
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of polyisocyanurate prepolymer and the resulting flexible elastomers with tunable mechanical properties†
Yunfei
Guo
a,
Julian
Kleemann
c,
Stefan
Bokern
c,
Andre
Kamm‡
c,
Rint P.
Sijbesma
 *ab and
Željko
Tomović
*ab
*ab and
Željko
Tomović
*ab
aDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: z.tomovic@tue.nl; r.p.sijbesma@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
cBASF Polyurethanes GmbH, Elastogranstraße 60, 49448 Lemförde, Germany
First published on 27th March 2023
Abstract
Polyurethane (PU) is used in a wide range of applications due to its diverse chemical and physical properties. To meet the increasing demands on thermal and mechanical properties of PU materials, polyisocyanurates (PIRs) have been introduced in PU materials as crosslinkers and due to their high decomposition temperature. We prepared a liquid PIR prepolymer with high PIR content by co-trimerization of 4,4′-methylene diphenyl diisocyanate (4,4′-MDI) and mono-isocyanates. The mono-isocyanate was synthesized via reaction between a 4,4′-MDI and a 2-ethyl-1-hexanol. The PIR prepolymer obtained was further reacted with long chain polyols and chain extenders in both solvent and solvent-free conditions, leading to PIR elastomers that exhibited good thermal stability with high char formation, and improved mechanical properties with much higher Young's modulus. This work demonstrates that the liquid PIR prepolymer can potentially be used in various large-scale industrial applications.
Introduction
Polyurethane (PU) is one of the most common polymers used to provide a wide range of materials, such as soft cushioning foams, rigid thermal insulation foams, elastomers, coatings and adhesives.1–5 In terms of chemical structures, polyurethanes include various isocyanate-based bonds such as urethane, urea, uretdione, biuret, allophanate and isocyanurate, among which the isocyanurate structure exhibits a higher thermal decomposition temperature.6–14 Therefore, the thermal stability of PU materials can be greatly enhanced by introducing isocyanurate or polyisocyanurate (PIR) structures, which is especially important for rigid thermal insulation foams.In addition to PU rigid foams, researchers have been developing PIR elastomers via trimerization of isocyanate prepolymers obtained from the reaction between excess of isocyanate and long chain polyols, or via in situ synthesis with isocyanates, polyols and chain extender reacting in the presence of a trimerization catalyst.15–20 However, the resulting PU materials in both cases have a relatively low PIR content due to the fast increase of viscosity and poor catalyst diffusion during trimerization reaction.
An alternative approach to obtain a PIR network with high concentration of isocyanurate structures is via direct trimerization of di- or polyisocyanate monomers, leading to a multi-functional PIR-containing isocyanate prepolymer.21–26 However, high crosslink density as well as rigid isocyanurate structures lead to brittle solid PIR networks. To improve the processability and reduce the brittleness of the materials, co-trimerization of mono- and di-functional isocyanates has been reported to generate a flexible PIR network. For instance, di-functional isocyanate hexamethylene diisocyanate (HDI) was co-trimerized with mono-functional isocyanates such as butyl isocyanate or phenyl isocyanate.22,27 By changing the ratio between HDI and mono-functional isocyanate, the mechanical properties of the polymer as well as the reaction kinetics can be adjusted. 4,4′-Methylene diphenyl diisocyanate (4,4′-MDI) was also co-trimerized with different mono-functional aliphatic or aromatic isocyanates, providing flexible PIR films with good thermal stability (Td5 > 400 °C).28,29 These studies provide a good way to prepare liquid PIR-containing isocyanate prepolymers containing controllable isocyanurate content and such prepolymer can be used to prepare PIR-PU elastomers in both solvent and solvent-free conditions. However, commercially available mono-functional isocyanates, such as phenyl and butyl isocyanates, are not suitable for industrial applications due to high toxicity caused by high vapor pressure.
In the current work, an alternative strategy for synthesis of non-volatile mono-functional isocyanate is explored. The reaction between diisocyanate and mono-functional alcohol is used for the preparation of a mixture containing di- and mono-isocyanates that can be used to prepare flexible PIR prepolymers (Scheme 1). In this work, a mixture of mono- and di-functional isocyanates was synthesized by reacting excess of 4,4′-MDI with 2-ethyl-1-hexanol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.25 molar ratio. The mixture was then co-trimerized and quenched by diethylene glycol bis-chloroformate (DGBCF) before solidification in order to get a liquid PIR prepolymer (Scheme 2). The PIR prepolymer was further used to prepare PIR elastomers by reacting the prepolymer with long chain polyol and chain extender in both solvent and bulk conditions. The elastomers obtained exhibit superior mechanical properties in addition to good thermal stability. The preparation of a liquid PIR prepolymer with high PIR content is a promising approach for the large-scale, solvent free production of thermally stable and mechanically strong PU materials, which is important for various real industrial applications.
:0.25 molar ratio. The mixture was then co-trimerized and quenched by diethylene glycol bis-chloroformate (DGBCF) before solidification in order to get a liquid PIR prepolymer (Scheme 2). The PIR prepolymer was further used to prepare PIR elastomers by reacting the prepolymer with long chain polyol and chain extender in both solvent and bulk conditions. The elastomers obtained exhibit superior mechanical properties in addition to good thermal stability. The preparation of a liquid PIR prepolymer with high PIR content is a promising approach for the large-scale, solvent free production of thermally stable and mechanically strong PU materials, which is important for various real industrial applications.
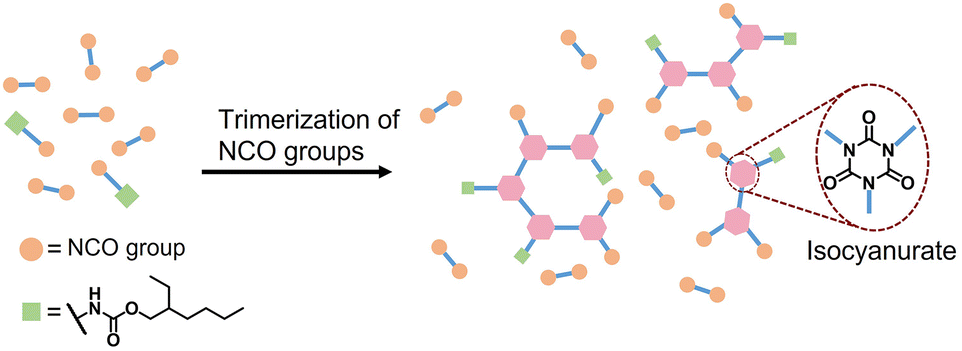 | ||
| Scheme 1 PIR prepolymer obtained via co-cyclotrimerization of di- and mono-functional isocyanates. | ||
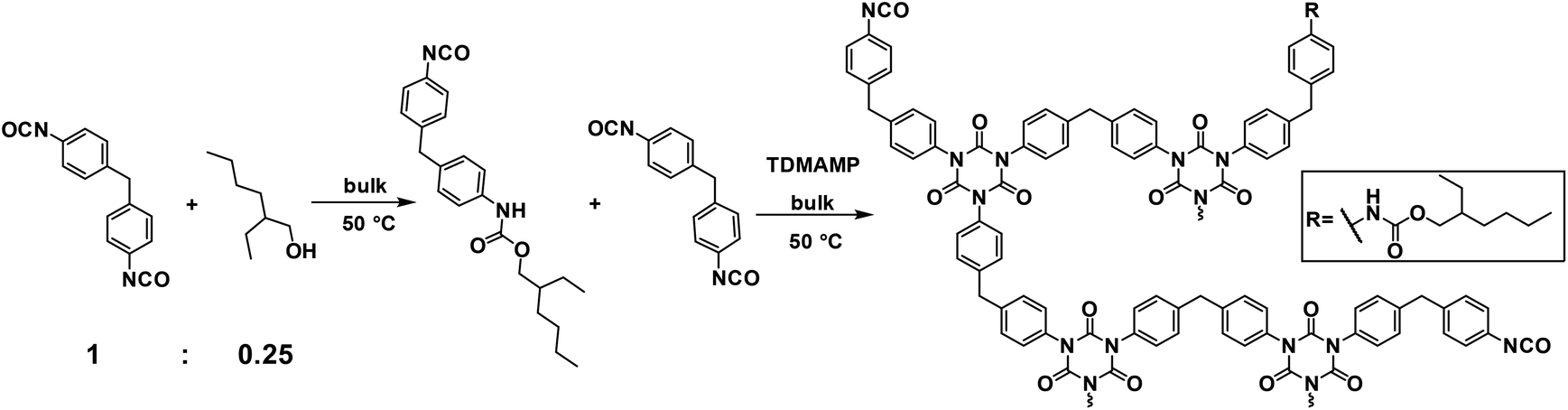 | ||
| Scheme 2 Synthesis of PIR prepolymer. | ||
Experimental
Materials
2-Ethyl-1-hexanol (≥99.6%), p-tolyl isocyanate (99%), 2,4,6-tris(dimethylaminomethyl)phenol (TDMAMP) (>95%), tetrabutylammonium acetate (97%), sodium p-toluenesulfinate (p-TolSO2Na) (95%), potassium acetate (≥99%), 2-{[2-(dimethylamino)ethyl]methylamino}ethanol (DMAEMAE) (98%), chromium(III) acetylacetonate (Cr(acac)3) (99.99% trace metals basis), 1,4-butanediol (BDO) (99%) and dibutyltin dilaurate (DBTL) (95%) were purchased from Sigma-Aldrich. Potassium 2-ethylhexanoate (>95%) was purchased from TCI. [HTBD][OAc] was synthesized following a literature procedure.30 1,4-Butanediol was dried over mol-sieves, all other reagents were used directly without treatment. 4,4′-Methylene diphenyl diisocyanate (4,4′-MDI), polymeric MDI Lupranate® M20 (M20), N,N′,N′′-tris(3-dimethylaminopropyl)hexahydro-1,3,5-triazine (TDMAPHT), diethylene glycol bis-chloroformate (DGBCF), 1,4-diazabicyclo[2.2.2]octane (DABCO)-based urethane gel catalyst Lupragen® N202, PolyTHF® 2000 with molecular weight of 2000 g mol−1 (OHv = 56 mg KOH per g), and Lupraphen® 6601/2 with molecular weight of 2000 g mol−1 (polyester polyol synthesized from adipic acid, 1,4-butanediol and mono ethylene glycol, OHv = 56 mg KOH per g) were kindly provided by BASF Polyurethanes GmbH. Polyols were dried at 80 °C under vacuum for 2 h before use. THF (containing BHT as stabilizer) and toluene were purchased from Biosolve and THF was dried over mol-sieves overnight before use.Synthesis of PIR prepolymer
