
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards high-performance polyurethanes: a mechanism of amine catalyzed aromatic imide formation from the reaction of isocyanates with anhydrides†
Yunfei
Guo
a,
Sebastian
Spicher
c,
Anna
Cristadoro
d,
Peter
Deglmann
c,
Rint P.
Sijbesma
 *ab and
Željko
Tomović
*ab
*ab and
Željko
Tomović
*ab
aDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: z.tomovic@tue.nl; r.p.sijbesma@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
cBASF SE, Carl-Bosch-Str. 38, 67056 Ludwigshafen am Rhein, Germany
dBASF Polyurethanes GmbH, Elastogranstraße 60, 49448 Lemförde, Germany
First published on 22nd March 2023
Abstract
Poly(urethane imide)s (PUIs) with improved thermal properties and flame retardancy can be made in a direct way by the reaction of isocyanates with anhydrides to give aromatic imides. We investigated the mechanism of this reaction in the presence of water with experimental studies and quantum chemical calculations. The catalytic cycle is driven by the urea obtained from the hydrolysis of isocyanates. We show that with a secondary amine as a pre-catalyst and tertiary amine as a co-catalyst, the reaction proceeds fast without a need for additional solvent. The insights in the underlying mechanism provided by the computational study have guided the development of a solvent-free synthetic method that provides a pathway to produce PUIs on an industrial scale.
Introduction
Polyurethanes (PU's) are among the most widely produced plastics worldwide.1 Showing versatile properties, PU's are used for construction, consumer products, furniture and in the automotive industry.2–5 Although many different PU materials are available, further improvement of the physical properties of the materials will make them suitable for an increased number of applications, providing new market opportunities. Aromatic polyimides present a class of well-known high-performance polymers with outstanding thermal, mechanical, and electrical properties.6–13 They have been used to improve the properties of a variety of polymer matrixes (e.g., poly(ether imide)s, poly(ester imide)s, poly(amide imide)s).10,14–18 Especially the co-polymerization of prepolymers containing aromatic polyimide structures with conventional PU raw materials results in significant improvements in thermal stability and flame retardancy and hence, poly(urethane imide)s are promising building blocks for the development of new thermoplastics,19–25 rigid foams26–30 and coatings.31–34Aromatic imides are usually introduced in poly(urethane-imide)s by the reaction of amines or isocyanates with anhydrides.9–11 It is generally accepted that the amine–anhydride reaction follows a two step mechanism. First, the nucleophilic amine attacks one of the carbonyl carbon atoms of the anhydride, forming an amic acid. Second, imidization of the amic acid leads to the release of a molecule of water.9,11,35 Whereas the mechanism of the reaction of anhydrides with amines is known, the much more complicated mechanism of imide formation from reaction of anhydrides with aromatic isocyanates is still under debate.11
Two distinct mechanisms have been proposed for the formation of imides from reaction of aromatic isocyanates with anhydrides. Based on Fourier-transform infrared spectroscopy (FT-IR) studies, several groups proposed that the reaction between aromatic isocyanate and anhydride forms a seven-membered-ring, followed by imidization with elimination of a CO2 molecule (Scheme 1a).36–40 Later, these findings were challenged by Carleton et al. who postulated that the spectroscopic evidence points to a hydrolysis product rather than an intermediate with a seven-membered ring, because they discovered that the polymerization rate increased with water concentration.28,41 Accordingly, they proposed that the aromatic imide is obtained from the reaction of anhydride with a urea (Scheme 1b), accelerated by water and other nucleophiles. However, no reliable kinetic or mechanistic studies are currently available to give conclusive evidence for either proposed mechanism.
 | ||
| Scheme 1 Proposed mechanisms of aromatic isocyanate–anhydride reaction proposed from literature: (a) 7-membered ring mechanism;36 (b) urea–dianhydride mechanism, where urea is formed from the intermediary amine hydrolysis product of isocyanate.41 | ||
A common feature in all experimental studies on aromatic isocyanate–anhydride reactions is the use of highly polar solvents such as NMP, DMAc, DMF.28,36–41 These solvents not only ensure the solubility of reactants and products, but also inevitably contain water, which catalyzes the reaction. Therefore, it is very difficult to prove a mechanism with a seven-membered-ring intermediate, because it will only occur in water-free conditions.
