
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilising the effect of reaction concentration to tune the physical properties of hyperbranched polymers synthesised using transfer-dominated branching radical telomerisation (TBRT)†
Savannah R.
Cassin
ab,
Stephen
Wright
ab,
Samuel
Mckeating
ab,
Oliver B.
Penrhyn-Lowe
ab,
Sean
Flynn
ab,
Sarah
Lomas
ab,
Pierre
Chambon
ab and
Steve P.
Rannard
 *ab
*ab
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
bMaterials Innovation Factory, University of Liverpool, Crown Street, L69 7ZD, UK
First published on 14th March 2023
Abstract
The formation of complex polymer architectures with novel physicochemical properties is of great importance in the development of next generation advanced materials. The recent reports of Transfer-dominated Branching Radical Telomerisation (TBRT) provide readily accessible routes to a range of previously inaccessible macromolecular architectures, utilising free radical reactions under telomerisation conditions. Herein, we describe the variation in the physical properties of hyperbranched polymers synthesised via TBRT, upon the manipulation of the reaction concentration at which the telomerisations were conducted. Through the careful control of multi-vinyl taxogen to telogen ratios, we have shown that the formation of a crosslinked network can be prevented at various reaction concentrations (0 wt% (bulk) – 90 wt% solvent), leading to materials with tuneable physical properties. The impact of excess solvent (≥70 wt% solvent) was exemplified by the significant variation of glass transition temperatures of the resulting materials and the reduction of telogen incorporated in the final structure, providing clear evidence of intramolecular cyclisation.
Introduction
Complex polymer architectures offer materials chemists the opportunity to access novel physical properties and behaviours. New, scalable polymerisation strategies for the synthesis of hyperbranched macromolecules are of wide interest in both academia and industry. Over the past decade, the homopolymerisation of multi-vinyl monomers (MVMs) has become a focus for several research groups as an approach for the preparation of a variety of hyperbranched polymers.1 A range of elegant homopolymerisation technologies have been developed using synthesis strategies ranging from catalytic chain transfer polymerisation,2 RAFT,3 initiator-fragment incorporation radical polymerisation4 and deactivation enhanced ATRP.5,6 These all effectively prepare hyperbranched polymers containing residual vinyl functionality, even when very high MVM consumption is seen. Incomplete reaction of vinyl groups is a crucial characteristic in these reports as it allows the avoidance of a crosslinked network by keeping vinyl group conversion lower than approximately 70 mol%. Unlike a linear polymerisation of mono-vinyl monomers, where a decrease in vinyl group concentration may be correlated to polymer formation, an MVM polymerisation may fully consume free monomer whilst leaving a significant number of vinyl groups unreacted.Recently we reported a novel synthetic approach that utilises free radical telomerisation7–9 conditions to create soluble, high molecular weight hyperbranched polymers with complete vinyl group conversion, namely Transfer-dominated Branching Radical Telomerisation (TBRT), Fig. 1.10–14 Telomerisation reactions are conventionally used to form very short chains with a number average degree of polymerisation (DPn) of less than approximately 5 units.
 | ||
| Fig. 1 Comparative schematic representation of (A) conventional free radical telomerisation of mono-vinyl taxogens with a thiol-based telogen forming linear telomers, and (B) transfer-dominated branching radical telomerisation (TBRT) of multi-vinyl taxogens with thiol-based telogens forming hyperbranched structures. | ||
Under telomerisation conditions, Fig. 1A, unsaturated substrates are termed taxogens and agents that control the telomerisation reaction are termed telogens (Tel); an MVM is, therefore, more correctly termed a multi-vinyl taxogen (MVT) during a TBRT reaction. The polyfunctional nature of MVTs ensures that, although the average kinetic chain length of the telomer distribution is extremely short, the population of telomers is conjoined through the MVT linking chemistry. Indeed, the chemistry within the MVT dominates the longest chain structures within the hyperbranched polymer architectures and the nominal repeating structures resemble those more conventionally formed by step-growth polymerisation, e.g. a dimethacrylate MVT will form a hyperbranched polyester under TBRT conditions, Fig. 1B.
Within this study, and many historical reports of telomerisation, thiols are utilised as telogens. The weight average molecular weight (Mw) of a TBRT polymer may be varied by the starting MVT/Tel molar ratio and optimisation of TBRT conditions will ensure that propagation of the vinyl groups is restricted to an average kinetic chain length <2 units via extensive transfer reactions, allowing avoidance of gelation at complete vinyl group conversion.10–15 Within a TBRT reaction the telogen is incorporated into the final hyperbranched structure at a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio with MVT, forming a thioether group that is pendant to the backbone, Fig. 1B & Scheme 1, and subsequently influencing the physical properties of the final branched polymer.12 This is fundamentally different to branched copolymerisations using thiol chain transfer agents and low concentrations of MVMs with reactions predominantly focussed on the formation of primary chains derived from mono-vinyl monomers (e.g. the Strathclyde approach).16–18
:1 molar ratio with MVT, forming a thioether group that is pendant to the backbone, Fig. 1B & Scheme 1, and subsequently influencing the physical properties of the final branched polymer.12 This is fundamentally different to branched copolymerisations using thiol chain transfer agents and low concentrations of MVMs with reactions predominantly focussed on the formation of primary chains derived from mono-vinyl monomers (e.g. the Strathclyde approach).16–18
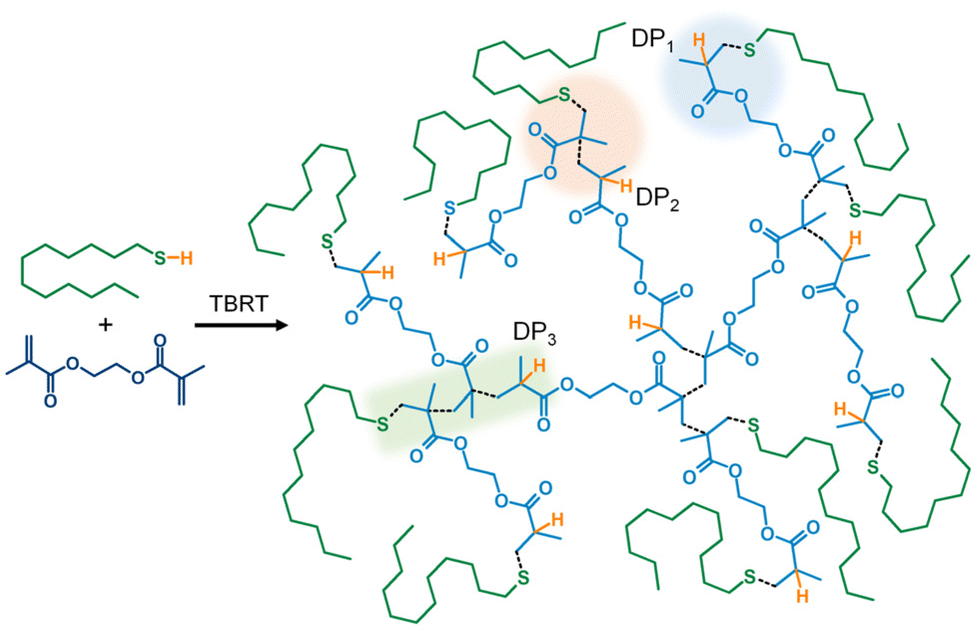 | ||
| Scheme 1 Synthesis of branched polyester by TBRT using ethylene glycol dimethacrylate (EGDMA) as the MVT, and 1-dodecanethiol (DDT) as the telogen. Black dotted bonds show bonds formed during the polymerisation. Substructures derived from addition of DDT to: one vinyl bond (DP1) highlighted by blue area; one double bond plus a single propagation step (DP2) highlighted by orange area; and one double bond plus two propagation steps (DP3) highlighted by green area. Low molecular weight polymer shown to allow detail to be seen. Compare with Fig. 1. | ||
A detailed understanding of new polymerisation approaches requires studies to determine how reaction conditions may influence the resulting materials. Many polymerisations are known to be susceptible to variations in synthesis conditions. For example, step-growth polymerisations of ABn or combinations of Am + Bn + C monomers may be subject to intramolecular cyclisation.19–21 Indeed, reaction concentration can enhance cyclisation within many linear step-growth polymerisations by intramolecular end-group reaction.22,23 Maximising intermolecular branching within the homopolymerisation of MVMs is key to the elimination of intramolecular cyclisation, and reaction concentration is a clear parameter that may strongly influence this balance.