4,4′-MDI (184.0 g, 0.74 mol) in a dry 3-neck flask equipped with a dropping funnel was stirred under Ar flow in 50 °C oil bath. 2-Ethyl-1-hexanol (24.0 g, 0.18 mol) was added in the dropping funnel and dropped into the flask at a speed of 1 drop every 10 s with the internal temperature kept under 55 °C. The reaction was immediately completed after addition of the alcohol and the mixture was calculated to have average molecular weight of 282.6 g mol−1 with fn of 1.75.The isocyanate mixture (169.9 g, 0.60 mol) was added to a dry beaker under Ar flow. Trimerization catalyst TDMAMP (0.1 g, 0.53 mmol) was dissolved in 6 mL THF and was quickly injected into the isocyanate mixture. The mixture was stirred at 50 °C in an oil bath with an anchor-shape stirrer and the internal temperature as well as the torque were monitored by the mechanical stirrer. After 200 min, the co-trimerization was quenched with DGBCF (0.1 g, 0.53 mmol) in 2 mL THF. The reaction was monitored with 13C NMR spectroscopy and the product was characterized with FT-IR spectroscopy (see ESI†). After that, the NCO content of the PIR prepolymer was titrated and the prepolymer was used for casting elastomers in solution.
For bulk-cast elastomers, the same procedure was used except that the catalyst amount was 0.1 mol%.
Synthesis of 2-ethylhexyl-p-tolylcarbamate
p-Tolyl isocyanate (1.6 g, 12.18 mmol), 2-ethyl-1-hexanol (1.8 g, 13.39 mmol) and toluene (5 mL) were added in a 3-neck flask equipped with a condenser under Ar atmosphere. The mixture was stirred and heated to 50 °C until the NCO stretching band at 2270 cm−1 disappeared according to FT-IR spectra. After that, solvent was evaporated, the mixture was purified by column chromatography and transparent liquid was obtained. 1H NMR (400 MHz, acetone-d6): δ 8.49 (s, 1H), 7.44 (d, J = 8.2 Hz, 2H), 7.09 (d, 2H), 4.21–3.89 (m, 2H), 2.26 (s, 3H), 1.59 (hept, 1H), 1.51–1.17 (m, 8H), 1.00–0.81 (m, 6H) ppm; 13C NMR (400 MHz, acetone-d6): δ 153.8, 136.9, 131.6, 129.1, 118.2, 66.4, 39.1, 30.2, 23.5, 22.8, 19.9, 13.5, 10.5 ppm.Preparation of PIR elastomers in solution
The preparation of PIR elastomer with PolyTHF® 2000 and 15 wt% BDO is used as an example to illustrate the synthetic route: the PIR prepolymer (2.9 g, NCO content = 16.0 wt%) was dissolved in 15 mL dry THF in a dry 1-neck flask at room temperature. The polyol component solution was prepared by dissolving PolyTHF® 2000 (2.1 g, 1.07 mmol), BDO (0.4 g, 4.18 mmol) and DBTL (7.0 mg, 0.01 mmol, 0.1 mol% to the NCO groups) in 5 mL dry THF in another dry 1-neck flask at room temperature. Then the polyol solution was added into the prepolymer solution and the mixture obtained was stirred for 1 min under Ar atmosphere. The solution was poured on a metal lid which was preheated at 50 °C in N2 oven and the elastomer was cured overnight. After that, the elastomer was dried at 80 °C vacuum oven for one day to remove solvent.The same procedures and the same conditions were used for the preparation of PIR elastomers, 4,4′-MDI based classical elastomers, and M20 based elastomers in solution.
Preparation of elastomers in bulk
The preparation of PIR elastomer with Lupraphen® 6601/2 and 15 wt% BDO is used as an example to illustrate the synthetic route: The PIR prepolymer was kept in a vacuum oven overnight at 50 °C. The polyol component that consisted of Lupraphen® 6601/2 (64.2 g, 0.03 mol), BDO (11.4 g, 0.13 mol) and Lupragen® N202 (0.1 g) was added in a sealed polypropylene cup and stirred at 1000 rpm under vacuum in a speedmixer for 10 min at 50 °C. PIR prepolymer (84.5 g, NCO content = 16.5 wt%) was added and the mixture was mixed in a speedmixer for 30 s under vacuum. After that, the mixture was poured on a preheated metal mold at 80 °C. The elastomers were further cured in 80 °C oven overnight.The same procedure and conditions were used for the preparation of PIR and M20 elastomers in bulk.
Characterization methods
Fourier-transform infrared spectroscopy (FT-IR)
FT-IR spectroscopy measurements were carried out in attenuated total reflection mode on a Spectrum Two (Perkin Elmer) spectrometer at room temperature. 8 scans were performed from 4000–450 cm−1.Nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectroscopy was performed using a Bruker UltraShield 400 MHz or Varian Mercury 400 MHz spectrometer at room temperature using acetone-d6 as solvent with TMS as internal standard with a delay time of 1 s and 32 scans per spectrum. 13C NMR spectroscopy measurements for monitoring the PIR reaction were performed using either Bruker UltraShield 400 MHz or Varian Mercury 400 MHz spectrometer at room temperature using acetone-d6 as solvent with TMS as internal standard with a delay time of 2 s and 256 scans per spectrum. Quantitative 13C NMR spectroscopy measurements for determining the urethane, allophanate and isocyanurate ratio were performed on a Varian Unit Inova 500 MHz spectrometer using acetone-d6 as solvent with TMS as internal standard at room temperature with a delay time of 12 s, 2048 scans per spectrum and Cr(acac)3 (20 mg mL−1) as relaxation agent.Determination of isocyanate content
The titration of NCO groups was performed on a 916 Ti-Touch titration machine (Metrohm) equipped with an electrode using tetraethylammonium bromide (0.4 mol L−1 in ethylene glycol) as electrolyte. The isocyanate was quenched with excess of dibutylamine and unreacted dibutylamine was titrated with 1 M HCl as the titrant. After getting the volume (V1) at the end of titration of sample as well as the volume (V0) of the blank titration at the same condition, the NCO content was calculated by the titration machine using the following equation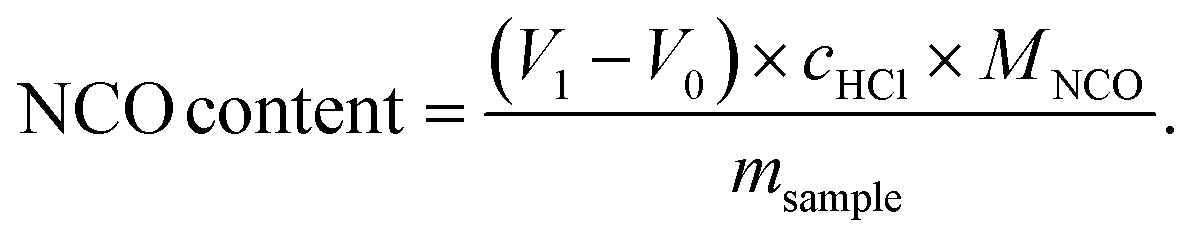
Gel permeation chromatography (GPC)
The average molecular weight of the PIR prepolymer was measured on a GPC system comprising a series of columns (one PSS-SDV 500 Å (5 μm) and three Agilent PL-Gel 500 Å (5 μm) columns) and a UV detector. THF was used as the eluent with a flow rate of 1 mL min−1 and toluene was used as internal standard. Calibration was done with monodisperse PMMA and Lupranate® M20 samples.Gel permeation chromatography (GPC) for studying selectivity of 4,4′-MDI and 2-ethyl-1-hexanol reaction was performed on a series of columns including four Agilent PL-Gel-Columns (1 × 50 Å–3 × 100 Å) equipped with a UV detector and a differential refractometer. THF was used as the eluent with a flow rate of 0.5 mL min−1. The system was calibrated by Basonat® HI 100 NH with molecular weight range 2200–168 g mol−1.
