmospheres. | ||
Microscale combustion calorimetry (MCC) is a thermal analysis method evaluating for flammability screening of polymers. The MCC method directly measures the heat of combustion of the gases evolved during controlled heating of milligram-sized samples.28 The MCC test was conducted three times for each sample and the average values of the flammability parameters of PE–Br–PO(OPh)2_50% and PE–Br–PO(OPh)2_80% including the heat release capacity (HRC), the peak heat release rate (pHRR), the total heat release (THR) and the temperature at pHRR (TpHRR) are listed in Table 4.
| Sample | HRC (J g−1 K) | pHRR (W g−1) | THR (kJ g−1) | T pHRR (°C) |
|---|---|---|---|---|
| HDPE28,29 | 1486 | 1351 | 43.5 | 504 |
| LDPE foam | 984.0 | 971.5 | 37.0 | 493.0 |
| PE–Br–PO(OPh)2_50% | 271.39 | 228.70 | 8.52 | 488.2 |
| PE–Br–PO(OPh)2_80% | 223.16 | 188.49 | 7.36 | 488.7 |
| PE–PO(OPh)2_100% | 218.37 | 185.79 | 11.10 | 496.2 |
The HDPE sample gives high values of HRC, pHRR and THR indicating a high release amount of the flammable products. More than 80% reduction in the values of HRC, pHRR and THR was observed from PE–Br–PO(OPh)2_50% and PE–Br–PO(OPh)2_80% compared to those of HDPE. These indicate that the incorporation of the phosphonate group increases the flame retardancy performance of polyethylene. Lower values of MCC parameters from PE–Br–PO(OPh)2_80% compared to those of PE–Br–PO(OPh)2_50% were observed suggesting the enhanced flame resistance correlating with higher phosphonate group incorporation level. The HRC value from the MCC test was utilised to predict the fire behaviour and flame resistance of flame-retardant polymers.28 The HRC of PE–Br–PO(OPh)2_50% and PE–Br–PO(OPh)2_80% are lower than 300 J g−1 K (HRC = 223–271 J g−1 K) suggesting the UL 94 V-0 rating.
Polymer compounding with LDPE and inorganic flame retardant additive
The mixtures of low-density polyethylene (LDPE) and PE–PO(OiPr)2 or PE–PO(OPh)2 in a ratio of 90:10, 95:5 and 99:1 wt% were prepared via melting in xylene at 120 °C for 5 minutes to obtain a clear solution. The polymer was precipitated with an addition of pentane, filtered and dried under vacuum at 90 °C for 18 h. Their thermal properties were studied using TGA performed under an air atmosphere. The TGA parameters and curves are listed in Table 5 and Fig. 3. LDPE was employed in this study due to its application as a protective jacket for wires and cables.30 However, LDPE is a highly flammable thermoplastic with melt dripping behaviour when burning.
 | ||
| Fig. 3 TGA (a and c) and DTG (b and d) curves of PE–PO(OR)2 and blended LDPE with PE–PO(OR)2 polymers under an air atmosphere. | ||
| Sample | Wt% | T 10% (°C) | T 50% (°C) | T max (°C) | Residue at 700 °C (%) | T m (°C) | Crystallinity (%) |
|---|---|---|---|---|---|---|---|
| a TGA tests were performed between 50–800 °C under an air atmosphere with a heating rate of 20 °C min−1. | |||||||
| LDPE | 100 | 299 | 355 | 335 | 0.0 | 110 | 47 |
| LDPE:PE–PO(OiPr)2 |
90:10 |
401 | 463 | 465 | 0.5 | 111 | 49 |
| 95:5 |
357 | 426 | 425 | 0.1 | 111 | 52 | |
| 99:1 |
385 | 426 | 444 | 0.3 | 110 | 38 | |
| LDPE:PE–PO(OPh)2 |
90:10 |
383 | 440 | 393, 454 | 0.4 | 111 | 41 |
| 95:5 |
389 | 427 | 426 | 0.3 | 110 | 42 | |
| 99:1 |
363 | 400 | 392 | 1.3 | 111 | 46 | |
| LDPE:ATH |
90:10 |
320 | 360 | 333 | 5.8 | 111 | 46 |
| LDPE:ATH:PE–PO(OiPr)2 |
80:10:10 |
372 | 473 | 310, 477 | 8.2 | 113 | 32 |
| LDPE:ATH:PE–PO(OPh)2 |
80:10:10 |
362 | 475 | 328, 476 | 7.1 | 111 | 42 |
As expected, reduced thermal stability was observed when blending PE–PO(OiPr)2 or PE–PO(OPh)2 with LDPE according to their decreased T10%, T50%, Tmax and % residual at 700 °C, compared with the pure phosphonate-functionalised polyethylene. The blended polymers; however, have higher T10%, T50% and Tmax compared to those of pure LDPE regardless of 10, 5 and 1 wt% substitution of LDPE with PE–PO(OiPr)2 or PE–PO(OPh)2. These signify that a low concentration of the phosphonate group on polyethylene can lead to enhanced thermal stability.