A better understanding of the mechanism of aromatic isocyanate–dianhydride reactions will allow further development of poly(urethane-imide) chemistry, by guiding optimization of the reaction conditions. Here, the reaction mechanism of imide formation from aromatic isocyanates and dianhydrides in the presence of catalytic amounts of water was investigated with nuclear magnetic resonance (NMR) spectroscopy, liquid chromatography-mass spectrometry (LC-MS), and quantum mechanical (QM) computations. The results reveal (i) that the preferred pathway involves formation of urea as a hydrolysis product, and (ii) in the reaction of excess isocyanate with anhydrides, urea instead of water is the actual catalyst in the reaction. Furthermore, we demonstrate that secondary amines are better pre-catalysts than water and that addition of a nucleophilic co-catalyst considerably reduces reaction time. These novel insights of the underlying mechanism will help to pave the way towards a completely solvent-free synthetic route to imides, which is important for developing poly(urethane-imide)s in industrial applications.
Results and discussion
Hydrolysis products in aromatic isocyanate–anhydride reactions
The reaction between phthalic anhydride and phenyl isocyanate (this combination giving rise to a soluble imide) in the presence of water was used to determine the hydrolysis products (Scheme 2). First, model reaction (2) was carried out in an NMR tube in DMSO-d6. The reaction was monitored with NMR spectroscopy at room temperature and the formation of N-phenylphthalimide was observed. However, neither the proton nor the carbon NMR spectra showed evidence of an intermediate with a seven-membered-ring (see ESI†). To shed some light on this mechanism, we also performed QM calculations for the seven-membered ring pathway. The results given in the ESI (Scheme S1†) show no indication of the formation of such an intermediate, due to a high kinetic barrier resulting from a very high activation energy for its formation. | ||
| Scheme 2 Formation of N-phenylphthalimide from reaction between phthalic anhydride and phenyl isocyanate. | ||
When water is present in the reaction mixture, isocyanate or anhydride can be hydrolyzed to urea or the diacid, respectively. To verify the formation of these hydrolysis products, model reaction (2) was further dissected by studying reactions (3a) and (3b), in which each one of the phthalic anhydride or phenyl isocyanate hydrolysis products was allowed to react with the other component in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (Scheme 3).
:1 molar ratio (Scheme 3).
 | ||
| Scheme 3 Formation of N-phenylphthalimide from reaction between (a) 1,3-diphenylurea and phthalic anhydride; (b) phthalic acid and phenyl isocyanate. | ||
According to 13C NMR spectra (Fig. 1a), full conversion to N-phenylphthalimide was achieved after reacting 1,3-diphenyl urea and phthalic anhydride (reaction (3a)) in DMF at 140 °C for 22 h, while no N-phenylphthalimide was obtained by carrying out the reaction at lower temperature (e.g., at 66 °C in refluxing THF). Reaction of phthalic acid with phenyl isocyanate (reaction (3b)) in refluxing THF only gave mono- and di-amides. Interestingly, the formation of phthalic anhydride (carbonyl carbon at 164.0 ppm) and 1,3-diphenylurea (carbonyl carbon at 153.5 ppm) were observed (Fig. 1b).42,43 In DMF at 140 °C, phthalic anhydride and diphenylurea were formed within 15 min, and a small amount of imide was produced. Comparison of reactions (3a) and (3b) suggests that urea is more readily formed than diacid, and that the reaction between urea and anhydride requires high temperatures.