Thermogravimetric analysis (TGA)
TGA measurement was performed on a TA Q500 or TA Q550 (TA instruments) under N2 atmosphere. Samples were heated from 28 to 600 °C at a rate of 10 °C min−1.Tensile testing
Tensile testing was performed on a EZ20 (Lloyd instrument) with a 500 N load cell. The tensile bars used had an effective length of 12 mm, width of 2 mm and thickness of 0.6 mm. The elongation rate used was 50 mm min−1.Dynamic mechanical analysis (DMA)
DMA measurement was measured on a DMA Q850 (TA Instruments) with a film tension setup. The test bars had a width of 5.3 mm and thickness of 0.6 mm. For each measurement, a temperature ramp from −80 to 180 °C was programmed with a heating rate of 3 °C min−1 at a frequency of 1.0 Hz. A preload force of 0.01 N, an amplitude of 100 μm and a force track of 115% were used. The storage and loss modulus were recorded as a function of temperature. The glass transition temperature (Tg) was determined from the peak maximum of the tan(δ).Results and discussion
Synthesis of PIR prepolymer
Statistically, the reaction between 4,4′-MDI with 2-ethyl-1-hexanol leads to the formation of di-urethanes besides mono-urethanes. To minimize formation of di-urethanes, the selectivity of the reaction was studied. MDI was reacted with 2-ethyl-1-hexanol in various molar ratios and the reaction was performed by slowly dropping the alcohol into the isocyanate at the relatively low temperature of 50 °C. The chemical composition of the obtained mixture was determined by gel permeation chromatography (GPC) (Fig. 1, Fig. S1 and Table S1†).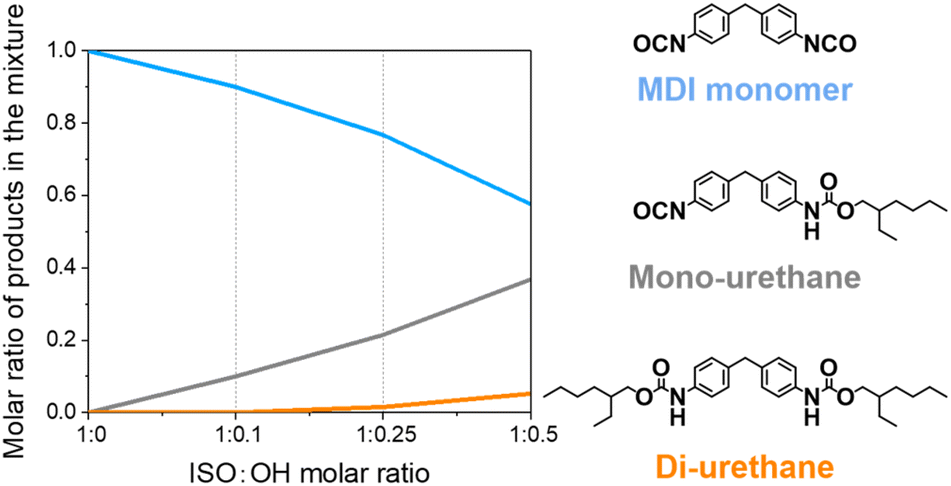 | ||
| Fig. 1 Product compositions of reaction mixtures obtained at various 4,4′-MDI to 2-ethyl-1-hexanol molar ratio. The reaction was carried out at 50 °C with 2-ethyl-1-hexanol being added dropwise to 4,4′-MDI. The ratio of different products was determined by GPC. | ||
With more mono-functional alcohol, the amount of di-urethane structures increased. In order to minimize the formation of di-urethanes which do not contribute to the network, and maximize the mono-functional isocyanate content which helps to maintain a flexible PIR network in a final product, the optimized isocyanate-to-alcohol molar ratio of 1:0.25 was chosen, resulting in a mixture containing around 75 mol% di-functional isocyanate and 25 mol% mono-functional isocyanate.
The mixture obtained was further trimerized using a selection of common trimerization catalysts, mainly based on tertiary amines and Lewis basic salts with nucleophilic anions (Scheme 3). Various reaction conditions, such as reaction temperature and catalyst concentration, were investigated. 2,4,6-Tris(dimethylaminomethyl)phenol (TDMAMP)6,31 was found to be the most suitable trimerization catalyst for PIR prepolymer preparation in terms of mildness of reaction conditions, and product processability and solubility. While some catalysts, for example, tetrabutylammonium acetate (TBAA),32,33 potassium 2-ethylhexanoate34–37 and 2-{[2-(dimethylamino)ethyl]methylamino}ethanol (DMAEMAE)38 were too active to be controlled, others, such as sodium p-toluenesulfinate (p-TolSO2Na)39 and potassium acetate40,41 had poor solubility in either solvents or isocyanate. N,N′,N′′-tris(3-dimethylaminopropyl)hexahydro-1,3,5-triazine (TDMAPHT)7,42,43 provided more allophanate and less isocyanurate than TDMAMP (Fig. S2 and S3†), and [HTBD][OAc]30 (a conjugate between 1,5,7-triazabicyclo [4.4.0] dec-5-ene and acetic acid), which catalyzes cyclotrimerization of isocyanates via synergistic hydrogen bonding functional catalytic mechanism, was deactivated most probably due to the presence of urethane groups in the mixture.
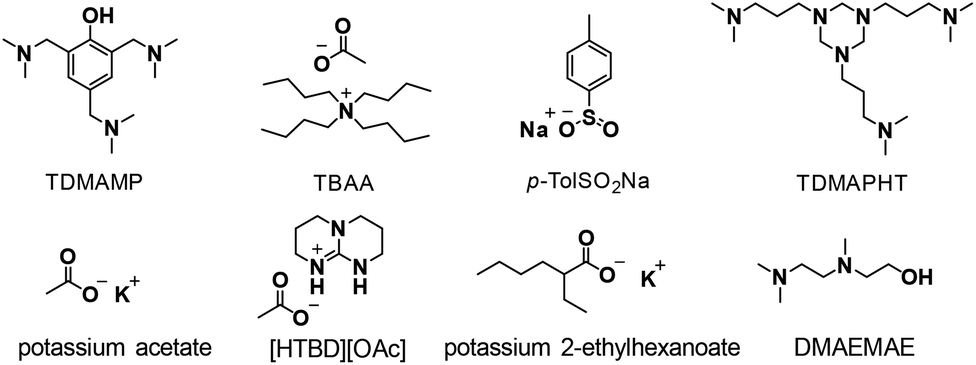 | ||
| Scheme 3 Structures of various cyclotrimerization catalysts. | ||
The isocyanate trimerization mechanism using TDMAMP as a catalyst was further studied by cyclotrimerization of p-tolyl isocyanate in the presence of one equivalent 2-ethylhexyl p-tolylcarbamate in deuterated acetone at 50 °C (Fig. S4†). In agreement with the mechanism proposed by Nabulsi and Schwetlick, the NMR spectroscopy studies show that in the presence of carbamate, the cyclotrimerization follows an allophanate pathway (Fig. 2 and 3).41,42 In this mechanism, the isocyanate first reacts with carbamate to form allophanate as a key intermediate. Next, the allophanate decomposes to anionic species B or C by elimination of carbamate or alcohol, finally followed by ring closure to form isocyanurate. However, in the model reaction, around 10 mol% of allophanate was still present after 34 h reaction due to the limited catalytic activity of the TDMAMP in highly diluted solution (Fig. S5†).