The inorganic flame retardant additive, Al(OH)3 or ATH, was blended with LDPE and PE–PO(OiPr)2 or PE–PO(OPh)2. ATH has been commercially used in polyethylene wire and cable formulations due to its low toxicity, cost efficiency, white colour and excellent flame retardancy and smoke suppression property.31 ATH function by liberating water vapour from endothermically decomposition reaction occurring between 220–400 °C. Water vapour can hinder the combustion process by diluting the flammable gases and shielding the polymer surface from oxygen. However, high loading of ATH of at least 35 wt% is required leading to degraded physical properties (increasing part density and brittleness). Therefore, synergistic flame retardant from the combination of low loading of ATH with phosphorus,30,32 silicon,32e,33 nitrogen32e,34 or boron35 containing flame retardant has been extensively studied.
The ratio of LDPE:ATH:phosphonate-functionalised polyethylene is 80:10:10 wt%. TGA and DTG curves of LDPE:ATH (90:10 wt%) show comparable thermal stability to those of pure LDPE (Fig. 4a and b). Higher T10%, T50%, Tmax and % residual at 700 °C (7.1–8.2%) was obtained from LDPE:ATH:PE–PO(OR)2 (80:10:10 wt%), compared to those with LDPE:ATH (90:10 wt%) and LDPE:PE–PO(OR)2 (90:10 wt%). The enhanced thermal stability can be attributed to the synergistic effect in the condensed phase.32b–d During the combustion process, phosphonate group on polyethylene degrades generating polyphosphoric acid. In the meantime, ATH releases water and formes aluminium oxide. Polyphosphoric acid then reacts with aluminium oxide to form aluminium metaphosphate (Al(PO3)3), increasing the density and isolation effect of the char layer and inhibiting the transmission of oxygen and heat.
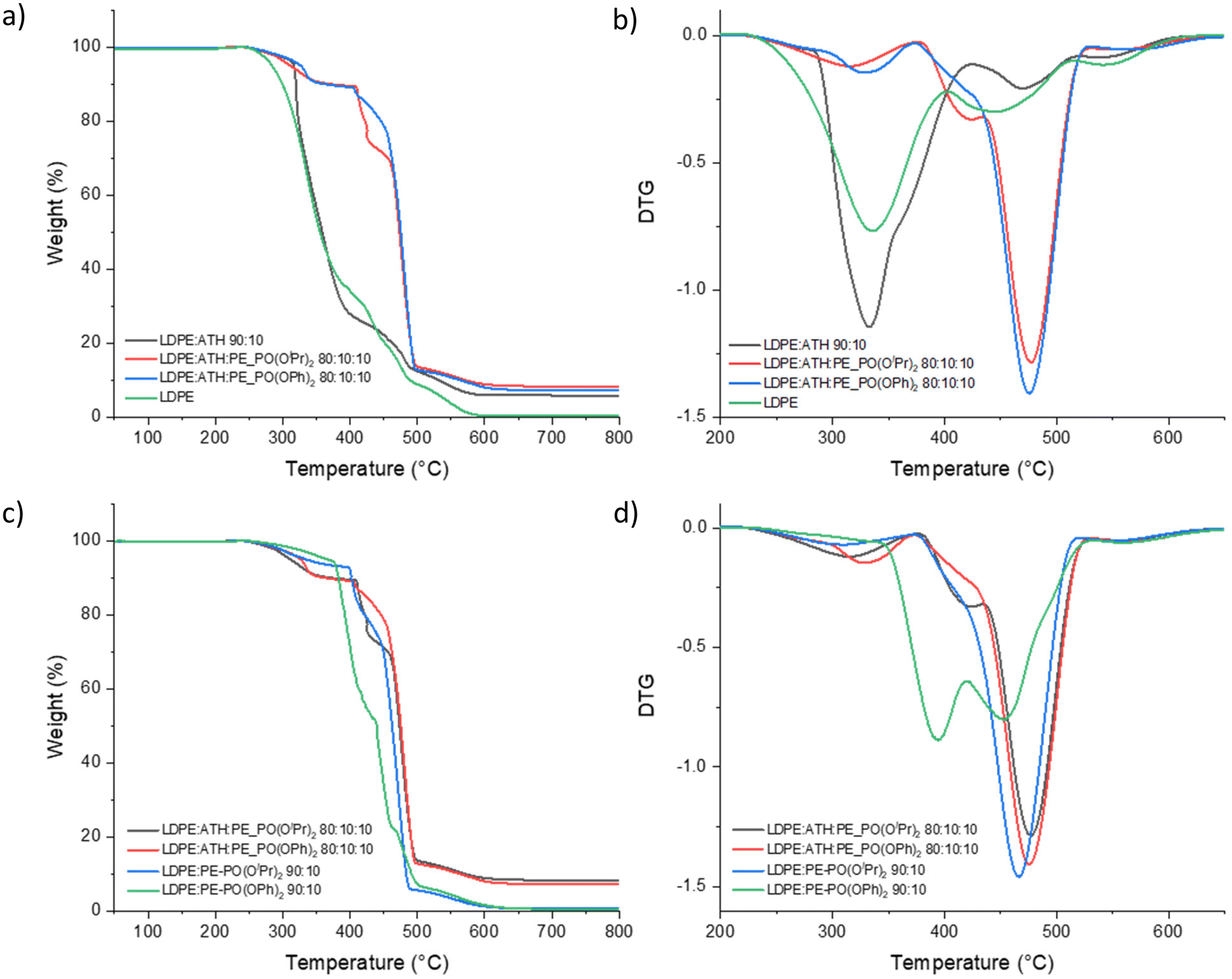 | ||
| Fig. 4 TGA (a and c) and DTG (b and d) curves of blended LDPE, ATH and PE–PO(OR)2 polymers under an air atmosphere. | ||
Conclusions