 | ||
| Fig. 1 13C NMR spectra (400 MHz, acetone-d6) of the product obtained from reaction between (a) 1,3-diphenylurea and phthalic anhydride in DMF at 140 °C; (b) phthalic acid and phenyl isocyanate in THF reflux or DMF at 140 °C. | ||
Nevertheless, the mechanism of the reaction between urea and anhydride needs further investigation because it is not clear how urea reacts with two molecules of anhydride under the release of one molecule of each CO2 and H2O. Therefore, LC-MS was used to identify the intermediates in the isocyanate–anhydride reaction in the presence of water. To speed up the reaction, a highly reactive anhydride, pyromellitic dianhydride (PMDA) was used, while the solubility of the imide product for LC-MS measurement was improved by using hexyl isocyanate. Model reaction (4) between PMDA and hexyl isocyanate was carried out in refluxing THF containing 1 mol% water (Scheme 4). After 21 h, new peaks in the 1H NMR spectrum between 3.65–3.80 ppm (Fig. 2a) indicated formation of dihexylurea and imide. In LC-MS of the mixture obtained (Fig. 2b and Table S1†), a peak with a signal at m/z = 548.25 was observed, which was assigned to the ring-opened product of the reaction of PMDA with 1,3-dihexylurea (VI and VII). This is a strong indication that a urea is involved as intermediate in the formation of imide from reaction of isocyanate with anhydride.
 | ||
| Scheme 4 Model reaction of pyromellitic anhydride and hexyl isocyanate in the presence of catalytic amount of water. The reaction was used to identify the intermediate during the isocyanate–anhydride reaction. | ||
 | ||
| Fig. 2 (a) 1H NMR spectrum (400 MHz, acetone-d6) and (b) LC-MS spectrum of the intermediates obtained in model reaction (4) (all detected structures in LC-MS spectrum are listed in Table S1†). | ||
Optimization of the aromatic isocyanate–anhydride reaction
Based on the observation of a signal with m/z = 548.25 in LC-MS, we proposed that during the aromatic isocyanate–anhydride reaction, the urea is deprotonated and attacks the positively charged carbonyl carbon of PMDA, forming an amic acid. After the ring closure of the amic acid and formation of an imide, the urea is released. To accelerate this reaction, we systematically investigated different pre-catalysts, other than water in combination with additional co-catalysts.Reaction conditions were optimized with PMDA as a substrate. PMDA is one of the most reactive dianhydrides due to its high electron affinity,9,44 and thus, it is commonly used to react with excess of isocyanates to prepare isocyanate prepolymers that contain imides. The imide-containing prepolymer is used to prepare different polyurethane materials such as compact materials and rigid foams with enhanced thermal properties. In our optimization experiments, PMDA was reacted with polymeric methylene diphenyl diisocyanate (PMDI), Lupranate® M20 (NCO = 31.5 wt%, fn = 2.7) in a 1:19 weight ratio to investigate and optimize the pre-catalyst as well as co-catalyst (Scheme 5). The dosing amount of the pre-catalyst and co-catalyst was based on the mole amount of PMDI. Model reaction (5) was carried out at 140 °C in bulk with 10 vol% triethyl phosphate (TEP) as an additive to avoid sublimation of PMDA. The reaction was followed with 13C NMR spectroscopy, monitoring the carbonyl carbon peaks of anhydride and imide products (Fig. S2†). When an excess of isocyanate reacted with PMDA in the presence of water, we have demonstrated that urea was the only hydrolysis product (Fig. S3 and S4†). This also supports the proposal that urea is the real catalyst of the reaction, with water only acting as a pre-catalyst.
 | ||
| Scheme 5 Model reaction between pyromellitic anhydride and polymeric methylene diphenyl diisocyanate in a 1:19 weight ratio. | ||
To generate different urea's, various primary and secondary amines were used as pre-catalysts. The reaction time to achieve full conversion is listed in Table 1. It was found that dibutylamine, a secondary amine, was able to accelerate the reaction.