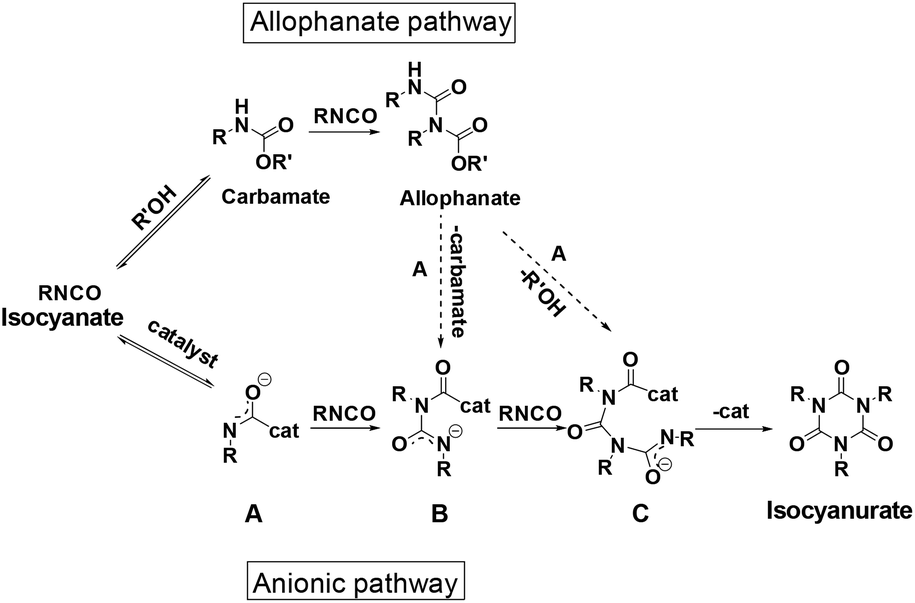 | ||
| Fig. 2 Proposed cyclotrimerization mechanism of isocyanates via allophanate and anionic pathways. | ||
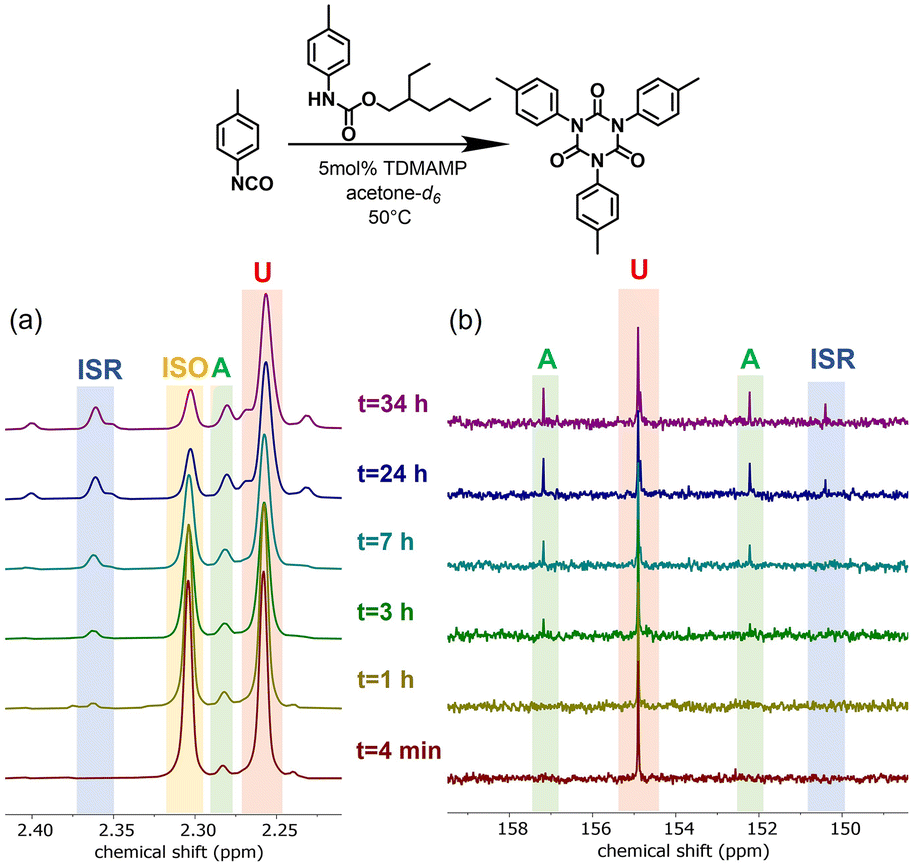 | ||
| Fig. 3 (a) 1H NMR spectra (400 MHz, acetone-d6) and (b) 13C NMR spectra (100 MHz, acetone-d6) of reaction between p-tolyl isocyanate and 2-ethylhexyl p-tolylcarbamate in 1:1 molar ratio at 50 °C using 5 mol% TDMAMP as catalyst. ISO (p-tolyl isocyanate); U (carbamate); A (allophanate); ISR (isocyanurate). The reaction was carried out in an NMR tube in deuterated acetone and monitored by (a) peaks between 2.2 and 2.4 ppm (methyl group protons), and (b) peaks between 148 and 158 ppm (carbonyl carbons). The molar ratio of U:A:ISR after 34 h reaction was 1:0.16:0.04 (see Fig. S5†). | ||
Based on the catalyst screening, the trimerization of mono- and di-functional isocyanate mixture was carried out at 50 °C in bulk with TDMAMP as the catalyst; the PIR reaction was quenched with DGBCF before solidification could take place (Fig. S6–S8†). The resulting PIR prepolymer was further characterized by quantitative 13C NMR spectroscopy to quantify the molar ratio of urethane (U), allophanate (A) and isocyanurate (ISR) with the help of chromium(III) acetylacetonate (Cr(acac)3) as relaxation agent.44–46 Carbonyl carbon peaks were assigned to the urethane, allophanate and isocyanurate carbonyl atoms and integrated to give a ratio of U:A:ISR of 1:0.5:1.0 (Fig. 4).26,47 In addition, NCO content was determined as 16.0 wt% by back-titration, and the average molar mass of the PIR prepolymer, 660 g mol−1, was determined by GPC measurement (Fig. S9†), from which the functionality fn of the PIR prepolymer was calculated to be 2.5 via the following equation:
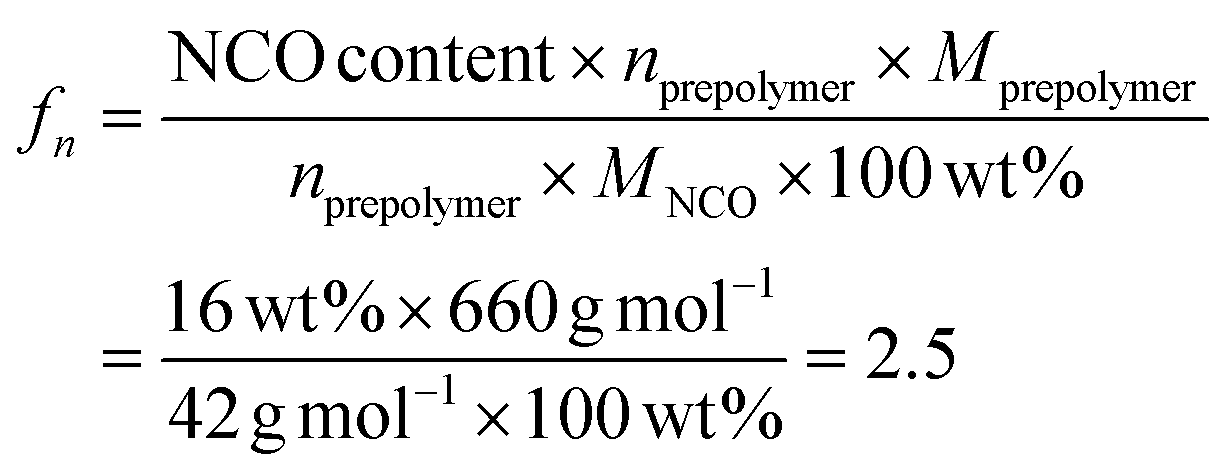
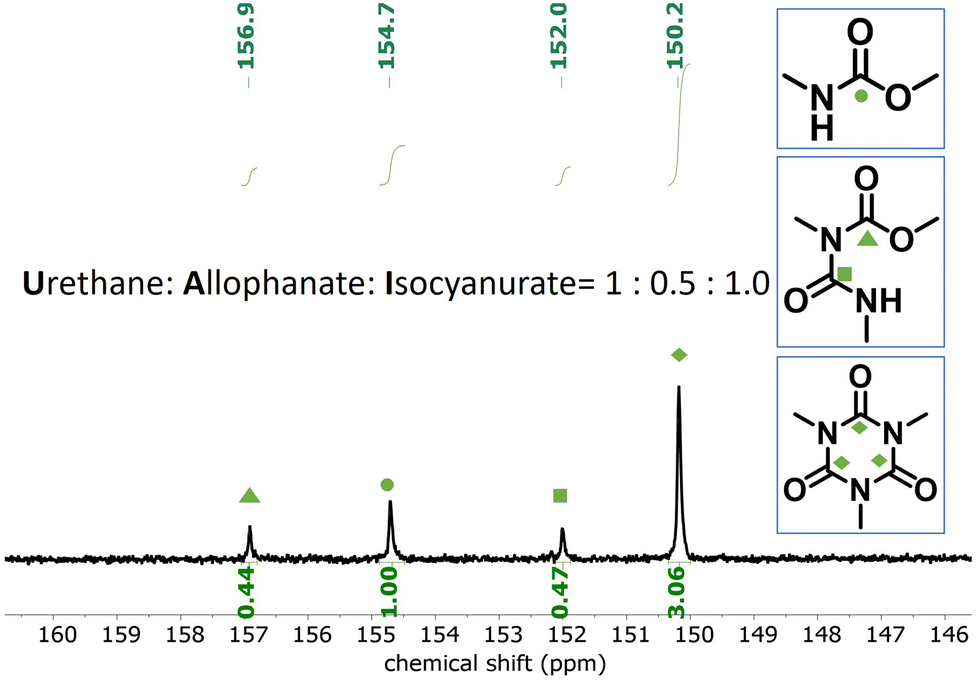 | ||
| Fig. 4 Quantitative 13C NMR spectra (125 MHz, acetone-d6) of the PIR prepolymer. The ratio of U:A:ISR was determined by integrals of the carbonyl carbon peaks. U (carbamate); A (allophanate); ISR (isocyanurate). | ||
Preparation of PIR elastomers in solution
PIR elastomers were prepared by the reaction of a PIR prepolymer with two commercially available polyether polyols or polyester polyols with molecular weight of 2000 g mol−1, PolyTHF® 2000 (PTHF) and Lupraphen® 6601/2 (PESOL), using different amounts of 1,4-butanediol (BDO) as chain extender (0, 5 wt%, 10 wt%, 15 wt% of the polyol component). The molar ratio of NCO:OH was kept constant at 1.05 (index 105) typically used in industrial applications,48,49 and the elastomers were cast from THF solution (Table S2†). All elastomers were transparent as shown in Fig. 5.
 | ||
| Fig. 5 Photographs of solvent casted PIR elastomers (left: PTHF; right: PESOL). | ||
In addition, a commercially available polymeric MDI with functionality of 2.7, Lupranate® M20 (M20), was used to compare with PIR prepolymer due to the similar average functionality. Using the same procedure and conditions, M20 elastomers were prepared by reacting M20 and PTHF or PESOL with either 0 or 15 wt% BDO.