The preparation of phosphonate-functionalised polyethylenes was carried out via reaction of poly(ethylene)-co-(11-bromo-1-undecene) and phosphite esters (P(OR)3, R = iPr and Ph) under neat conditions. The presence of the phosphonate groups was indicated by 1H and 31P{1H} NMR and FT-IR spectroscopy and DSC. The thermal stability of the phosphonate-containing polyethylene was investigated by TGA. Higher thermal stability was obtained from the phosphonate-containing polyethylene compared to those of neat LDPE evaluated by higher Tmax and T50% and higher char residue at 700 °C. Two decomposition stages were observed from TGA curves of polyethylene containing phosphonate groups, while a single decomposition step was demonstrated on the TGA curve of LDPE. The phosphonate functionalised polyethylene show enhanced flame resistance indicated by more than 80% reduction in HRC, pHRR, THR and TpHRR from MCC compared to those from HDPE. The polymer compounding of LDPE with PE–PO(OiPr)2 or PE–PO(OPh)2 (90:10, 95:5 and 99:1 wt%) exhibited higher thermal stability than pure LDPE evidenced by higher T10%, T50% and Tmax determined from TGA. However, a decrease in thermal stability was observed when compared with pure phosphonate functionalised polyethylene. The improvement of thermal stability was observed from the blending of LDPE, PE–PO(OR)2 and ATH (80:10:10 wt%) compared to pure LDPE or LDPE:ATH (90:10 wt%) due to the synergistic effect in the condensed phase.
Author contributions
Nichabhat Blake: Conceptualisation, methodology, formal analysis, investigation and writing – original draft. Zoë R. Turner, Jean-Charles Buffet and Dermot O'Hare: Conceptualisation, writing – review & editing, supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. B., J.-C. B. and Z. R. T. would like to thank SCG Chemicals Co., Ltd (Thailand) for financial support and an SCG Research Fellowship (Z. R. T.).References
- (a) Y. Liu, D.-Y. Wang, J.-S. Wang, Y.-P. Song and Y.-Z. Wang, Polym. Adv. Technol., 2008, 19, 1566–1575 CrossRef CAS; (b) E. Rezvani Ghomi, F. Khosravi, Z. Mossayebi, A. Saedi Ardahaei, F. Morshedi Dehaghi, M. Khorasani, R. E. Neisiany, O. Das, A. Marani, R. A. Mensah, L. Jiang, Q. Xu, M. Försth, F. Berto and S. Ramakrishna, Molecules, 2020, 25, 5157 CrossRef PubMed.