Next, using dibutylamine as a pre-catalyst, basic or nucleophilic tertiary amines were used as co-catalysts to deprotonate urea (Table 2). With the help of co-catalysts, the reaction time was significantly reduced, particularly when co-catalysts with little steric hindrance were used. However, the more nucleophilic the co-catalyst was, the more likely it was to catalyze the cyclotrimerization of isocyanates after the isocyanate–dianhydride reaction (entries 5-8 to 5-10).45
| Entry | Co-catalyst | Reaction timea |
|---|---|---|
| a The time when there was only di-imide carbonyl peak in 13C NMR spectra was defined as reaction time. | ||
| 5-4 | N,N-Diisopropylethylamine | 8 h |
| 5-5 | Tributylamine | 8 h |
| 5-6 | N,N-Dimethylcyclohexylamine | 6 h |
| 5-7 | 4-Methylmorpholine | >18 h |
| 5-8 | 4-(Dimethylamino)pyridine | Isocyanurate formation |
| 5-9 | 1,4-Diazabicyclo[2.2.2]octane | Isocyanurate formation |
| 5-10 | 1,5,7-Triazabicyclo[4.4.0]dec-5-en | Isocyanurate formation |
Based on results shown in Table 2, N,N-dimethylcyclohexylamine (DMCHA) was chosen as the optimal co-catalyst and various secondary amines were evaluated as a pre-catalyst (Table 3). N-Methylaniline turned out to be a better secondary amine pre-catalyst than dibutylamine. When N-methylaniline was used as a pre-catalyst and DMCHA was used as a co-catalyst, the reaction time was greatly shortened from 24 h (entry 5-1) to 4 h (entry 5-11).
| Entry | Pre-catalyst | Reaction timea |
|---|---|---|
| a The time when there was only di-imide carbonyl peak in 13C NMR spectra was defined as reaction time. | ||
| 5-11 | N-Methylaniline | 4 h |
| 5-12 | 4-Methoxy-N-methylaniline | 6 h |
| 5-13 | N-Methyl-o-toluldine | 7 h |
| 5-14 | Imidazole | 7 h |
| 5-15 | Diphenylamine | 6 h |
| 5-16 | ε-Caprolactam | 5 h |
| 5-17 | Phthalimide | 8 h |
Based on these experimentally findings, we conclude that water or amines function as pre-catalysts in the aromatic isocyanate–anhydride reaction, while the urea obtained out of it is the real catalyst. Additional experiments were performed by replacing secondary amines with urea as catalysts in model reaction (5); also here, imide structures were obtained in the end (see ESI†).
Computational studies of aromatic isocyanate–anhydride reaction with secondary amines as pre-catalysts
To obtain further insights into the thermodynamics and kinetics of the isocyanate–anhydride reaction, computer simulations were additionally applied. Quantum chemical calculations serve nowadays as an indispensable tool to reveal reaction mechanism at an atomic level. Therefore, in state-of-the-art computational workflows, efficient screening techniques are combined with highly accurate density functional theory (DFT) methods for postprocessing.46 In this work, we employ the widely used CREST algorithm combined with extended tight-binding QM methods (GFN2-xTB) to explore the low-energy conformational space.46 Conformational screening was followed by DFT re-optimization (TPSS-D3/def2-TZVP)47–50 and final single-point energies were computed with the M06-2X51 density functional approximation in a large def2-QZVP basis set. Bulk phenyl isocyanate was taken as the solvent and accounted for implicitly by COSMO-RS52,53 theory (Fig. 3). For further computational details see Experimental section.
 | ||
| Fig. 3 Computational workflow combining efficient SQM and DFT methods to generate conformers and compute free energies in solution. | ||
The free energy diagram of the imide-forming reaction is given in Fig. 4 (black line). As an upstream process, N-methylaniline is added to phenyl isocyanate to form 1-methyl-1,3-diphenyl-urea, or simply urea from now on. In the transition state TS0 (see Fig. S5a†), water acts as a hydrogen shuttle and forms a six-membered ring. With secondary amines, the urea formation is 30 kJ mol−1 higher in energy, underlining the assumption that water is the pre-catalyst for fast urea generation. Urea is then deprotonated by DMCHA, yielding the effective catalyst of the imide-forming reaction mechanism. Please note, from this point on, the reaction path occurs via charged species. In a nucleophilic addition, the urea anion ring-opens the anhydride A to form intermediate B. The reaction is facilitated by hydrogen-bonding between DMCHA+ and PMDA within the transition state TS1 (Fig. S5b†), showing an activation barrier (ΔG‡) of 9 kJ mol−1. As none of the two nitrogen atoms within this intermediate are good nucleophiles, ring-closure at this stage of the reaction cycle is not possible. Instead, the carboxylate group of B adds one more equivalent of phenyl isocyanate to form the second intermediate Cvia TS2 (Fig. S5c†) with a ΔG‡ of 63 kJ mol−1. In the rate determining step of the reaction, CO2 is released via TS3 that occurs via a four-membered ring (Fig. S5d†). The activation barrier for the CO2 release amounts to 52 kJ mol−1. It is now the nucleophilic nitrogen atom in D that initializes the ring closure by formation of the tetrahedral intermediate E. Elimination of the catalytically acting urea anion finally yields the desired imide F.
 | ||
| Fig. 4 Free energy diagram of the conversion of pyromellitic anhydride and phenyl isocyanate to form an (di)imide at the M06-2X/def2-QZVP+COSMO-RS(phenyl isocyanate)//TPSS-D3(COSMO(∞))/def2-TZVP level of theory. All free energies are given in kJ mol−1. | ||
Our quantum chemical computer simulations prove that water only acts as a pre-catalyst, while urea, or to be more precise its deprotonated form, acts as the actual catalyst. All findings can be summarized in a single catalytic cycle shown in Fig. 5 (black lines).
 | ||
| Fig. 5 Catalytic cycle of the aromatic isocyanate–dianhydride reaction using N-methylaniline as a pre-catalyst and N,N-dimethylcyclohexylamine as a co-catalyst. The computed activation free energies (ΔG‡) for all transition states and reaction free energies (ΔG) are given in kJ mol−1. | ||
Further theoretical insights were needed to explain a fast initial release of CO2 (minutes) that was observed experimentally, followed by a slower release of CO2 over the next few hours. NMR studies suggested that the symmetric anhydride PMDA reacts in a two-step process via a one-sided imide to a di-imide (Fig. 6). Therefore, the QM simulation of the reaction cycle of imide formation was repeated, starting from the half-imide–half-anhydride structure (Fig. 4 and 5 red line). The change in the electronic structure caused by the half-imide formation leads to an upshift in energy of the reaction path compared to PMDA. Especially at the highest energy point (TS3), according to transition state theory,54 a relative increase of 8 kJ mol−1 causes the reaction to last more than ten times longer (hours instead of minutes). This good agreement between computer simulations and experimental findings further establishes the correctness of the identified reaction mechanism.
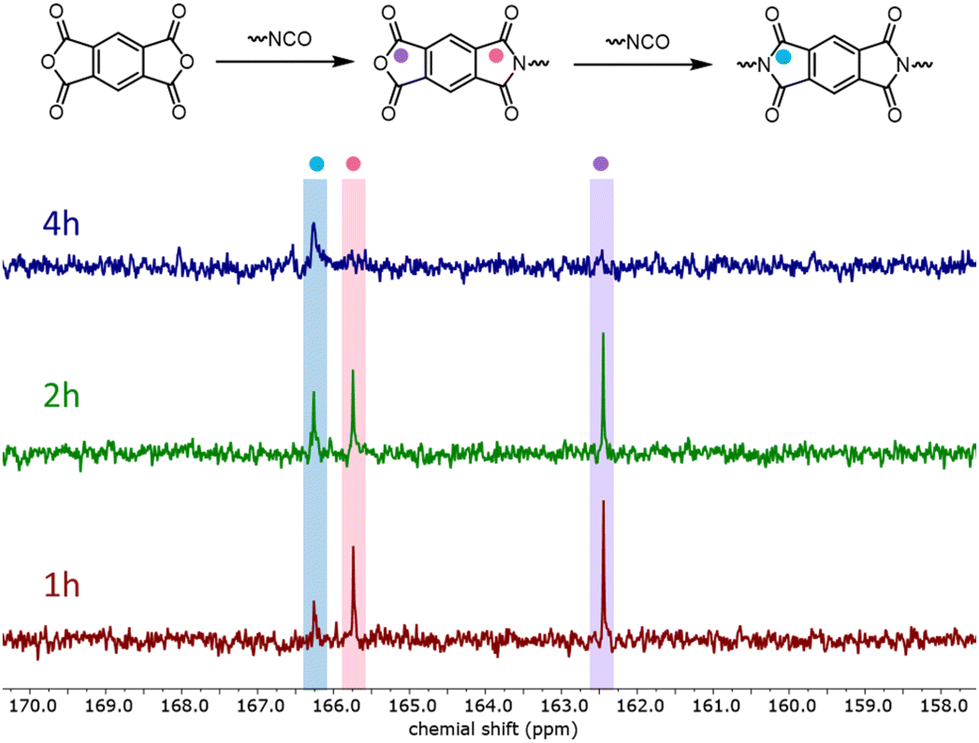 | ||
| Fig. 6 13C NMR spectra (400 MHz, acetone-d6) of model reaction (5) using N-methylaniline as a pre-catalyst and N,N-dimethylcyclohexylamine as a co-catalyst. | ||
There is a discrepancy between experimental observations and absolute theoretically computed activation barriers. With optimal catalysts (N-methylaniline, DMCHA) and additives (TEP) the reaction finishes within four hours, whereas it would take days to overcome the highest activation barrier (TS3) of 168 kJ mol−1 with respect to the catalytic resting state. It can be expected that the applied implicit COSMO(-RS) solvation model does not accurately describe the charge separation leading to free anions. Especially because of the high temperatures (140 °C), the validity of the applied solvation model is questionable. Since the importance of additive TEP is experimentally known, yet the implicit incorporation of 10 vol% within COSMO-RS shows no significant energy lowering of the ionic species, we added an explicit TEP molecule for stabilization. Exemplary, the deprotonation of urea by DMCHA was explicitly solvated with TEP, lowering the free energy of the resulting complex by −10 kJ mol−1. Since the explicit incorporation of TEP within the entire catalytic cycle is computationally too demanding, we assume that this energy lowering can be transferred to all ionic species along the reaction path. This simple example of explicit solvent addition already significantly reduces the divergence between experiment and theory. It highlights the importance of an accurate description of solvation effects and shows that in current solvent models there is still room for improvement if ionic species are concerned.
Conclusion
We investigated the reaction mechanism of aromatic isocyanates and dianhydrides in the presence of water to form imides. Our study revealed that during the reaction urea, formed as the hydrolysis product from isocyanate, acted as the actual catalyst in the reaction. Secondary amines, with the help of tertiary amine bases, were further discovered as better pre-catalysts (or, rather, co-catalysts) than water. The reaction time strongly decreased from 24 h with water as a pre-catalyst to 4 h with N-methylaniline and N,N-dimethylcyclohexylamine as pre-catalyst and co-catalyst respectively. Further, the combination of experimental and computational results revealed exclusive insights in the reaction of aromatic isocyanates and dianhydrides, starting from the deprotonated urea, which ring-opens the anhydrides to form an amic acid intermediate. After this step, the carboxylate of the acid intermediate reacts with one more isocyanate molecule, followed by release of a CO2 molecule and ring closure, forming the imide structure.Our current study highlights how the combination of state-of-art experimental and computational techniques allows for unique mechanistic insights and is able to answer questions that have been open for debate for quite a while. These exclusive insights into the underlying mechanism demonstrate the possibility of producing poly(urethane-imide)s under completely solvent-free conditions with a short reaction time. This is especially important for industrial applications in terms of green chemistry and low costs. Our results provide convincing evidence that the revealed mechanism of aromatic imide formation can also be applied in the reaction between various aliphatic isocyanates and dianhydrides to form aliphatic imides. These aliphatic imides can potentially help to improve the thermal stability of materials that are synthesized from aliphatic isocyanates such as PU coatings, sealants, and adhesives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Julian Kleemann (BASF Polyurethanes GmbH) for discussions and supports. The authors also acknowledge financial support from BASF Polyurethanes GmbH.Notes and references
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- N. V. Gama, A. Ferreira and A. Barros-Timmons, Materials, 2018, 11, 1841 CrossRef PubMed.