The thermal stability of the PIR elastomers was measured with thermogravimetric analysis (TGA), the results of which are shown in Fig. 6 and Table 1. With a higher BDO content, the decomposition temperatures of PIR elastomers at 5% weight loss (Td5) and 10% weight loss (Td10) slightly decreased due to the presence of more urethane bonds in the material. However, as the amount of PIR structures and the aromatic content also increased, higher char formation was obtained at 596 °C. The decomposition temperatures and char formation of M20-based elastomers were slightly higher than PIR elastomers, which may be due to a higher crosslink density. On the other hand, all PIR elastomers showed better thermal stability than classical linear 4,4′-MDI-based elastomers which are synthesized from 4,4′-MDI, polyol and BDO without trimerization (Fig. S10 and Table S3†).
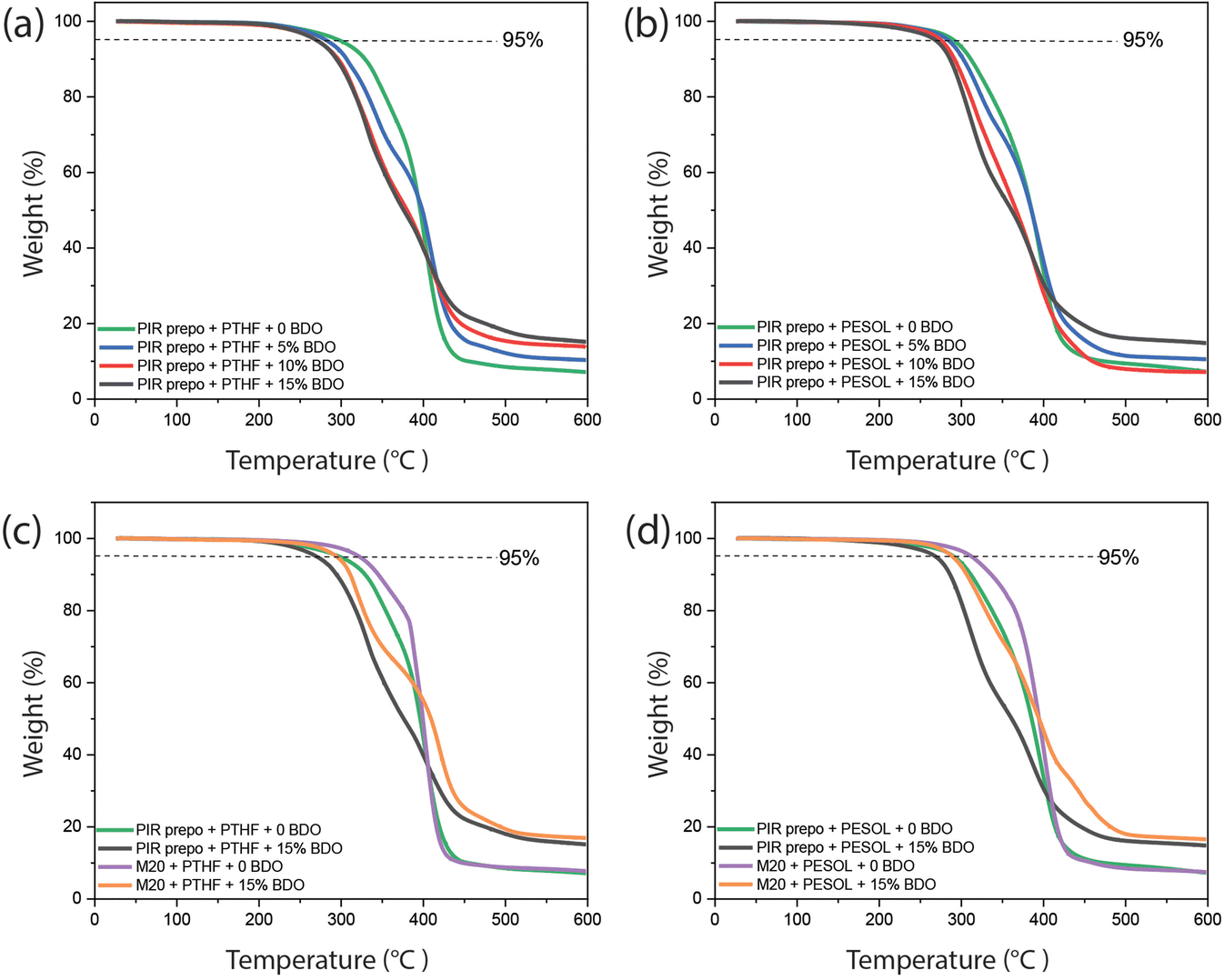 | ||
| Fig. 6 TGA curves of (a) PTHF-containing and (b) PESOL-containing PIR elastomers with different amount of BDO; (c) PTHF-containing and (d) PESOL-containing PIR and M20 elastomers with 0 and 15 wt% BDO. | ||
| Sample | Aromatic contenta (wt%) | T d5 (°C) | T d10 (°C) | Char formation (%) |
|---|---|---|---|---|
| a The aromatic content is calculated based on the weight percentage of aromatic isocyanate in the elastomers (see ESI†). | ||||
| PIR prepo + PTHF | 19 | 297.8 | 329.0 | 7.2 |
| PIR prepo + PTHF + 5 wt% BDO | 32 | 277.7 | 306.6 | 10.4 |
| PIR prepo + PTHF + 10 wt% BDO | 41 | 270.0 | 296.2 | 14.0 |
| PIR prepo + PTHF + 15 wt% BDO | 47 | 270.1 | 294.2 | 15.2 |
| PIR prepo + PESOL | 19 | 290.8 | 311.4 | 7.3 |
| PIR prepo + PESOL + 5 wt% BDO | 32 | 284.1 | 302.2 | 10.6 |
| PIR prepo + PESOL + 10 wt% BDO | 41 | 275.1 | 291.5 | 7.2 |
| PIR prepo + PESOL + 15 wt% BDO | 47 | 268.5 | 286.2 | 14.9 |
| M20 + PTHF | 12 | 321.8 | 345.1 | 7.8 |
| M20 + PTHF + 15 wt% BDO | 37 | 294.3 | 311.0 | 16.9 |
| M20 + PESOL | 12 | 312.4 | 336.2 | 7.5 |
| M20 + PESOL + 15 wt% BDO | 37 | 289.1 | 306.5 | 16.6 |
A significant difference in mechanical properties was found between PIR elastomers and M20 elastomers (Fig. 7 and Table 2). Compared to M20 elastomers with the same amount of BDO (yellow curve), the tensile strength and Young's modulus of PIR elastomer (black curve) was much higher due to a higher aromatic content and the presence of PIR structures. In addition, with alkyl chains that improved flexibility of the PIR network, the elongation at break of the PIR elastomers was also higher than in the corresponding M20 elastomers. The only exception is that the PIR elastomer containing PTHF and 15 wt% BDO has a slightly lower elongation at break than M20 elastomer with PTHF and with the same amount of BDO. The higher Young's modulus leads to lower elongation at break as shown in Table 2. Although the elastomer based on PIR prepolymer has around 12 times higher Young's modulus relative to M20 based elastomer, the elongation at break is still very high due to the presence of flexible alkyl chains. The same trend is also visible for the PESOL-containing elastomers. The obtained absolute values are the result of a complex interplay of different parameters, such as polarity or compatibility of PTHF with hard phase, Tg of elastomers and hard phase content in the PIR prepolymer.