- V. I. Babushok, P. Deglmann, R. Krämer and G. T. Linteris, Combust. Sci. Technol., 2017, 189, 290–311 CrossRef CAS PubMed.
- (a) R. J. Law, A. Covaci, S. Harrad, D. Herzke, M. A. E. Abdallah, K. Fernie, L.-M. L. Toms and H. Takigami, Environ. Int., 2014, 65, 147–158 CrossRef CAS PubMed; (b) M. Venier, A. Salamova and R. A. Hites, Acc. Chem. Res., 2015, 48, 1853–1861 CrossRef CAS PubMed; (c) I. T. Cousins, C. A. Ng, Z. Wang and M. Scheringer, Environ. Sci.: Processes Impacts, 2019, 21, 781–792 RSC; (d) P. Schmid, M. Kohler, A. C. Gerecke, E. Gujer, M. Zennegg and M. Wolfensberger, Chimia, 2003, 57, 509–513 CrossRef CAS.
- (a) L. Chen and Y.-Z. Wang, Polym. Adv. Technol., 2010, 21, 1–26 CrossRef; (b) I. van der Veen and J. de Boer, Chemosphere, 2012, 88, 1119–1153 CrossRef CAS PubMed; (c) S. Hörold, in Polymer Green Flame Retardants, ed. C. D. Papaspyrides and P. Kiliaris, Elsevier, Amsterdam, 2014, pp. 221–254 Search PubMed.
- (a) M. M. Velencoso, A. Battig, J. C. Markwart, B. Schartel and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 10450–10467 CrossRef CAS PubMed; (b) B. Schartel, Materials, 2010, 3, 4710–4745 CrossRef CAS PubMed.
- S. Rabe, Y. Chuenban and B. Schartel, Materials, 2017, 10, 455 CrossRef PubMed.
- (a) J. Green, J. Fire Sci., 1992, 10, 470–487 CrossRef CAS; (b) J. Green, J. Fire Sci., 1996, 14, 426–442 CrossRef CAS.
- (a) O. P. Korobeinichev, A. A. Paletsky, L. V. Kuibida, M. B. Gonchikzhapov and I. K. Shundrina, Proc. Combust. Inst., 2013, 34, 2699–2706 CrossRef CAS; (b) K. Salasinska, K. Mizera, M. Celiński, P. Kozikowski, M. Borucka and A. Gajek, Fire Saf. J., 2020, 115, 103137 CrossRef CAS; (c) F. Xie, Y.-Z. Wang, B. Yang and Y. Liu, Macromol. Mater. Eng., 2006, 291, 247–253 CrossRef CAS; (d) Y. Z. Wang, in Advances in Fire Retardant Materials, ed. A. R. Horrocks and D. Price, Woodhead Publishing, 2008, pp. 67–94 Search PubMed; (e) Q. Wang, J. P. Undrell, Y. Gao, G. Cai, J.-C. Buffet, C. A. Wilkie and D. O'Hare, Macromolecules, 2013, 46, 6145–6150 CrossRef CAS; (f) Y. Luo, Y. Xie, R. Chen, R. Zheng, H. Wu, X. Sheng, D. Xie and Y. Mei, Front. Chem. Sci. Eng., 2021, 15, 1332–1345 CrossRef CAS.
- (a) H. Wang, H. Niu and J.-Y. Dong, Polymer, 2017, 126, 109–115 CrossRef CAS; (b) J. R. Ebdon, D. Price, B. J. Hunt, P. Joseph, F. Gao, G. J. Milnes and L. K. Cunliffe, Polym. Degrad. Stab., 2000, 69, 267–277 CrossRef CAS; (c) T. Xu, L. Zhang, Z. Cheng and X. Zhu, Sci. China: Chem., 2015, 58, 1633–1640 CrossRef CAS; (d) M. Yılmaz, A. Akar, N. Köken and N. Kızılcan, J. Appl. Polym. Sci., 2020, 137, 49023 CrossRef; (e) D. Price, L. K. Cunliffe, K. J. Bullet, T. R. Hull, G. J. Milnes, J. R. Ebdon, B. J. Hunt and P. Joseph, Polym. Adv. Technol., 2008, 19, 710–723 CrossRef CAS; (f) W. Hu, J. Zhan, N. Hong, T. R. Hull, A. A. Stec, L. Song, J. Wang and Y. Hu, Polym. Adv. Technol., 2014, 25, 631–637 CrossRef CAS.