Under relatively dilute reaction conditions, pendant vinyl groups on growing macromolecules may experience a relatively high local vinyl group concentration due to the proximity of nearby pendant groups, thereby encouraging cycle formation. These reactions do not contribute to increasing molecular weight but will influence the polymer topology. Several computational studies, including Monte Carlo simulations, have aimed to provide a view of complex polymerisation reactions and cycle formation.24–27
Conversely, under concentrated reaction conditions intermolecular branching would be expected to be favoured. The critical overlap concentration, c*, of a polymer is related to the molecular volume and radius of gyration and has been used to predict the dominant interactions in various MVM homopolymerisations.28,29 A concentration below c* implies the dominant interactions are intramolecular, since the growing polymer chains do not interpenetrate/overlap under dilute conditions. Alternatively, a value above c* (high polymer concentration, or solids content) implies an increase in intermolecular branching. For free radical polymerisations, an increase in chain concentration by solvent reduction may lead to a loss of kinetic control due to an increase in the rate of propagation, resulting in a higher flux of free radical species and possible gelation when MVMs are present.
We recently described the impact of varying MVT dimensions on cyclisation within TBRT reactions11 and due to the novel nature of TBRT, here we aim to study the impact of reaction concentration on this new polymerisation strategy. To enable direct comparison across several reaction concentrations a single MVT/Tel combination has been selected, namely ethylene glycol dimethacrylate (EGDMA) as the MVT, and 1-dodecanethiol (DDT) as the telogen. Ethyl acetate (EtOAc) has been selected as the reaction solvent and all reactions have been conducted at a temperature of 70 °C using azobisisobutyronitrile (AIBN) as the radical source.
Results and discussion
Exploring the impact of high concentration conditions within TBRT
A range of TBRT reactions targeting p(DDT-EGDMA) were conducted with solvent content ranging from 0 wt%–50 wt% as detailed in Table 1. DDT and EGDMA are not entirely miscible at without heating, forming a visibly turbid mixture at ambient temperature and low solvent conditions. Upon heating, all reaction conditions became homogeneous, including TBRT polymerisations conducted in the absence of solvent (0 wt% EtOAc). As mentioned earlier, TBRT reactions are governed by the dominant chain transfer of thiyl radicals with an excess of thiol and the conditions required to obtain soluble polymer requires empirical study for each MVT and telogen combination in a chosen solvent. By varying the initial [MVT]0/[Tel]0 ratio for each reaction condition, a limiting gel point ratio can be identified. At this point, the telogen is unable to suppress gelation resulting in the formation of a crosslinked network. As can be seen in Table 1, reactions conducted in 50 wt% EtOAc were able to form soluble high molecular weight branched polymers, with complete reaction of vinyl functionality, at [EGDMA]0/[DDT]0 values <0.875 but the same reaction carried out in the absence of EtOAc (0 wt%) rapidly gelled (<1 hour) under these conditions. As mentioned earlier, this is to be expected as the probability of intermolecular reactions increases with increasing concentration.25| EtOAc (wt%) | [EGDMA]0 /[DDT]0a |
1H NMR (CDCl3) | TD-SEC (THF/TEA)d | |||||
|---|---|---|---|---|---|---|---|---|
| Vinyl conversionb (%) | [EGDMA]F /[DDT]Fc |
M w (g mol−1) | M n (g mol−1) | Đ | α | dn/dc | ||
| a Calculated based on feedstock reagent masses added to reaction flasks. b Determined by 1H NMR of crude sample after 24 h in CDCl3 (ESI S1–S6†). c Determined by 1H NMR of purified and dried material in CDCl3 (ESI Fig. S1–S6†). d Determined by triple-detection size exclusion chromatography using a 2% v/v TEA/THF eluent system. | ||||||||