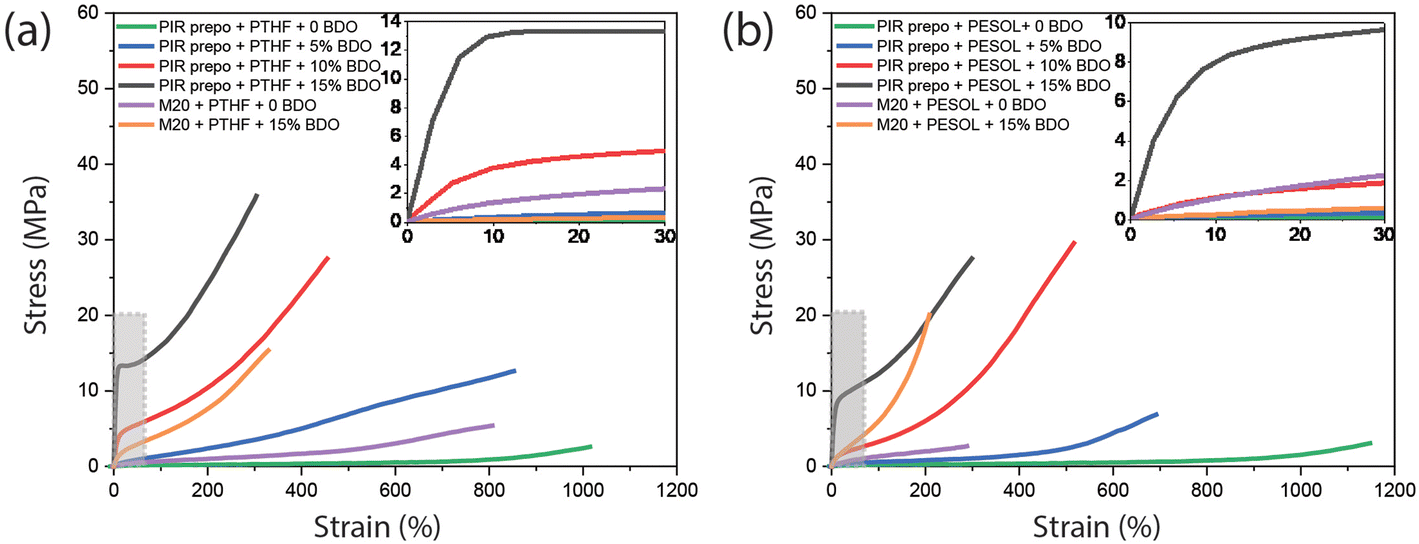 | ||
| Fig. 7 Tensile test of PIR and M20 elastomers based on (a) PTHF and (b) PESOL, and different amounts of BDO. The inserts show the slopes of the curves which are correlated to Young's modulus of the elastomers. | ||
| Sample | Tensile strength (MPa) | Elongation at break (%) | Young's modulusa (MPa) |
|---|---|---|---|
| a The Young's modulus is calculated by the initial linear slope of the tensile curve. | |||
| PIR prepo + PTHF | 2.5 | 1000 | 0.3 |
| PIR prepo + PTHF + 5 wt% BDO | 13.5 | 845 | 2.9 |
| PIR prepo + PTHF + 10 wt% BDO | 26.8 | 453 | 47.9 |
| PIR prepo + PTHF + 15 wt% BDO | 36.3 | 310 | 173.9 |
| PIR prepo + PESOL | 2.6 | 1190 | 0.3 |
| PIR prepo + PESOL + 5 wt% BDO | 6.6 | 703 | 1.1 |
| PIR prepo + PESOL + 10 wt% BDO | 28.2 | 500 | 8.3 |
| PIR prepo + PESOL + 15 wt% BDO | 28.7 | 314 | 81.1 |
| M20 + PTHF | 5.4 | 807 | 0.9 |
| M20 + PTHF + 15 wt% BDO | 15.4 | 329 | 14.7 |
| M20 + PESOL | 2.7 | 288 | 1.9 |
| M20 + PESOL + 15 wt% BDO | 20.1 | 208 | 12.2 |
Dynamic mechanical analysis (DMA) measurements were performed on the polymers and the maximum in the loss tangent tan(δ) was used to determine Tg. The results are shown in Fig. 8 and Table 3. The PIR structures and aromatic content increased with BDO content, and the resulting Tg as well as the storage modulus at rubbery plateau of the PIR elastomers increased. The PIR elastomer based on PTHF without BDO had a narrow tan(δ) peak (green curve in Fig. 8a) with a maximum at −49 °C, which is strongly influenced by the Tg of PTHF polyol. When there was no BDO in the elastomer, cold crystallization with subsequent melting of PTHF was found between −30 to 30 °C.50 The presence of BDO limited the crystallization tendency of PTHF, thus no cold crystallization peaks were found in the curves of elastomers containing BDO. The broad tan(δ) peaks of PIR elastomers containing PTHF and BDO indicate that there might be phase separation between PTHF and hard segments in the elastomers. On the other hand, due to the good compatibility of the PIR prepolymer and the polyester polyol, only one narrow tan(δ) peak was observed in each PESOL-containing elastomer which increased with higher PIR and aromatic content. In comparison to PIR elastomers, M20 elastomers have lower Tg, which was caused by the absence of PIR structures and lower aromatic content.