- J. Gao, W. Cai, Y. Hu and C. Chen, Polym. Chem., 2019, 10, 1416–1422 RSC.
- D. Price, K. Pyrah, T. R. Hull, G. J. Milnes, J. R. Ebdon, B. J. Hunt, P. Joseph and C. S. Konkel, Polym. Degrad. Stab., 2001, 74, 441–447 CrossRef CAS.
- A. Keyes, H. E. Basbug Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y.-S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 58, 12370–12391 CrossRef CAS PubMed.
- (a) H. Hagihara, K. Tsuchihara, J. Sugiyama, K. Takeuchi and T. Shiono, Macromolecules, 2004, 37, 5145–5148 CrossRef CAS; (b) J.-I. Imuta, N. Kashiwa and Y. Toda, J. Am. Chem. Soc., 2002, 124, 1176–1177 CrossRef CAS PubMed; (c) H. Terao, S. Ishii, M. Mitani, H. Tanaka and T. Fujita, J. Am. Chem. Soc., 2008, 130, 17636–17637 CrossRef CAS PubMed.
- (a) J. Chen, A. Motta, B. Wang, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 7030–7034 CrossRef CAS PubMed; (b) L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS PubMed.
- (a) B. P. Carrow and K. Nozaki, Macromolecules, 2014, 47, 2541–2555 CrossRef CAS; (b) R. Nakano and K. Nozaki, J. Am. Chem. Soc., 2015, 137, 10934–10937 CrossRef CAS PubMed; (c) Y. Ota, S. Ito, M. Kobayashi, S. Kitade, K. Sakata, T. Tayano and K. Nozaki, Angew. Chem., Int. Ed., 2016, 55, 7505–7509 CrossRef CAS PubMed; (d) L. Guo, W. Liu and C. Chen, Mater. Chem. Front., 2017, 1, 2487–2494 RSC; (e) L. Guo, S. Dai, X. Sui and C. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS; (f) Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- (a) T. P. Coogan, D. M. Latta, E. T. Snow, M. Costa and A. Lawrence, CRC Crit. Rev. Toxicol., 1989, 19, 341–384 CrossRef CAS PubMed; (b) K. K. Das, R. C. Reddy, I. B. Bagoji, S. Das, S. Bagali, L. Mullur, J. P. Khodnapur and M. S. Biradar, J. Basic Clin. Physiol. Pharmacol., 2019, 30, 141–152 CAS; (c) E. Denkhaus and K. Salnikow, Crit. Rev. Oncol. Hematol., 2002, 42, 35–56 CrossRef CAS PubMed; (d) Nat. Catal., 2019, 2, 735–735 Search PubMed.
- (a) W. Zhao, B. Wang, B. Dong, H. Liu, Y. Hu, M. S. Eisen and X. Zhang, Organometallics, 2020, 39, 3983–3991 CrossRef CAS; (b) X. Wang, Y. Wang, X. Shi, J. Liu, C. Chen and Y. Li, Macromolecules, 2014, 47, 552–559 CrossRef CAS; (c) S. Dai and C. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281–13285 CrossRef CAS PubMed.
- X.-Y. Wang, Y.-Y. Long, Y.-X. Wang and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3421–3428 CrossRef CAS.
- J. Chen, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 14726–14735 CrossRef CAS PubMed.
- (a) S. Bruzaud, H. Cramail, L. Duvignac and A. Deffieux, Macromol. Chem. Phys., 1997, 198, 291–303 CrossRef CAS; (b) T. Toda, I. Miura, M. Miya and K. Takenaka, Catalysts, 2019, 9, 660 CrossRef.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS.
- (a) W. Nzahou Ottou, S. Norsic, F. D'Agosto and C. Boisson, Macromol. Rapid Commun., 2018, 39, 1800154 CrossRef PubMed; (b) C. Negrell-Guirao, G. David, B. Boutevin and K. Chougrani, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3905–3910 CrossRef CAS; (c) R. Tayouo, G. David, B. Améduri, J. Rozière and S. Roualdès, Macromolecules, 2010, 43, 5269–5276 CrossRef CAS; (d) Z. Yu, W.-X. Zhu and I. Cabasso, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 227–230 CrossRef CAS.
- X. Chen, Y. Hu and L. Song, Polym. Eng. Sci., 2008, 48, 116–123 CrossRef CAS.
- (a) K. Wazarkar, M. Kathalewar and A. Sabnis, Polym. Compos., 2017, 38, 1483–1491 CrossRef CAS; (b) M. Hussain, R. J. Varley, Z. Mathys, Y. B. Cheng and G. P. Simon, J. Appl. Polym. Sci., 2004, 91, 1233–1253 CrossRef CAS; (c) L.-P. Gao, D.-Y. Wang, Y.-Z. Wang, J.-S. Wang and B. Yang, Polym. Degrad. Stab., 2008, 93, 1308–1315 CrossRef CAS.
- K. Sałasińska, M. Borucka, M. Celiński, A. Gajek, W. Zatorski, K. Mizera, M. Leszczyńska and J. Ryszkowska, Adv. Polym. Technol., 2018, 37, 2394–2410 CrossRef.
- (a) B. Yang, L. Wang, Y. Guo, Y. Zhang, N. Wang, J. Cui, J. Guo and L. Tian, Polym. Adv. Technol., 2020, 31, 472–481 CrossRef CAS; (b) Z. Bai, L. Song, Y. Hu and R. K. K. Yuen, Ind. Eng. Chem., 2013, 52, 12855–12864 CrossRef CAS.
- R. E. Lyon, R. N. Walters and S. I. Stoliarov, Polym. Eng. Sci., 2007, 47, 1501–1510 CrossRef CAS.
- Q. Tai, Y. Hu, R. K. K. Yuen, L. Song and H. Lu, J. Mater. Chem., 2011, 21, 6621–6627 RSC.
- N. H. Thi, T. N. Nguyen, H. T. Oanh, N. T. T. Trang, D. Q. Tham, H. T. Nguyen, T. Van Nguyen and M. H. Hoang, J. Appl. Polym. Sci., 2021, 138, 50317 CrossRef CAS.
- E. A. Coleman, in Applied Plastics Engineering Handbook, ed. M. Kutz, William Andrew Publishing, 2nd edn, 2017, pp. 489–500, DOI:10.1016/B978-0-323-39040-8.00021-3.
- (a) Z.-S. Xu, L. Yan and L. Chen, Procedia Eng., 2016, 135, 631–636 CrossRef; (b) L. Yan, Z. Xu, X. Wang, N. Deng and Z. Chu, J. Coat. Technol. Res., 2018, 15, 1357–1369 CrossRef CAS; (c) P. Khalili, K. Y. Tshai, D. Hui and I. Kong, Composites, Part B, 2017, 114, 101–110 CrossRef CAS; (d) Z. Qin, D. Li, Q. Li and R. Yang, Mater. Des., 2016, 89, 988–995 CrossRef CAS; (e) Q. Li, P. Jiang, Z. Su, P. Wei, G. Wang and X. Tang, J. Appl. Polym. Sci., 2005, 96, 854–860 CrossRef CAS; (f) Y. Wang, L. Zhang, Y. Yang and X. Cai, J. Therm. Anal. Calorim., 2016, 125, 839–848 CrossRef CAS; (g) D. Yang, Y. Hu, H. Li, L. Song, H. Xu and B. Li, J. Therm. Anal. Calorim., 2015, 119, 619–624 CrossRef CAS.
- P. Wei, Z. Han, X. Xu and Z. Li, J. Fire Sci., 2006, 24, 487–498 CrossRef CAS.
- Z.-Y. Wang, Y. Liu and Q. Wang, Polym. Degrad. Stab., 2010, 95, 945–954 CrossRef CAS.
- (a) L. Ai, S. Chen, L. Yang and P. Liu, Fibers Polym., 2021, 22, 354–365 CrossRef CAS; (b) C. Hoffendahl, G. Fontaine, S. Duquesne, F. Taschner, M. Mezger and S. Bourbigot, Polym. Degrad. Stab., 2015, 115, 77–88 CrossRef CAS; (c) S. Bourbigot, M. L. Bras, R. Leeuwendal, K. K. Shen and D. Schubert, Polym. Degrad. Stab., 1999, 64, 419–425 CrossRef CAS; (d) N. A. Isitman and C. Kaynak, J. Fire Sci., 2013, 31, 73–84 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer syntheses and characterisations, NMR and IR spectroscopy, DSC, TGA, GPC, powder XRD and MCC. See DOI: https://doi.org/10.1039/d3py00143a |
| This journal is © The Royal Society of Chemistry 2023 |