- D. Randall and S. Lee, The Polyurethanes Book, Wiley, 2003 Search PubMed.
- B. Eling, Ž. Tomović and V. Schädler, Macromol. Chem. Phys., 2020, 2000114, 1–11 Search PubMed.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- P. M. Hergenrother, High Perform. Polym., 2003, 15, 3–45 CrossRef CAS.
- C. David, Thermal degradation of polymers, 1975, vol. 14 Search PubMed.
- J. M. Dodda and P. Bělský, Eur. Polym. J., 2016, 84, 514–537 CrossRef CAS.
- C. E. Sroog, J. Polym. Sci., Macromol. Rev., 1976, 11, 161–208 CrossRef CAS.
- G. S. Liou and H. J. Yen, Polyimides, 2012, vol. 5 Search PubMed.
- M. Ghosh, Polyimides: Fundamentals and Applications, CRC Press, 2018 Search PubMed.
- A. Sezer Hicyilmaz and A. Celik Bedeloglu, SN Appl. Sci., 2021, 3, 1–22 Search PubMed.
- X. J. Liu, M. S. Zheng, G. Chen, Z. M. Dang and J. W. Zha, Energy Environ. Sci., 2022, 15, 56–81 RSC.
- L. Li, Y. Xu, J. Che and Z. Ye, Polym. Adv. Technol., 2019, 30, 120–127 CrossRef CAS.
- K. Cao, Y. Guo, M. Zhang, C. B. Arrington, T. E. Long, R. R. Odle and G. Liu, Macromolecules, 2019, 52, 7361–7368 CrossRef CAS.
- P. Banu and G. Radhakrishnan, J. Polym. Sci. Part A Polym. Chem., 2003, 42, 341–350 CrossRef.
- G. Zhu, H. Lao, F. Feng, M. Wang, X. Fang and G. Chen, Eur. Polym. J., 2022, 179, 111558 CrossRef CAS.
- S. D. Kim, B. Lee, T. Byun, I. S. Chung, J. Park, I. Shin, N. Y. Ahn, M. Seo, Y. Lee, Y. Kim, W. Y. Kim, H. Kwon, H. Moon, S. Yoo and S. Y. Kim, Sci. Adv., 2018, 4, eaau1956 CrossRef CAS PubMed.
- Q. Tang, Y. Song, J. He and R. Yang, J. Appl. Polym. Sci., 2014, 131, 9524–9533 Search PubMed.
- M. F. Lin, Y. C. Shu, W. C. Tsen and F. S. Chuang, Polym. Int., 1999, 48, 433–445 CrossRef CAS.
- M. P. Sokolova, A. N. Bugrov, M. A. Smirnov, A. V. Smirnov, E. Lahderanta, V. M. Svetlichnyi and A. M. Toikka, Polymers, 2018, 10, 1222 CrossRef PubMed.
- P. R. Nair, C. P. R. Nair and D. J. Francis, J. Appl. Polym. Sci., 1998, 70, 1483–1491 CrossRef.
- K. Asai, S. I. Inoue and H. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 715–723 CrossRef CAS.
- T. Philip Gnanarajan, N. Padmanabha Iyer, A. Sultan Nasar and G. Radhakrishnan, Eur. Polym. J., 2002, 38, 487–495 CrossRef CAS.
- J. Liu and M. Dezhu, J. Appl. Polym. Sci., 2002, 84, 2206–2215 CrossRef CAS.
- A. Müller-Cristadoro and F. Prissok, WO 2014023796 A1, 2014.
- H. Tian, Y. Yao, S. Zhang, Y. Wang and A. Xiang, Polym. Test., 2018, 67, 68–74 CrossRef CAS.
- W. J. Farrissey, J. S. Rose and P. S. Carleton, J. Appl. Polym. Sci., 1970, 14, 1093–1101 CrossRef CAS.
- J. Kashiwame and K. Ashida, J. Appl. Polym. Sci., 1994, 54, 477–486 CrossRef CAS.
- K. Xi, D. J. Shieh, L. Wu and S. Singh, WO 2021030115 A1, 2021.
- A. K. Mishra, D. K. Chattopadhyay, B. Sreedhar and K. V. S. N. Raju, Prog. Org. Coat., 2006, 55, 231–243 CrossRef CAS.
- A. K. Mishra, D. K. Chattopadhyay, B. Sreedhar and K. V. S. N. Raju, J. Appl. Polym. Sci., 2006, 102, 3158–3167 CrossRef CAS.
- D. K. Chattopadhyay, A. K. Mishra, B. Sreedhar and K. V. S. N. Raju, Polym. Degrad. Stab., 2006, 91, 1837–1849 CrossRef CAS.
- M. Meena, A. Kerketta, M. Tripathi, P. Roy and J. Jacob, J. Appl. Polym. Sci., 2022, 139, e52508 CrossRef CAS.
- Z. Xu, Z. L. Croft, D. Guo, K. Cao and G. Liu, J. Polym. Sci., 2021, 59, 943–962 CrossRef CAS.
- R. A. Meyers, J. Polym. Sci., Part A: Polym. Chem., 1969, 7, 2757–2762 CrossRef.
- M. Barikani and S. M. Ataei, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2245–2250 CrossRef CAS.
- M. Barikani and S. Mehdipour-ataei, J. Appl. Polym. Sci., 2000, 77, 1102–1107 CrossRef CAS.
- J. Y. Jeon and T. M. Tak, J. Appl. Polym. Sci., 1996, 62, 763–769 CrossRef CAS.
- C. Chidambareswarapattar, Z. Larimore, C. Sotiriou-Leventis, J. T. Mang and N. Leventis, J. Mater. Chem., 2010, 20, 9666–9678 RSC.
- P. S. Carleton, W. J. Farrissey and J. S. Rose, J. Appl. Polym. Sci., 1972, 16, 2983–2989 CrossRef CAS.
- W. R. Sorenson, J. Org. Chem., 1959, 24, 978–980 CrossRef CAS.
- A. Fry, J. Am. Chem. Soc., 1953, 75, 2686–2688 CrossRef CAS.
- R. F. Mundhenke and T. S. Willis, High Perform. Polym., 1990, 2, 57–66 CrossRef CAS.
- Y. Guo, M. Muuronen, P. Deglmann, F. Lucas, R. P. Sijbesma and Ž. Tomović, J. Org. Chem., 2021, 86, 5651–5659 CrossRef CAS PubMed.
- S. Grimme, F. Bohle, A. Hansen, P. Pracht, S. Spicher and M. Stahn, J. Phys. Chem. A, 2021, 125, 4039–4054 CrossRef CAS PubMed.
- J. Tao, J. Perdew, V. Staroverov and G. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A. Constantin and J. Sun, Phys. Rev. Lett., 2009, 103, 10–13 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00109a |
| This journal is © The Royal Society of Chemistry 2023 |