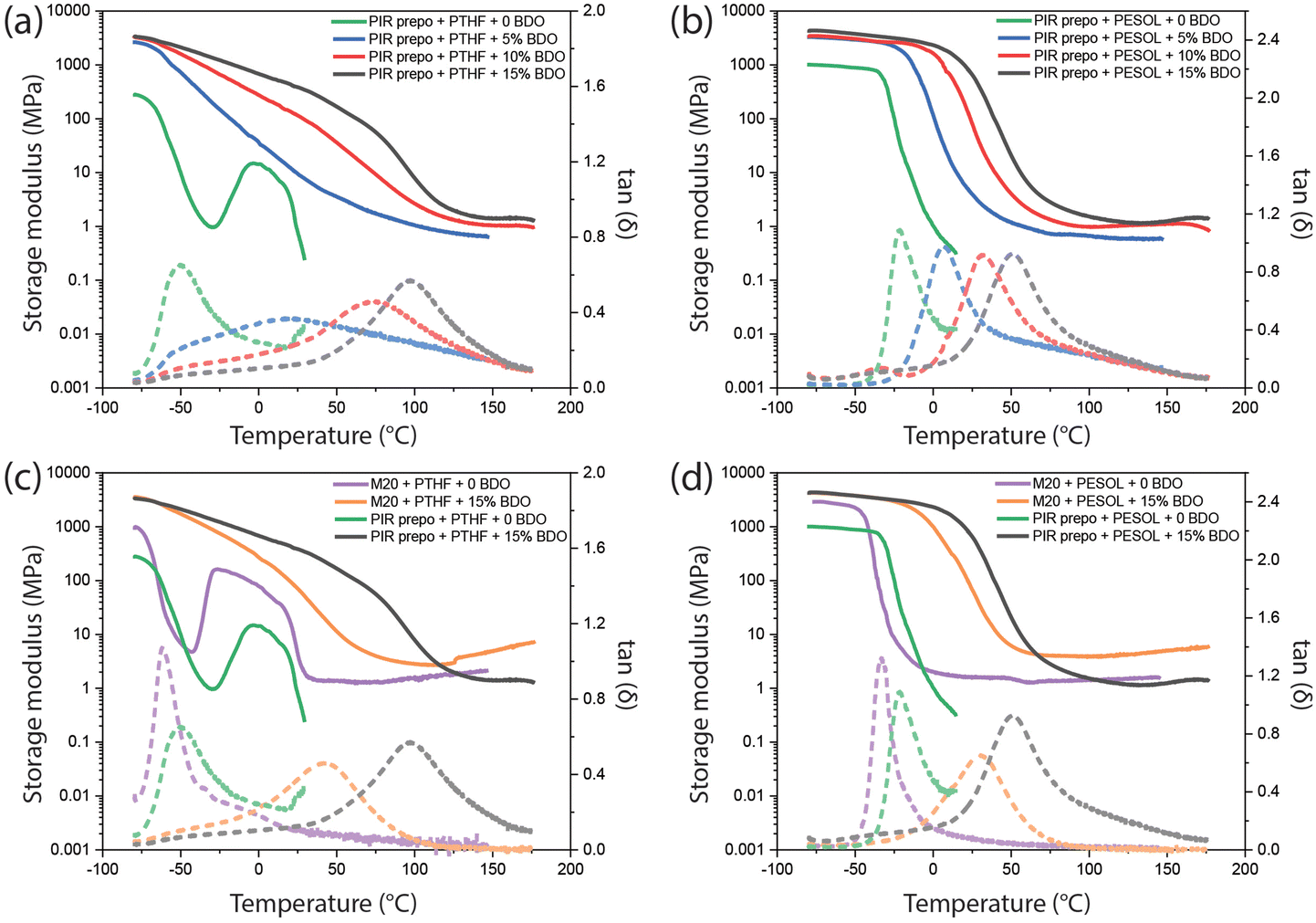 | ||
| Fig. 8 DMA results of (a) PTHF-containing and (b) PESOL-containing PIR elastomers with different amount of BDO; (c) PTHF-containing and (d) PESOL-containing PIR and M20 elastomers with 0 and 15 wt% BDO. Solid line: storage modulus, dash line: tan(δ). | ||
| Sample | T g (°C) |
|---|---|
| PIR prepo + PTHF | −49 |
| PIR prepo + PTHF + 5 wt% BDO | −55, −20 |
| PIR prepo + PTHF + 10 wt% BDO | −54, 74 |
| PIR prepo + PTHF + 15 wt% BDO | −52, 95 |
| PIR prepo + PESOL | −22 |
| PIR prepo + PESOL + 5 wt% BDO | 7 |
| PIR prepo + PESOL + 10 wt% BDO | 32 |
| PIR prepo + PESOL + 15 wt% BDO | 51 |
| M20 + PTHF | −61 |
| M20 + PTHF + 15 wt% BDO | 43 |
| M20 + PESOL | −35 |
| M20 + PESOL + 15 wt% BDO | 30 |
Preparation of PIR elastomers in bulk
PIR elastomers were also prepared by reacting a PIR prepolymer with PESOL without solvent (Table S4†). A PIR prepolymer with NCO content of 16.5 wt% for bulk casting of elastomers was synthesized via the same procedure as that for solution casting of elastomers. The PIR prepolymer and the polyol component (PESOL, BDO and urethane catalyst) were mixed and the elastomers were prepared at 80 °C in bulk. The trend of increasing char formation and Tg with increasing PIR and aromatic content observed in solvent-cast elastomers was also observed in bulk-cast elastomer (Fig. 9). Similar trends were observed in mechanical properties of elastomers using either of the two preparation methods. When BDO content was increased from 10 wt% to 15 wt%, the Young's modulus of PIR elastomers increased from 2.6 MPa to 116.7 MPa respectively, while the bulk-cast M20 elastomers with PESOL and 15 wt% BDO had Young's modulus of only 20.6 MPa. Despite the same trend of mechanical properties, the bulk-cast PIR elastomers with 15 wt% BDO prepared from bulk has higher Young's modulus compared to that prepared from solution (116.7 MPa vs. 81.1 MPa).51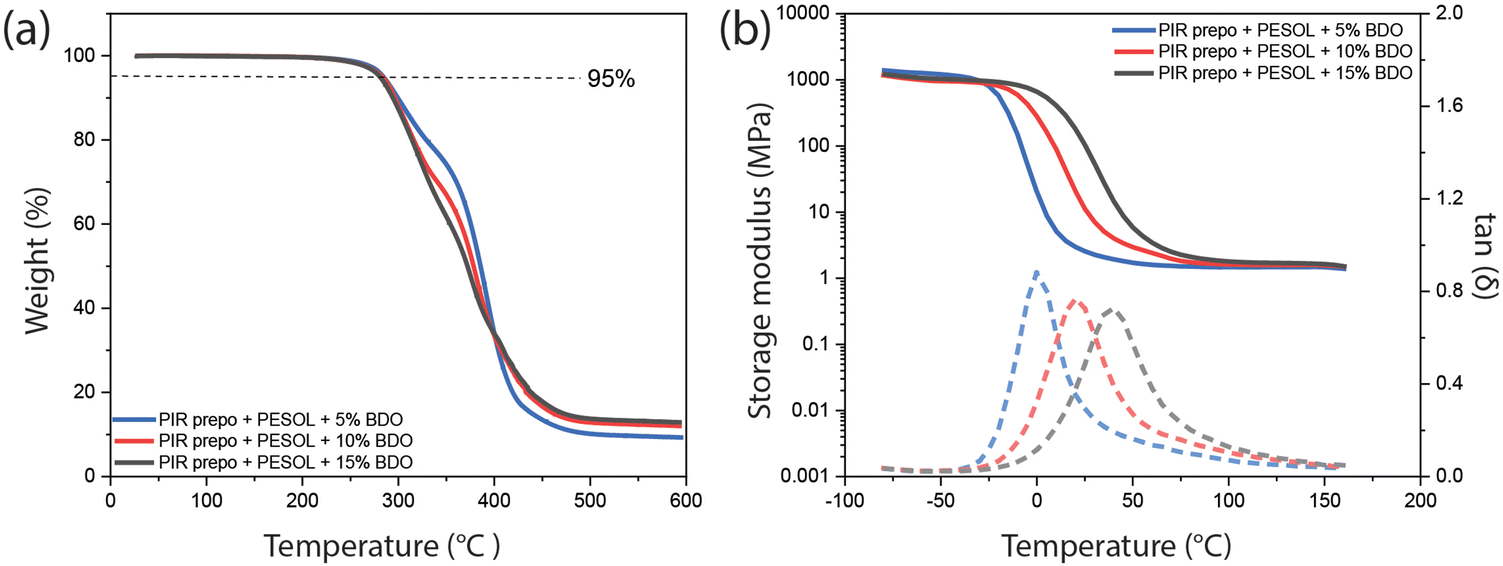 | ||
| Fig. 9 (a) TGA and (b) DMA results of the PIR elastomers cast in bulk with PESOL as a polyol and different amount of BDO. Solid line: storage modulus, dash line: tan(δ). | ||
Conclusions
In this work, PIR prepolymers were synthesized from co-trimerization of mono- and di-isocyanates, and isocyanurate containing elastomers were prepared from PIR prepolymers. TDMAMP turned out to be the most suitable catalyst for the preparation of prepolymers due to high isocyanurate content after isocyanate trimerization, mild reaction conditions and the possibility to be quenched after a certain reaction time. In the final products, the PIR elastomers, char formation at 596 °C, Tg as well as Young's modulus increased with increasing PIR and aromatic content. In addition, the mechanical properties of PIR elastomers (stress at break, elongation and Young's moduli) are much better than the elastomers cast from commercially available polymeric MDI.This study provides an elegant synthetic pathway to obtain liquid, flexible elastomer networks with high PIR content and good thermal stability as well as superior mechanical properties. It is expected that by substituting 2-ethyl-1-hexanol with other mono-functional alcohols, or by adjusting the molar ratio of mono- and di-functional isocyanates, the chemical, physical and mechanical properties, such as polarity, rigidity and thermal stability of the PIR prepolymer can be tuned. The PIR prepolymer with versatile properties will be suitable for diverse large-scale industrial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Frederic Lucas (BASF SE) for GPC measurement and Nadine Schliebe (BASF Polyurethanes GmbH) for experimental support. The authors also acknowledge financial support from BASF Polyurethanes GmbH.References
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- D. Randall and S. Lee, The Polyurethanes Book, Wiley, 2003 Search PubMed.