| 0 | 0.350 | >99 | 1.04 | 30150 |
2450 | 12.3 | 0.238 | 0.089 |
| 0 | 0.400 | >99 | 1.01 | 56800 |
2850 | 19.9 | 0.243 | 0.091 |
| 0 | 0.425 | >99 | 0.94 | 80800 |
2400 | 33.7 | 0.271 | 0.087 |
| 0 | 0.450 | >99 | 0.98 | 292350 |
3500 | 83.5 | 0.260 | 0.086 |
| 0 | 0.500 | Gel | — | — | — | — | — | — |
| 10 | 0.450 | >99 | 1.05 | 127650 |
12650 |
10.1 | 0.294 | 0.089 |
| 10 | 0.475 | >99 | 1.07 | 133900 |
6350 | 21.1 | 0.271 | 0.089 |
| 10 | 0.500 | >99 | 0.94 | 327300 |
2550 | 128 | 0.300 | 0.088 |
| 10 | 0.525 | >99 | 1.05 | 949100 |
4650 | 204 | 0.321 | 0.090 |
| 10 | 0.550 | Gel | — | — | — | — | — | — |
| 20 | 0.500 | >99 | 1.00 | 63000 |
3250 | 19.4 | 0.285 | 0.088 |
| 20 | 0.525 | >99 | 1.08 | 107550 |
3200 | 33.6 | 0.287 | 0.090 |
| 20 | 0.550 | >99 | 1.03 | 402050 |
4100 | 98.1 | 0.307 | 0.088 |
| 20 | 0.575 | >99 | 1.03 | 757200 |
6950 | 109 | 0.306 | 0.092 |
| 20 | 0.600 | >99 | 0.95 | 1239000 |
12100 |
102 | 0.342 | 0.090 |
| 20 | 0.625 | Gel | — | — | — | — | — | — |
| 30 | 0.550 | >99 | 0.78 | 56150 |
1300 | 43.2 | 0.299 | 0.088 |
| 30 | 0.600 | >99 | 0.85 | 143950 |
1350 | 107 | 0.314 | 0.090 |
| 30 | 0.625 | >99 | 1.00 | 656100 |
2950 | 222 | 0.368 | 0.087 |
| 30 | 0.650 | >99 | 0.85 | 1194000 |
9200 | 130 | 0.336 | 0.093 |
| 30 | 0.700 | Gel | — | — | — | — | — | — |
| 40 | 0.650 | >99 | 0.91 | 46050 |
2050 | 22.5 | 0.288 | 0.094 |
| 40 | 0.675 | >99 | 0.98 | 242250 |
1350 | 179 | 0.334 | 0.088 |
| 40 | 0.700 | >99 | 1.05 | 371900 |
4450 | 83.6 | 0.325 | 0.092 |
| 40 | 0.725 | >99 | 1.07 | 834000 |
10300 |
81.0 | 0.325 | 0.092 |
| 40 | 0.750 | >99 | 1.00 | 1495000 |
16900 |
88.5 | 0.314 | 0.089 |
| 40 | 0.775 | Gel | — | — | — | — | — | — |
| 50 | 0.500 | >99 | 0.93 | 12800 |
2100 | 6.10 | 0.232 | 0.094 |
| 50 | 0.750 | >99 | 1.04 | 129350 |
4550 | 28.4 | 0.294 | 0.089 |
| 50 | 0.800 | >99 | 0.98 | 354250 |
5850 | 60.6 | 0.334 | 0.099 |
| 50 | 0.825 | >99 | 1.03 | 510450 |
7100 | 71.9 | 0.323 | 0.094 |
| 50 | 0.850 | >99 | 1.02 | 1822000 |
23650 |
77.0 | 0.340 | 0.093 |
| 50 | 0.875 | Gel | — | — | — | — | — | — |
The 33 polymers formed using different [EGDMA]0/[DDT]0 ratios and 6 reaction concentrations, Table 1, were purified by precipitation into cold MeOH and analysed by 1H nuclear magnetic resonance spectroscopy (NMR) and triple detection size exclusion chromatography (TD-SEC) Table 1, ESI Fig. S1–S6 & S13–S19.†
Molecular weight analysis of the purified polymers showed several interesting insights. When conducting TBRT at different solvent concentrations, (e.g. 10 wt% and 50 wt% EtOAc) with identical [EGDMA]0/[DDT]0 ratios (e.g. 0.500), Mw decreases with increasing dilution (increasing solvent). This is in agreement with increased dilution leading to a decrease in intermolecular reaction as the spatial distribution of growing macromolecules increases. Uniquely within a TBRT reaction, the formation of DP1 units (where telogen adds to a single vinyl group and no propagation has occurred) is critical Fig. 1B, ESI Scheme S2.†
This enables full reaction of vinyl groups and allows the avoidance of gelation. As the reaction concentration decreases, the growing branched macromolecules are increasingly separated, thereby facilitating DP1 formation at high MVM consumption and limiting vinyl group propagation, leading to a subsequent impact on intermolecular branching.
Exploring the impact of dilute reaction conditions within TBRT
As stated above, progressive dilution of a branching polymerisation is expected to decrease the probability of intermolecular reaction, essentially to zero at infinite dilution,13 whilst intramolecular cyclisation should become more likely.30,31 To continue the investigation of the impact of reaction concentration on TBRT polymerisations, a series of reactions with systematically increasing dilution (60 wt%–90 wt% EtOAc) were conducted whilst again altering the [EGDMA]0/[DDT]0 ratios to identify the limiting gel point values, Table 2, Fig. 2.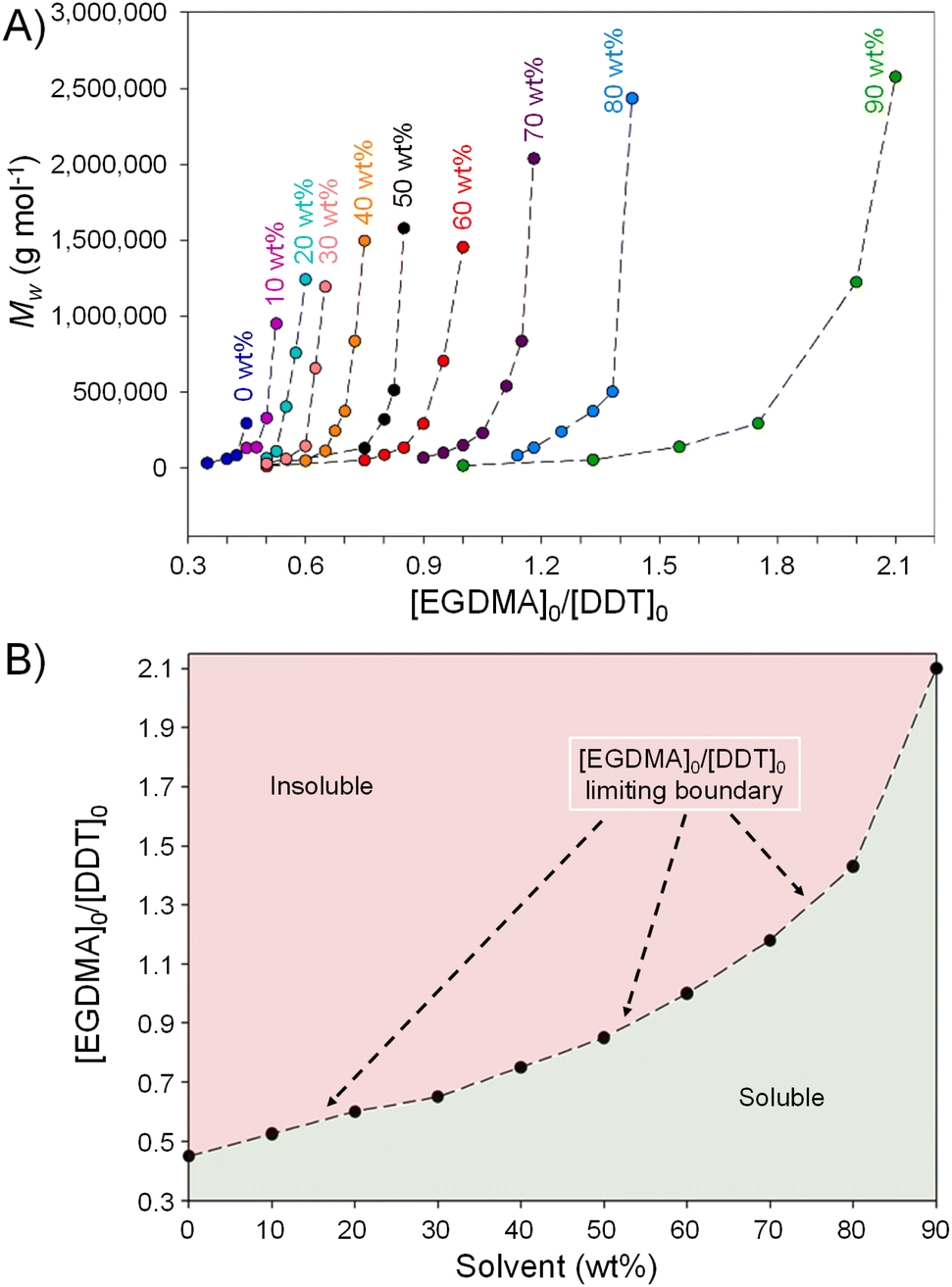 | ||
| Fig. 2 Correlation of (A) weight average molecular weight with starting reaction ratios of ethylene glycol dimethacrylate (EGDMA), as multi-vinyl taxogen, and 1-dodecanethiol (DDT), as telogen, and (B) limiting gel point values of [EGDMA]0/[DDT]0 with increasing ethyl acetate used as solvent in the reaction. | ||
| EtOAc (wt%) | [EGDMA]0 /[DDT]0a |
1H NMR (CDCl3) | TD-SEC (THF/TEA)d | |||||
|---|---|---|---|---|---|---|---|---|
| Vinyl conversionb (%) | [EGDMA]F /[DDT]Fc |
M w (g mol−1) | M n (g mol−1) | Đ | α | dn/dc | ||
| a Calculated based on feedstock reagent masses added to reaction flasks. b Determined by 1H NMR of crude sample after 24 h in CDCl3. c Determined by 1H NMR of purified and dried material in CDCl3 (ESI Fig. S20–S23†). d Determined by triple-detection size exclusion chromatography using a 2% v/v TEA/THF eluent system (ESI Fig. S25–S28†). e Data for reactions containing 50 wt% EtOAc are included from Table 1 to aid comparison. | ||||||||
| 50e | 0.500 | >99 | 0.93 | 12800 |
2100 | 6.10 | 0.232 | 0.094 |
| 50e | 0.750 | >99 | 1.04 | 129350 |
4550 | 28.4 | 0.294 | 0.089 |
| 50e | 0.800 | >99 | 0.98 | 354250 |
5850 | 60.6 | 0.334 | 0.099 |
| 50e | 0.825 | >99 | 1.03 | 510450 |
7100 | 71.9 | 0.323 | 0.094 |
| 50e | 0.850 | >99 | 1.02 | 1822000 |
23650 |
77.0 | 0.340 | 0.093 |
| 50e | 0.875 | Gel | — | — | — | — | — | — |
| 60 | 0.750 | >99 | 0.68 | 48900 |
1450 | 33.7 | 0.276 | 0.088 |
| 60 | 0.800 | >99 | 1.00 | 84700 |
2600 | 32.6 | 0.265 | 0.088 |
| 60 | 0.850 | >99 | 1.00 | 132650 |
2400 | 55.3 | 0.318 | 0.090 |
| 60 | 0.900 | >99 | 0.98 | 290500 |
2200 | 132 | 0.310 | 0.090 |
| 60 | 0.950 | >99 | 0.97 | 703900 |
2750 | 256 | 0.352 | 0.089 |
| 60 | 1.000 | >99 | 1.06 | 1433000 |
9250 | 155 | 0.301 | 0.092 |
| 60 | 1.025 | Gel | — | — | — | — | — | — |
| 70 | 1.000 | >99 | 0.94 | 147900 |
4000 | 37.0 | 0.295 | 0.097 |
| 70 | 1.050 | >99 | 1.08 | 229100 |
3600 | 63.6 | 0.304 | 0.097 |
| 70 | 1.110 | >99 | 1.10 | 537200 |
3650 | 147 | 0.322 | 0.101 |
| 70 | 1.150 | >99 | 1.17 | 834600 |
4250 | 196 | 0.300 | 0.096 |
| 70 | 1.180 | >99 | 1.18 | 2035000 |
21250 |
95.7 | 0.346 | 0.100 |
| 70 | 1.250 | Gel | — | — | — | — | — | — |
| 80 | 1.138 | >99 | 1.02 | 85600 |
4000 | 21.4 | 0.281 | 0.093 |
| 80 | 1.180 | >99 | 1.17 | 131650 |
4450 | 29.6 | 0.276 | 0.094 |
| 80 | 1.250 | >99 | 1.19 | 237600 |
4350 | 54.6 | 0.289 | 0.092 |
| 80 | 1.330 | >99 | 1.19 | 371700 |
3650 | 102 | 0.293 | 0.095 |
| 80 | 1.380 | >99 | 1.15 | 499400 |
5350 | 93.3 | 0.322 | 0.098 |
| 80 | 1.430 | >99 | 1.25 | 2433000 |
22450 |
108 | 0.329 | 0.101 |
| 80 | 1.540 | Gel | — | — | — | — | — | — |
| 90 | 1.000 | >99 | 1.28 | 14000 |
2350 | 5.96 | 0.206 | 0.105 |
| 90 | 1.330 | >99 | 1.43 | 51300 |
3600 | 14.3 | 0.229 | 0.118 |
| 90 | 1.550 | >99 | 1.52 | 137550 |
7450 | 18.5 | 0.218 | 0.098 |
| 90 | 1.750 | >99 | 1.61 | 291850 |
7050 | 41.4 | 0.259 | 0.097 |
| 90 | 2.000 | >99 | 1.66 | 1223000 |
40200 |
30.4 | 0.261 | 0.108 |
| 90 | 2.100 | >99 | 1.83 | 2662000 |
37850 |
70.3 | 0.204 | 0.098 |
| 90 | 2.220 | Gel | — | — | — | — | — | — |
As above, reactions were conducted using identical initiator to vinyl group ratios and reaction temperature. Below the limiting gel point ratios, all reactions proceeded to near complete vinyl conversion (>99%), and soluble highly branched polymers were obtained. Analysis by 1H NMR and TD-SEC respectively, Table 2, ESI Fig. S20–S23 & S25–S28,† allowed identification of trends.
Increasing dilution continued to lead to higher limiting [EGDMA]0/[DDT]0 gel point ratios ranging from 0.875 for reactions conducted at 50 wt% solvent to a highly surprising [EGDMA]0/[DDT]0 = 2.10 at 90 wt% solvent (10 wt% solids). Clearly reaction conducted >70 wt% solvent could utilise an excess of MVT, hence significantly decreasing the amount of telogen required to control the polymerisation and avoid gelation.
As seen previously, Mw values of the purified polymers increased for each [EGDMA]0/[DDT]0 within each solvent condition with values >2000000 g mol−1 being achieved from 70 wt% EtOAc and >2600000 g mol−1 under very high dilution conditions (90 wt% EtOAc), Table 2 & Fig. 2A. A clear boundary of [EGDMA]0/[DDT]0 values across the reaction conditions was seen, above which insoluble polymer was formed, and below which soluble branched polymer was obtained at vinyl group conversions of >99%, Fig. 2B. The boundary, therefore, represents the reaction conditions that define a TBRT reaction for this MVT/Tel/solvent selection. Below the boundary, the ratio of vinyl groups and telogen is sufficient to ensure that the dominance of the transfer reactions allows complete reaction of MVT to polymer and complete consumption of vinyl functionality without gelation.
Impact of dilution on branching and cyclisation of polymers synthesised using TBRT
We recently described the quantification of branching within TBRT polymers using inverse-gated 13C NMR spectroscopy.13 Analogous structures to those found in hyperbranched polymers derived from AB2 polymerisations, notably terminal, linear, and branched groups, can be defined within TBRT polymers by identifying the extent of propagation within the telomer subunits; terminal groups = DP1, linear groups = DP2, and branched >DP2. The highest molecular weight polymers formed under each solvent condition, Table 1, were subjected to this analysis and were found to be nearly identical in their architecture as defined by the ratio of subunits, ESI Table S1 and Fig. S7–S12,† with only minor variations between samples. Additionally, the hydrodynamic radii (RH) of the polymers analysed via TD-SEC are extremely similar over the broad molecular weight distributions and appear to be independent of reaction concentration within this range, ESI Fig. S19.†Inverse-gated 13C NMR analysis was also conducted across the polymers synthesised under dilute conditions at their limiting [EGDMA]0/[DDT]0 ratios (i.e. the highest molecular weights achieved), Table 2, ESI Fig. S24,† however, determination of linear, branched and terminal units was not possible for these materials. Interestingly, a clear difference in the 13C spectra was observed with polymers synthesised under 80 wt% and 90 wt% EtOAc conditions, displaying significant suppression of resonances assigned to EGDMA residues, ESI Fig. S24,† relative to materials generated under concentrated conditions. This suggests limited mobility of a high proportion of EGDMA residues.
Under ideal conditions, TBRT polymers will incorporate MVT and telogen into the final branched polymer products in equimolar ratios (i.e. [EGDMA]F/[DDT]F = 1), leading to a nominal repeat unit that combines both structures equally, Fig. 1B. This has been established experimentally10–13 when high molecular weight polymers are formed, and is mathematically expected through the simple arithmetic progression of [EGDMA]F/[DDT]F defined by n/(n + 1) (where n = number of MVT residues in the polymer structure).10 Deviation from unity indicates the presence of low molecular weight species if [EGDMA]F/[DDT]F < 1, due to nature of the progression approaching unity only at high values of n. Values of [EGDMA]F/[DDT]F > 1 indicate structures that are deficient in telogen residues, due to cycle formation.13
Direct observation of cycles through 1H NMR has proven difficult due to the inability to resolve the difference between cyclised and uncyclised ester protons in the resulting polymer structure, ESI Scheme S2.† Within an ideal structure, however, the number of cycles, c, can be determined through the relationship:
| c = (n + 1) − t | (1) |
 | (2) |
At high values of n, this can be simplified further to;
 | (3) |
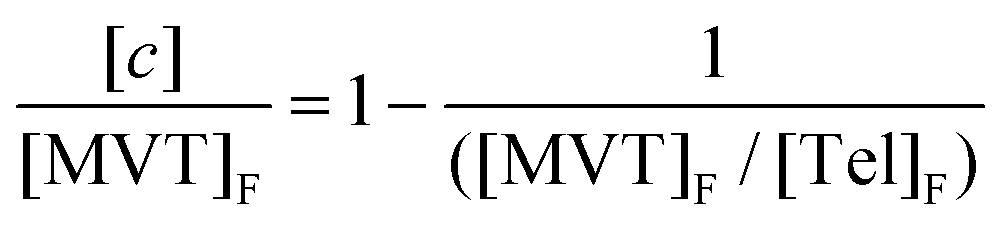 | (4) |
As an example, when c = n/2, [EGDMA]F/[DDT]F = 2, i.e. the number of cycles is equal to half of the number of MVTs within the average polymer; theoretically this value would approach infinity if all MVT vinyl groups were able to form cycles.32
As seen in Tables 1 & 2 and Fig. 3 (showing values from the limiting gel point [MVT]0/[Tel]0 ratios), relatively concentrated reaction conditions (0–60 wt% solvent) maintained [EGDMA]F/[DDT]F values close to unity; small variations are expected from NMR errors and purification. As the TBRT reactions are further diluted (high solvent wt%), [EGDMA]F/[DDT]F rises rapidly, with values as high as 1.83 at 90 wt% solvent (10 wt% solids), Fig. 3. This is a considerable variation from the theoretically ideal polymer structure. Using the considerations discussed above, it is apparent that the number of cycles present in the average polymer structure is equivalent to approximately 45% of its EGDMA residues.13
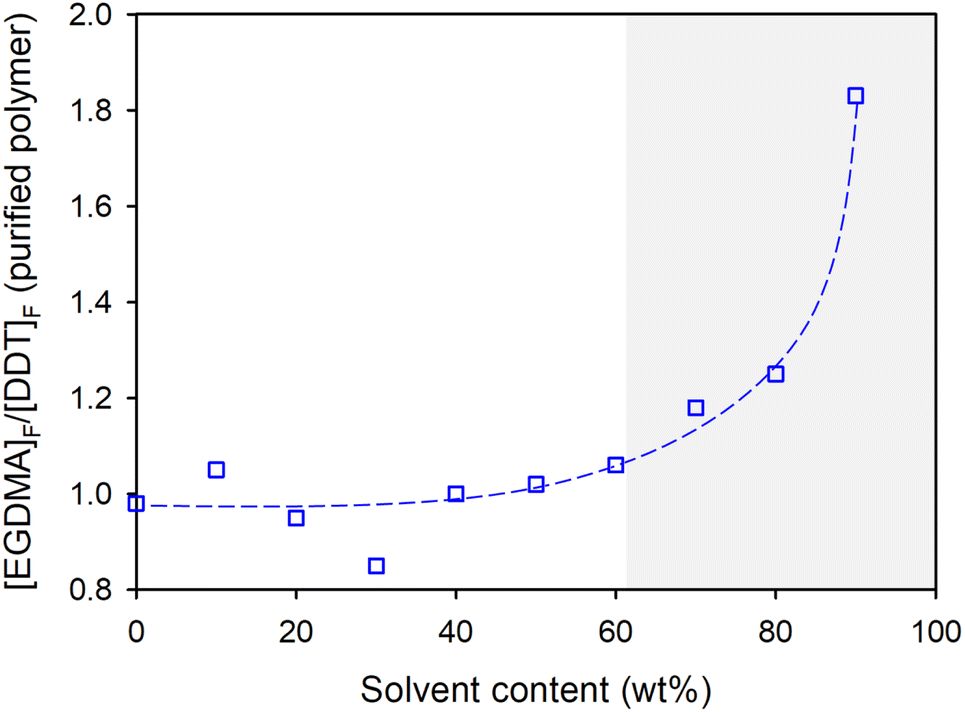 | ||
| Fig. 3 Variation of [EGDMA]F/[DDT]F of p(DDT-EGDMA) polymers at the limiting gel point [EGDMA]0/[DDT]0 ratios identified for each reaction concentration. | ||
TBRT polymerisations are controlled by the dominant chain transfer between thiyl radicals and thiols, thiyl radical reaction with vinyl groups, and thiol chain transfer with carbon centred radicals to minimise propagation.10 At dilutions below the critical overlap concentration the proximity of growing polymer structures is minimised,32 and the combination of DP1 structure formation and intramolecular cyclisation is expected to dominate vinyl group reaction within TBRT. The presented data suggests that for EGDMA/DDT TBRT reactions conducted in EtOAc, c* can be estimated at approximately 60 wt% solvent (40 wt% solids).
Previous reports of the quantification of intramolecular cycles within branched polymers derived from modified Strathclyde strategies, have utilised disulphide dimethacrylate (DSDMA).30 Inspired by this study, TBRT of DSMDA and DDT was attempted at 50 wt% EtOAc using the same scale and conditions as [EGDMA]0/[DDT]0 = 0.75. The reaction achieved >99% vinyl conversion, without gelation, after 24 hours, ESI Fig. S29,† but TD-SEC analysis of the purified material revealed the sample comprised low molecular weight species and a relatively high Mark–Houwink Sakurada α value (0.407) compared to the analogous EGDMA material (α = 0.294), ESI Fig. S30.† It is likely that the TBRT conditions that create a thiyl radical rich reaction mixture also leads to disulphide cleavage,33,34 unlike the low radical concentrations expected in the RAFT conditions previously reported;30 non-radical cleavage mechanisms have not been ruled out.35
Impact of dilution on the glass transition temperature of polymers synthesised using TBRT
The presence of a considerable number of cycles within recovered and purified TBRT polymers formed at increasing dilution, will have a clear implication for physical properties. The impact of restricted backbone segmental motion on glass transition temperature (Tg)36–38 was studied using differential scanning calorimetry, Fig. 4. It is worth noting that the polymer samples studied display high dispersities, Tables 1 & 2; it has been suggested that in these cases, the conventional Flory–Fox relationship of Tg and molecular weight may be best represented as inversely proportional to (Mn·Mw)1/2, as the presence of a high molecular weight fraction within the molecular weight distribution may dominate physical and thermal properties.39,40 | ||
| Fig. 4 Physical property comparison for p(DDT-EGDMA) TBRT polymers synthesised under different reaction conditions. (A) Differential scanning calorimetry of polymers formed at their limiting [EGDMA]0/[DDT]0 gel point values at 50 wt%–90 wt% solvent; (B) images showing a correlation of physical form with [EGDMA]F/[DDT]F values for polymers formed at their limiting [EGDMA]0/[DDT]0 gel point values (tacky semi-solid materials at 50 wt% and 60 wt% solvent, progressively becoming hard solids from 70 wt% through to 90 wt% solvent); (Ci) variation of physical form within samples formed at different [EGDMA]0/[DDT]0 values within reaction conducted at 90 wt% solvent; and (Cii) Physical form of a p(DDT-EGDMA) polymer synthesised at 50 wt% (liquid), and comparison with a lower molecular weight p(DDT-EGDMA) synthesised at 90 wt% solvent (hard solid; see red boxes) but with a higher [EGDMA]F/[DDT]F value. | ||
For this evaluation, samples produced at the limiting [EGDMA]0/[DDT]0 ratios across the different solvent conditions were selected, as the highest molecular weight materials obtained in each case. For polymers synthesised at high concentration between 0 wt% and 60 wt% EtOAc, very similar thermograms were observed, Fig. 4Ai, and all polymers yielded Tg values between −45 °C and −48 °C, ESI Fig. S31.†
At a solvent concentration of 70 wt%, a considerable shift of Tg was observed to 17 °C (approximately 63 °C increase), Fig. 4Aii & 5, with further increases to values of 28 °C and 41 °C with increasing solvent (higher dilution), Fig. 4Aii–iii & 5. Importantly, the variation of RH across the molecular weight distribution for the highest molecular weight sample formed at 60 wt% solvent showed no appreciable deviation from those formed between 0 wt% and 50 wt% solvent. A systematic reduction in RH was, however, seen for samples formed using 70 wt%–90 wt% solvent, further suggesting a more compact structure due to cyclisation and correlating directly with the sudden change in observed Tg values, ESI Fig. S32.†
The change in physical properties was clear within the visual observation of the samples where liquid/soft solid behaviour was seen for those polymers synthesised under relatively concentrated conditions and hard solid material formed at dilutions higher than 60 wt% solvent, Fig. 4B. The sudden increase in Tg for polymers formed under dilute conditions >60 wt% solvent, Fig. 5, correlates with the significant increase in [EGDMA]F/[DDT]F values, Fig. 3, and can be related to increased cycle formation. Interestingly, within a single reaction concentration and varying [EGDMA]0/[DDT]0 values, the same relationship between [EGDMA]F/[DDT]F and physical properties can be observed visually, Fig. 4Ci, where high values equate to hard solid samples and decreasing values correlate with soft solids and viscous liquid samples.
 | ||
| Fig. 5 Variation of glass transition temperature (Tg) of p(DDT-EGDMA) polymers at the limiting [EGDMA]0/[DDT]0 values identified for each reaction concentration. | ||
It is well known that molecular weight has a considerable impact on Tg up to a plateau where Tg = Tg∞ that is associated with a theoretical infinite molecular weight. Considering the polymer samples formed at the limiting gel point [EGDMA]0/[DDT]0 values across all solvent concentrations, Mw values vary from 292350 g mol−1 to 2662000 g mol−1, whilst Mn spans a range from 3500 g mol−1 to 37350 g mol−1, Tables 1 & 2. The Tg values of samples produced in solvent conditions up to 60 wt% are identical (within error), Fig. 5, and correspond to a considerable span of molecular weights (Mn = 3500 g mol−1 to 23650 g mol−1; Mw = 29200 g mol−1 to 182200 g mol−1). It is reasonable to assume that the consistent values of Tg across this range of molecular weights indicates the samples are at the Tg∞ value for branched p(DDT-EGDMA) with limited cyclisation. To support this assertion further, a comparison can be made between p(DDT-EGDMA) synthesised using 90 wt% solvent and an [EGDMA]0/[DDT]0 value of 1.33, and a sample formed using 50 wt% solvent and a [EGDMA]0/[DDT]0 = 0.75, Table 2. These samples have comparable Mn values, 3600 g mol−1 and 4550 g mol−1 respectively, but very different Mw values of 51300 g mol−1 and 129350 g mol−1 respectively. Despite having higher Mn and Mw values, the sample synthesised at 50 wt% solvent ([EGDMA]F/[DDT]F value = 1.04) is a viscous liquid, whilst the lower molecular weight sample formed at 90 wt% solvent is a hard solid, Fig. 4Ci & Cii. The difference between measured [EGDMA]F/[DDT]F values of 1.04 (cycles are approximately equivalent to 4% of the EGDMA residues) and 1.43 (cycles are approximately equivalent to 30% of the EGDMA residues) are clearly indicating the importance of cyclisation in directing the physical properties of the final sample.
Consideration of macromolecular topology
Although some of the aspects of the results shown here overlay with theoretical conditions or postulations regarding the polymerisation of multi-vinyl monomers, TBRT has not been available for study in this way until recently, and one surprising result is the variation of thermal properties that are achievable with the same starting combination of MVT and telogen. Within this study p(DDT-EGDMA) is formed in all cases, but cyclisation leads to materials with a range of Tg that spans >80 °C without any further reaction components or processing.Consideration of the breadth of [EGDMA]F/[DDT]F values achieved does also allow some potential insights on cycle formation, Fig. 6. For example, it is very difficult to establish the true nature of the cycles that are formed using techniques employed here; however, as [EGDMA]F/[DDT]F increases, the cycles may span the polymer architecture in the form of macrocycles, Fig. 6Bi, or be very small and highly localised, Fig. 6Bii, but still exhibit the same [EGDMA]F/[DDT]F values. As the number of cycles increases within a fixed number of EGDMA residues, creating a concomitant increase in observed [EGDMA]F/[DDT]F values, it becomes clear that a higher number of cycles necessitates that the ring size decreases considerably. At relatively modest values of [EGDMA]F/[DDT]F, the presence of both macrocycles and local cycles are to be expected, Fig. 6C, however, at higher values, the presence of macrocycles is highly unlikely, and a predominantly cyclised structure is expected, Fig. 6D. This may explain the limited resolution within the NMR spectroscopy studies of p(DDT-EGDMA) formed at high dilution and the sudden and dramatic increase in Tg. The highly restricted backbone motion would have a strong influence on segmental motion in solution (NMR relaxation) and in the solid state. The timing of cycle formation during polymer synthesis is subject to ongoing studies.
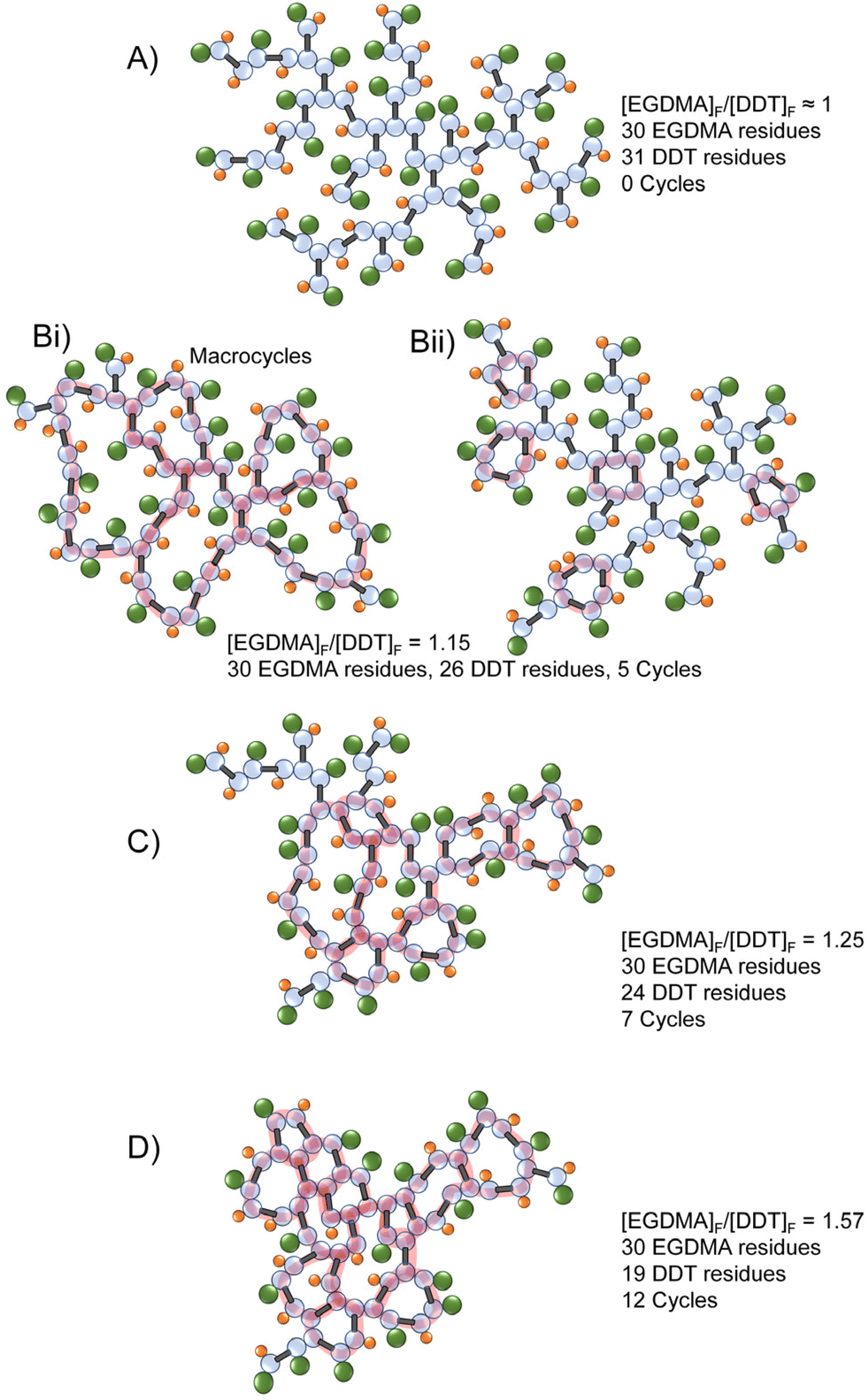 | ||
| Fig. 6 Schematic representations of TBRT polymers with varying [MVT]F/[Tel]F values. A fixed number of MVT (EGDMA) residues is used to demonstrate the impact of increased cyclisation. Cycles are highlighted in red as a visual aid. | ||
Conclusions
TBRT is a new polymerisation technique that offers considerable flexibility and scope for materials design. It is important to understand the influence of reaction conditions on the outcome of the recovered polymers, and here we have clearly shown that the concentration of the reaction is a key consideration. Not only does it dictate the starting ratios of MVT and telogen that can be utilised, it also impacts the achievable molecular weights and, importantly, the topology of the branched polymer architecture which in turn dictates the physical properties of the recovered materials. Additionally, relatively small differences in reaction concentration, above a critical value, may lead to dramatic differences in the resulting polymer sample. The extensive study of TBRT presented here has been collated using just one MVT and one telogen. The opportunity to tune polymer properties for different combinations is considerable and the insights provided within the current report will hopefully facilitate further progress in the advancement of this novel chemistry.Author contributions
SRC was responsible for conceptualisation, methodology, experimentation, investigation, data curation, formal analysis, visualisation and writing of the original draft. SW, SMck, OBPL, SF and SL all contributed to validation. PC contributed to supervision, project administration, validation, and manuscript review. SPR was responsible for funding acquisition, conceptualisation of the original research programme, methodology, validation, visualisation, supervision, project administration and manuscript review and editing.Conflicts of interest
SPR, SRC, SW and PC are co-inventors on patents that protect the TBRT chemistry; these patents have been licensed to Scott Bader and form the basis of Polymer Mimetics Ltd (Company number 12598928).Acknowledgements
This manuscript is based on work published in the PhD thesis of SRC. The authors are grateful for support from the Engineering & Physical Sciences Research Council, especially grant EP/R010544/1. SRC is grateful for financial support from Itaconix. SW is grateful for financial support from Synthomer. SMck is grateful for financial support from Unilever. OBPL is grateful for financial support from the University of Liverpool. SL is grateful for financial support from Scott Bader. The authors would like to thank the Materials Innovation Factory (University of Liverpool) for access to analytical equipment and NMR spectroscopy instrumentation.References
- Y. Gao, D. Zhou, J. Lyu, A. Sigen, Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS.
- C. J. Atkins, D. K. Seow, G. Burns, J. S. Town, R. A. Hand, D. W. Lester, N. R. Cameron, N. R. Cameron, D. M. Haddleton, A. M. Eissa and A. M. Eissa, Polym. Chem., 2020, 11, 3841–3848 RSC.
- M. L. Koh, D. Konkolewicz and S. Perrier, Macromolecules, 2011, 44, 2715–2724 CrossRef CAS.
- T. Sato, T. Miyagi, T. Hirano and M. Seno, Polym. Int., 2004, 53, 1503–1511 CrossRef CAS.
- W. Wang, Y. Zheng, E. Roberts, C. J. Duxbury, L. Ding, D. J. Irvine and S. M. Howdle, Macromolecules, 2007, 40, 7184–7194 CrossRef CAS.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef PubMed.
- B. Boutevin, G. David and C. Boyer, Adv. Polym. Sci., 2007, 206, 31–135 CrossRef CAS.
- C. Boyer, C. Loubat, J. J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5146–5160 CrossRef CAS.
- C. Boyer, G. Boutevin, J. J. Robin and B. Boutevin, Polymer, 2004, 45, 7863–7876 CrossRef CAS.
- S. R. Cassin, P. Chambon and S. P. Rannard, Polym. Chem., 2020, 11, 7637–7649 RSC.
- O. B. Penrhyn-Lowe, S. Flynn, S. R. Cassin, S. Mckeating, S. Lomas, S. Wright, P. Chambon and S. P. Rannard, Polym. Chem., 2021, 12, 6472–6483 RSC.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, Polym. Chem., 2022, 13, 2295–2306 RSC.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, RSC Adv., 2021, 11, 24374–24380 RSC.
- S. Flynn, O. B. Penrhyn-Lowe, S. Mckeating, S. Wright, S. Lomas, S. R. Cassin, P. Chambon and S. P. Rannard, RSC Adv., 2022, 12, 31424–31431 RSC.
- O. B. Penrhyn-Lowe, S. R. Cassin, P. Chambon and S. Rannard, Nanoscale Adv., 2022, 4, 4051–4058 RSC.
- P. Besenius, S. Slavin, F. Vilela and D. C. Sherrington, React. Funct. Polym., 2008, 68, 1524–1533 CrossRef CAS.
- S. Graham, S. P. Rannard, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2007, 17, 545–552 RSC.
- M. Chisholm, N. Hudson, N. Kirtley, F. Vilela and D. C. Sherrington, Macromolecules, 2009, 42, 7745–7752 CrossRef CAS.
- J. K. Gooden, M. L. Gross, A. Mueller, A. D. Stefanescu and K. L. Wooley, J. Am. Chem. Soc., 1998, 39, 10180–10186 CrossRef.
- Q. Ban and J. Kong, Polym. Chem., 2016, 7, 4717–4727 RSC.
- Q. Ban, Y. Li, Y. Qin, Y. Zheng and J. Kong, Eur. Polym. J., 2021, 155, 110539 CrossRef CAS.
- H. R. Kricheldorf and G. Schwarz, Macromol. Rapid Commun., 2003, 24, 359–381 CrossRef CAS.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- J. Lyu, Y. Gao, Z. Zhang, U. Greiser, P. Polanowski, J. K. Jeszka, K. Matyjaszewski, H. Tai and W. Wang, Macromolecules, 2018, 51, 6673–6681 CrossRef CAS.
- P. Polanowski, J. K. Jeszka, W. Li and K. Matyjaszewski, Polymer, 2011, 52, 5092–5101 CrossRef CAS.
- I. Bannister, N. C. Billingham and S. P. Armes, Soft Matter, 2009, 5, 3495–3504 RSC.
- O. Okay, M. Kurz, K. Lutz and W. Funke, Macromolecules, 1995, 28, 2728–2737 CrossRef CAS.
- Y. Li, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 5577–5581 CrossRef CAS.
- Y. Zheng, H. Cao, B. Newland, Y. Dong, A. Pandit and W. Wang, J. Am. Chem. Soc., 2011, 133, 13130–13137 CrossRef CAS PubMed.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- R. Wang, Y. Luo, B. G. Li and S. Zhu, Macromolecules, 2009, 42, 85–94 CrossRef CAS.
- M. Antonietti and C. Rosenauer, Macromolecules, 1991, 24, 3434–3344 CrossRef CAS.
- A. V. Tobolsky and B. Baysal, J. Am. Chem. Soc., 1953, 75, 1757 CrossRef CAS.
- K. Shanmugananda Murthy, K. Ganesh and K. Kishore, Polymer, 1996, 37, 5541–5543 CrossRef.
- R. Singh and G. M. Whitesides, Tech. Protein Chem., 1995, 6, 259–266 Search PubMed.
- Y. Ohta, Y. Kamijyo, A. Yokoyama and T. Yokozawa, Polymers, 2012, 4, 1170–1182 CrossRef.
- W. Gong, Y. Mai, Y. Zhou, N. Qi, B. Wang and D. Yan, Macromolecules, 2005, 38, 9644–9649 CrossRef CAS.
- A. Khalyavina, L. Häußler and A. Lederer, Polymer, 2012, 53, 1049–1053 CrossRef CAS.
- T. Alhilfi, P. Chambon and S. P. Rannard, J. Polym. Sci., Part A: Polym. Chem., 2020, 58, 1426–1438 CAS.
- T. Ogawa, J. Appl. Polym. Sci., 1992, 44, 1869–1871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and characterisation. See DOI: https://doi.org/10.1039/d3py00046j |
| This journal is © The Royal Society of Chemistry 2023 |