- B. Eling, Ž. Tomović and V. Schädler, Macromol. Chem. Phys., 2020, 2000114, 1–11 Search PubMed.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- P. I. Kordomenos and J. E. Kresta, Macromolecules, 1981, 14, 1434–1437 CrossRef CAS.
- P. I. Kordomenos, J. E. Kresta and K. C. Frisch, Macromolecules, 1987, 20, 2077–2083 CrossRef CAS.
- H. E. Reymore, P. S. Carleton, R. A. Kolakowski and A. A. R. Sayigh, J. Cell. Plast., 1975, 11, 328–344 CrossRef CAS.
- C. L. Wang, D. Klempner and K. C. Frisch, J. Appl. Polym. Sci., 1985, 30, 4337–4344 CrossRef CAS.
- I. C. Kogon, J. Am. Chem. Soc., 1956, 78, 4911–4914 CrossRef CAS.
- D. W. Duff and G. E. Maciel, Macromolecules, 1991, 24, 651–658 CrossRef CAS.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- Q. Xu, T. Hong, Z. Zhou, J. Gao and L. Xue, Fire Mater., 2018, 42, 119–127 CrossRef CAS.
- C. Dick, E. Dominguez-Rosado, B. Eling, J. J. Liggat, C. I. Lindsay, S. C. Martin, M. H. Mohammed, G. Seeley and C. E. Snape, Polymer, 2001, 42, 913–923 CrossRef CAS.
- R. Samborska-Skowron and A. Balas, Polym. Adv. Technol., 2002, 13, 653–662 CrossRef CAS.
- P. J. Driest, D. J. Dijkstra, D. Stamatialis and D. W. Grijpma, Macromol. Rapid Commun., 2019, 40, 1–6 CrossRef PubMed.
- H. E. Reymore, R. J. Lockwood and H. Ulrich, J. Cell. Plast., 1978, 14, 332–340 CrossRef CAS.
- N. Sasaki, T. Yokoyama and T. Tanaka, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 1765–1779 CrossRef CAS.
- P. J. Driest, D. J. Dijkstra, D. Stamatialis and D. W. Grijpma, Acta Biomater., 2020, 105, 87–96 CrossRef CAS PubMed.
- P. J. Driest, I. E. Allijn, D. J. Dijkstra, D. Stamatialis and D. W. Grijpma, Polym. Int., 2020, 69, 131–139 CrossRef CAS.
- S. Dabi and A. Zilkha, Eur. Polym. J., 1982, 18, 549–553 CrossRef CAS.
- S. Dabi and A. Zilkha, Eur. Polym. J., 1980, 16, 831–833 CrossRef CAS.
- M. Wejchan-Judek, I. Polus, B. Doczekalska and H. Pertek, Polymery, 2001, 46, 131–132 CrossRef CAS.
- J. Zeng, Y. Yang, Y. Tang, X. Xu, X. Chen, G. Li, K. Chen, H. Li, P. Ouyang, W. Tan, J. Ma, Y. Liu and R. Liang, Ind. Eng. Chem. Res., 2022, 61, 2403–2416 CrossRef CAS.
- J. E. Kresta and C. S. Shen, Polym. Bull., 1979, 1, 325–328 CrossRef CAS.
- D. W. Duff and G. E. Maciel, Macromolecules, 1990, 23, 3069–3079 CrossRef CAS.
- K. H. Hsieh and J. E. Kresta, in Cyclopolymerization and Polymers with Chain-Ring Structures, 1982, pp. 311–324 Search PubMed.
- M. Moritsugu, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4365–4367 CrossRef CAS.
- M. Moritsugu, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5186–5191 CrossRef CAS.
- L. Wu, W. Liu, J. Ye and R. Cheng, Catal. Commun., 2020, 145, 106097 CrossRef CAS.
- J. A. E. Hagquist, K. J. Reid, A. Giorgini and N. Hill, US 5556934 A, 1996 Search PubMed.
- Y. Guo, M. Muuronen, P. Deglmann, F. Lucas, R. P. Sijbesma and Ž. Tomović, J. Org. Chem., 2021, 86, 5651–5659 CrossRef CAS PubMed.
- M. Siebert, R. Sure, P. Deglmann, A. C. Closs, F. Lucas and O. Trapp, J. Org. Chem., 2020, 85, 8553–8562 CrossRef CAS PubMed.
- B. G. Jozef, H. Eric, R. Stijn, V. Marc and V. H. G. Guido, US 20080227929 A1, 2008 Search PubMed.
- J. J. Burdeniuc, T. Panitzsch and J. E. Dewhurst, US 8530534 B2, 2013 Search PubMed.
- I. Krakovský and M. Špírková, Collect. Czech. Chem. Commun., 1993, 58, 2663–2672 CrossRef.
- M. Špírková, M. Kubín, P.
![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) pa
pa![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ek, I. Krakovský and K. Du
ek, I. Krakovský and K. Du![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ek, J. Appl. Polym. Sci., 1994, 53, 1435–1446 CrossRef.
ek, J. Appl. Polym. Sci., 1994, 53, 1435–1446 CrossRef. - I. S. Bechara and F. P. Carroll, J. Cell. Plast., 1980, 16, 89–101 CrossRef CAS.
- F. M. Moghaddam, M. G. Dekamin, M. S. Khajavi and S. Jalili, Bull. Chem. Soc. Jpn., 2002, 75, 851–852 CrossRef CAS.
- P. J. Driest, V. Lenzi, L. S. A. Marques, M. M. D. Ramos, D. J. Dijkstra, F. U. Richter, D. Stamatialis and D. W. Grijpma, Polym. Adv. Technol., 2017, 28, 1299–1304 CrossRef CAS.
- A. Al Nabulsi, D. Cozzula, T. Hagen, W. Leitner and T. E. Müller, Polym. Chem., 2018, 9, 4891–4899 RSC.
- K. Schwetlickt and R. Noack, J. Chem. Soc., Perkin Trans. 2, 1995, 395–402 RSC.
- S.-W. Wong and K. C. Frisch, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2867–2875 CrossRef CAS.
- Z. Zhou, Y. He, X. Qiu, D. Redwine, J. Potter, R. Cong and M. Miller, Macromol. Symp., 2013, 330, 115–122 CrossRef CAS.
- S. Braun, H. O. Kalinowski and S. Berger, 100 and More Basic NMR Experiments: A Practical Course, Wiley, 1996 Search PubMed.
- L. A. Harris, J. D. Goff, A. Y. Carmichael, J. S. Riffle, J. J. Harburn, T. G. St Pierre and M. Saunders, Chem. Mater., 2003, 15, 1367–1377 CrossRef CAS.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J. P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- K. C. Frisch and D. Klempner, Advances in Urethane: Science & Technology, CRC Press, 2020, vol. XIV Search PubMed.
- J. S. Dick and R. A. Annicelli, Rubber Technology: Compounding and Testing for Performance, Hanser Publishers, 2009 Search PubMed.
- W. Ziegler, P. Guttmann, S. Kopeinig, M. Dietrich, S. Amirosanloo, G. Riess and W. Kern, Polym. Test., 2018, 71, 18–26 CrossRef CAS.
- P. A. Gunatillake, G. F. Meijs, E. Rizzardo, R. C. Chatelier, S. J. McCarthy, A. Brandwood and K. Schindhelm, J. Appl. Polym. Sci., 1992, 46, 319–328 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00124e |
| ‡ Present address: Department of Mechanical Engineering, Kiel University of Applied Sciences, Grenzstrasse 3, 24149 Kiel, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |