
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable vinyl polymer particles by radical aqueous emulsion copolymerization of methyl methacrylate and 5,6-benzo-2-methylene-1,3-dioxepane†
Paul
Galanopoulo‡
 a,
Laura
Sinniger‡
a,
Noémie
Gil
b,
Didier
Gigmes
b,
Catherine
Lefay
b,
Yohann
Guillaneuf
b,
Maëlle
Lages
c,
Julien
Nicolas
c,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
a,
Laura
Sinniger‡
a,
Noémie
Gil
b,
Didier
Gigmes
b,
Catherine
Lefay
b,
Yohann
Guillaneuf
b,
Maëlle
Lages
c,
Julien
Nicolas
c,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
aUniv Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials (CP2M), 43 Bd du 11 novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
bAix Marseille Univ, CNRS, Institut de Chimie Radicalaire, UMR 7273, Marseille, France
cUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, 17 avenue des Sciences, 91400 Orsay, France
First published on 13th February 2023
Abstract
With an increasing interest in the synthesis of (bio)degradable polymers, the synthesis of (bio)degradable vinyl polymer particles is attracting growing attention. In this work, degradable poly(methyl methacrylate) (PMMA) particles were synthesized by radical emulsion polymerization in water. A cyclic ketene acetal, 5,6-benzo-2-methylene-1,3-dioxepane (BMDO) was used as a co-monomer to introduce cleavable ester functions into the PMMA backbone. The stability of BMDO in aqueous media was studied and the degradation mechanism discussed in order to better comprehend the use of BMDO in emulsion and understand the incorporation of BMDO into the copolymer chains. Stable latexes containing various amounts of BMDO inserted units (from 2 to 6.4 mol%) were obtained. The degradation of the resulting copolymers was carried out in a basic medium and an average molar mass loss of 90% was observed.
Introduction
Vinyl polymers are widely used in many applications and are thus massively produced in industry. Their carbon–carbon backbone cannot be degraded easily, which is a valuable property for most applications. However, it also limits their use in certain fields where (bio)degradability is sought, notably in biomedical applications for, e.g., nanomedicines or implantable devices.1 In addition, considering the current quest for more environmentally friendly polymer products, including possible recycling, the synthesis of (bio)degradable vinyl polymers is a key challenge.2–4 Degradable polymers can be synthesized by inserting cleavable units distributed in the backbone. Under external stimuli such as pH, light, temperature or enzymatic activity, the targeted units cleave, leading to low molar mass oligomers. Ideally, the ultimate goal would be to insert enough cleavable bonds to achieve complete degradation without affecting the intended properties of the final materials.One strategy is based on the use of cyclic ketene acetals (CKAs) as comonomers, which allow the preparation by radical ring-opening polymerization (rROP) of vinyl copolymers containing cleavable ester functions.5–7 The polymers obtained can be thus subjected to hydrolytic or enzymatic degradation leading to a decrease in their molar mass.8 Besides, the use of CKAs in radical copolymerization is, a priori, less restrictive than the ring-opening anionic polymerization of cyclic monomers for introducing ester cleavable bonds into polymers.
The copolymerization of different CKAs with a variety of vinyl monomers has been widely studied in homogeneous conditions,8,9 the most reported ones being 2-methylene-1,3-dioxepane (MDO) and 5,6-benzo-2-methylene-1,3-dioxepane (BMDO, Scheme 1). The latter has been successfully copolymerized with styrene,5,10n-butyl acrylate,11 as well as methacrylic compounds such as poly(ethylene glycol) methacrylate (PEGMA)12 and methyl methacrylate (MMA).13 MDO has also been copolymerized with a variety of vinyl co-monomers including styrene or MMA.14 However, in these cases, low incorporation of MDO units was observed except when vinyl acetate (VAc) was used.14
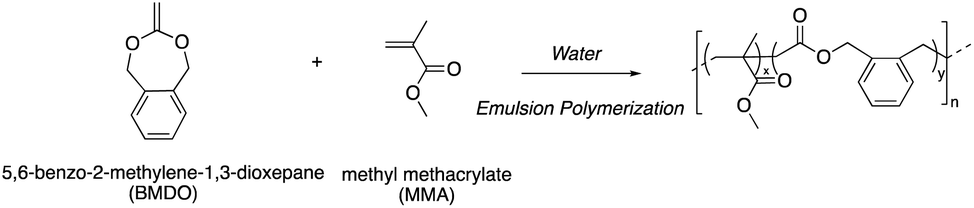 | ||
| Scheme 1 Aqueous radical emulsion copolymerization of BMDO and MMA. | ||
Nonetheless, these copolymerizations have been extensively investigated in bulk and in solution. In contrast, only a very few examples concerning the use of CKAs in an aqueous dispersed media have been reported,15–17,27 despite the tremendous importance of polymerization in dispersed media. This can be explained by the high sensitivity of CKAs to protic species since CKAs are known to rapidly hydrolyze in aqueous media.18,27
Miniemulsion copolymerization in water of BMDO with either MMA or styrene was described in 2012.17 The ultimate objective justifying the use of a miniemulsion polymerization process was the encapsulation of drugs in a degradable polymer matrix. Polymer particles were obtained, and the formation of copolymers based on MMA and BMDO claimed, while their degradation was not evaluated. In addition, the known sensitivity of BMDO to protic species11 did not appear to be an impediment to employ a broad range of surfactants and miniemulsion polymerization conditions. This process is indeed meant to protect the comonomers from the aqueous environment by trapping them into nanometric monomer droplets in which the polymerization will, in theory, exclusively take place.19
Emulsion polymerization provides a more universal and industry-relevant approach to polymerization in aqueous dispersed media. However, the polymerization starts in water and the hydrophobic comonomers are required to diffuse through the aqueous phase from large micrometric monomer droplets to nanometric particles. The challenge for the copolymerization of CKA with monomers commonly used in aqueous emulsion thus lies in finding suitable conditions for successful copolymerization while maintaining good control on the insertion of the ester functions, with the aim of ensuring homogeneous degradation of the copolymer. Very recently, the copolymerization of VAc with MDO was studied in aqueous emulsion polymerization.15,16 Indeed, as mentioned above, MDO was successfully copolymerized with VAc in homogenous medium (determined reactivity ratios: rMDO = 0.47 and rVAc = 1.53) leading to degradable polymers.20 P(VAc-co-MDO) copolymers were obtained in semi-batch emulsion conditions at 40 °C using redox initiators. The key to the success of this synthesis is the very specific formulation employed to rapidly nucleate particles in which MDO can rapidly escape from the aqueous phase. This however requires very fine adjustments in terms of pH and feeding rates, as well as the additional use of hydrophilic charged comonomers, which may not be suitable for a wide range of applications and may generate reproducibility issues.27
Copolymerization of BMDO with MMA, which also present favorable reactivity ratios (rBMDO = 0.53 and rMMA = 1.96),13 has never been investigated in aqueous emulsion copolymerization. In this paper, we look at developing very simple protocols for the aqueous emulsion copolymerization of BMDO with MMA to yield degradable vinyl polymer particles.
Materials and methods
Materials
Methyl methacrylate (MMA, Aldrich, 99%), potassium persulfate (KPS, Aldrich, 99%), sodium dodecyl sulfate (SDS, Aldrich, 99%), sodium bicarbonate (Aldrich, Bioreagent), potassium carbonate (K2CO3, Aldrich, 99%), potassium hydroxide (KOH, Aldrich, 85%), dichloromethane (ACS reagent, Aldrich), methanol (VWR) and tetrahydrofuran (THF, Fisher, HPLC grade) were used as received. 5,6-Benzo-2-methylene-1,3-dioxepane (BMDO) was synthesized as reported in the literature.5 Water was deionized before use (Purelab Classic UV, Elga LabWater).Experimental procedures
Analytical techniques
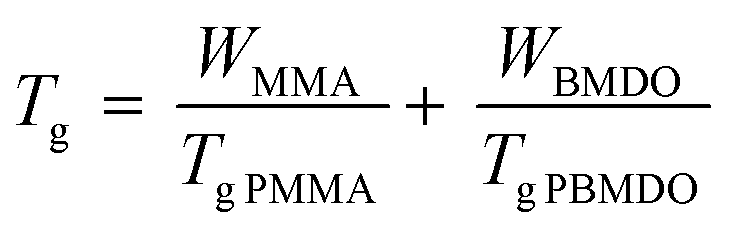 | (1) |
Results and discussion
Emulsion copolymerization of MMA and BMDO
P(MMA-co-BMDO) copolymer latexes were successfully synthesized under very conventional emulsion polymerization conditions, i.e. using SDS as surfactant and KPS as thermal initiator. NaHCO3 was also added to the medium to start at a basic pH (∼8.8) since we reckoned that BMDO hydrolysis would be kept to a minimum under these conditions.18A first synthesis was conducted with 5 mol% of BMDO at 70 °C (E1BMDO, Table 1). The conversion of MMA was monitored by 1H NMR, following the evolution of the signals characteristic of the vinyl protons of MMA (5.5 and 6 ppm) in relation to the signal characteristic of methoxy protons present in the copolymer (3.6 ppm). For E1BMDO (Table 1), the MMA conversion reached 99% (Fig. 1) and a stable latex was obtained. As mentioned in the introduction, CKAs are sensitive to protic species and thus easily hydrolyzed in water. We thus conducted a brief study of BMDO stability on storage and two distinct degradation products were identified as detailed in the ESI (D1 and D2 in Fig. S1 and S2†), at least one of them (D1) being volatile (see ESI†). Subsequently, we also studied its stability in aqueous dispersed media. However, due to the complexity of the NMR spectra associated with the presence of volatile compounds, the quantification of the different species was difficult. Likewise, the BMDO conversion was thus delicate to evaluate considering a similar potential degradation during polymerization, which is indeed shown by the presence of D2 product when monitoring E1BMDO reaction by 1H NMR (Fig. 1). It was thus not possible to determine BMDO conversion by 1H NMR nor gravimetrically. Compared to the homopolymerization of MMA (E1MMA, Table 1), the addition of BMDO induced a decrease of the polymerization rate (Fig. 1), despite the lower particle size (45 vs. 67 nm, respectively, Table 1) and thus the higher number of particles (Np) obtained in the presence of BMDO. The threefold increase of Np cannot be clearly explained at this stage. The lower polymerization rate could be linked to the decrease of the apparent rate constant of propagation 〈kp〉 considering the low kp values of BMDO.21 The monomer concentration into the particles could also be lower, and so could be the average number of radicals per particle.
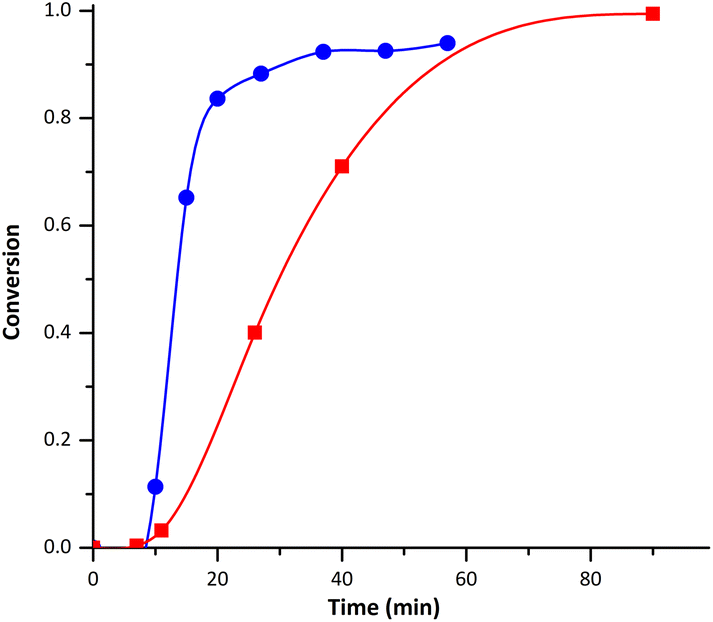 | ||
| Fig. 1 Evolution of MMA conversion with time during emulsion (co)polymerization of MMA and BMDO for experiments E1MMA (blue) and E1BMDO (red). | ||
| Run |
f
BMDO,0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
Base | t (min) | Conv.b (%) |
F
BMDOc (%) |
Z
aved (nm) |
PDId |
T
ge (°C) |
M
nf (kg mol−1) |
Đf |
|---|---|---|---|---|---|---|---|---|---|---|
| Experiments were conducted at 70 °C targeting a solid content of 10 wt%, with [SDS] = 14 mmol L−1, [Base] = 8 mmol L−1, [KPS] = 4 mmol L−1. Most of the experiments were performed with NaHCO3, leading to an initial pH (pH0) around 8.8 and a final pH (pHf) close to 8. For E2BMDO, K2CO3 was used instead (pH0 = 11.0/pHf = 7.7), while E3BMDO was run without a base (pH0 = 6.0/pHf = 2.3).a Molar fraction of BMDO in the initial monomer mixture.b Conversion of MMA determined by gravimetric analysis in the case of E1MMA and by 1H NMR for the copolymerizations.c Average molar fraction of BMDO in the copolymer calculated according by 1H NMR.d Intensity-average diameter and polydispersity index of the final particles from DLS.e Glass transition temperature estimated from DSC analyses.f Experimental number-average molar mass, Mn, and dispersity, Đ = Mn/Mw, of the copolymer determined by SEC-THF using a conventional calibration based on PMMA standards. n.d.: not determined. | ||||||||||
| E1MMA | 0 | NaHCO3 | 60 | 94 | — | 67 | 0.03 | 125 | 660 | 2.6 |
| E1BMDO | 5 | NaHCO3 | 90 | 99 | 3.8 | 45 | 0.07 | 76 | 61.5 | 5.8 |
| E2BMDO | 5 | K2CO3 | 90 | 94 | 4.6 | 39 | 0.06 | 56 | 27.8 | 3.3 |
| E3BMDO | 5 | None | 90 | 73 | 0.05 | 60 | 0.05 | n.d. | n.d. | n.d. |
| E4BMDO | 2 | NaHCO3 | 90 | 98 | 2.0 | 45 | 0.07 | 87 | 98.2 | 5.9 |
| E5BMDO | 10 | NaHCO3 | 90 | 98 | 6.4 | 47 | 0.06 | 67 | 28.5 | 1.9 |
1H NMR analysis was then run on the dried extract of both latexes. The assignments proposed in Fig. 2 were based on a previously published 1H NMR analysis of a copolymer obtained by bulk atom transfer radical copolymerization of MMA and BMDO.13 The signal at 5.0 ppm is characteristic of protons next to the ester functions (signal 2, Fig. 2). The presence of BMDO inserted units is further confirmed by the multiplicity of the signals associated to the protons a and c of the MMA units. Indeed, it is possible to distinguish the different triads in the copolymer according to the full investigation of the microstructure of P(MMA-co-BMDO) that has already been reported.13
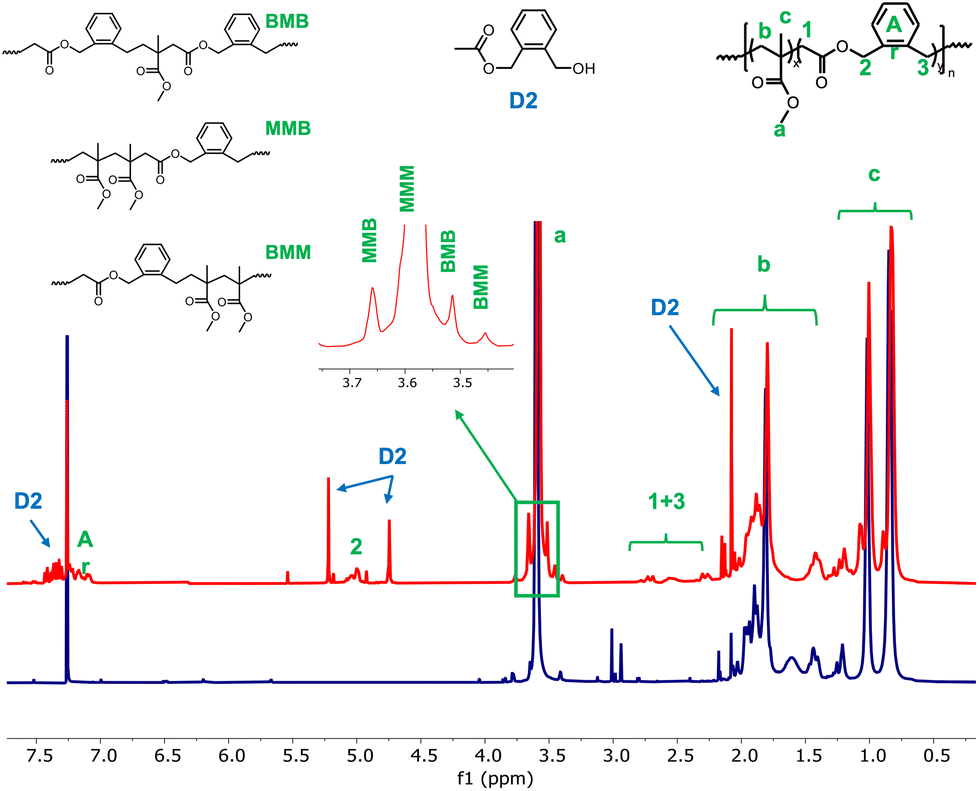 | ||
| Fig. 2 1H NMR spectra of the dried extract of latexes E1BMDO (red) and E1MMA (blue) (CDCl3, 256 scans). Assignments were made according to a previous report.13 B stands for BMDO units and M for MMA units in a triad. D2 corresponds to one of the identified degradation products of BMDO (see ESI, Fig. S1†). | ||
Notably, while signal a appears as a single resonance in the spectrum of PMMA, it is displayed as four distinct resonances in the spectrum of the dry extract of E1BMDO (Fig. 2). This change confirms the formation of triads containing two MMA units and one BMDO unit. As expected, c is already displayed as three distinct resonances in the case of PMMA. The incorporation of BMDO is also shown by the appearance of several other signals in the 0.6–1.3 ppm region, characteristic of the methyl protons from a MMA unit in triads containing BMDO units (Fig. 2).13
The composition of the copolymer was determined by integrating 2 at 5.0 ppm corresponding to two protons of a BMDO unit and signal a corresponding to the methoxy protons of MMA units (Fig. 2). According to the integration, 3.8 mol% of BMDO were incorporated into the PMMA chains. The rather low fraction of BMDO in the copolymer (FBMDO) leads to small intensity and poorly defined signals and does not allow for an accurate signal integration. The FBMDO values determined by NMR may therefore be slightly flawed. Nevertheless, the molar fraction of BMDO in the copolymer is lower than the initial molar fraction of BMDO in the monomer mixture. As mentioned above, a small part of BMDO degrades thus preventing full incorporation of the BMDO into the copolymer chains. In addition, copolymerization of CKAs with vinylic monomers generally leads to compositional drift and thus requires high amount of CKA in the initial feed.8 For instance, the bulk radical copolymerization of MMA with BMDO (fBMDO,0 = 0.5) conducted at 50 °C using AIBN leads to a copolymer containing only 13 mol% of BMDO units after 45 h of reaction,5 in agreement with the reactivity ratios reported for these monomers (rBMDO = 0.53; rMMA = 1.96).10 In emulsion polymerization, the reactivity ratios combined with the higher solubility of MMA in water probably lead to the formation of polymer chains richer in MMA units at the beginning the polymerization, or even to PMMA chains. Degradation of BMDO during polymerization, even if limited, could also lead to complete depletion of BMDO, resulting in the formation of PMMA chains.
SEC analysis performed on the dry extract gave additional information on the impact of BMDO during emulsion copolymerization with MMA. Indeed, the molar mass of the copolymer (Mn = 61500 g mol−1) is significantly lower than that of the PMMA homopolymer (Mn = 660 kg mol−1) obtained from the emulsion polymerization of MMA under the same conditions. Additionally, the SEC trace is seemingly broader, as evidenced by an increased dispersity (5.8 for E1BMDO vs. 2.1 for E1MMA, Table 1). This feature may be the result of side reactions, including chain transfer, originated from BMDO itself13 and/or its degradation products. Together with the impact of 〈kp〉, all these events could also explain why the polymerization is slower in the presence of BMDO, despite lower particle sizes.
The multi-wavelength UV detector of the SEC can bring additional information on the chain composition (Fig. 3a). The carbonyl groups of the MMA units absorb at 217 nm and the aromatic protons from BMDO at 260 nm. The UV analysis was thus used to study whether BMDO was evenly distributed within the copolymer chains. The blue and red chromatograms (Fig. 3b) do not fully overlap, indicating a slight difference in composition of the chains depending on the molar mass. The higher molar mass chains appear to contain more MMA units than the lower molar mass ones. In other words, lower molar mass chains are richer in BMDO units than higher molar mass chains. Most of the distribution nevertheless exhibit BMDO inserted units.
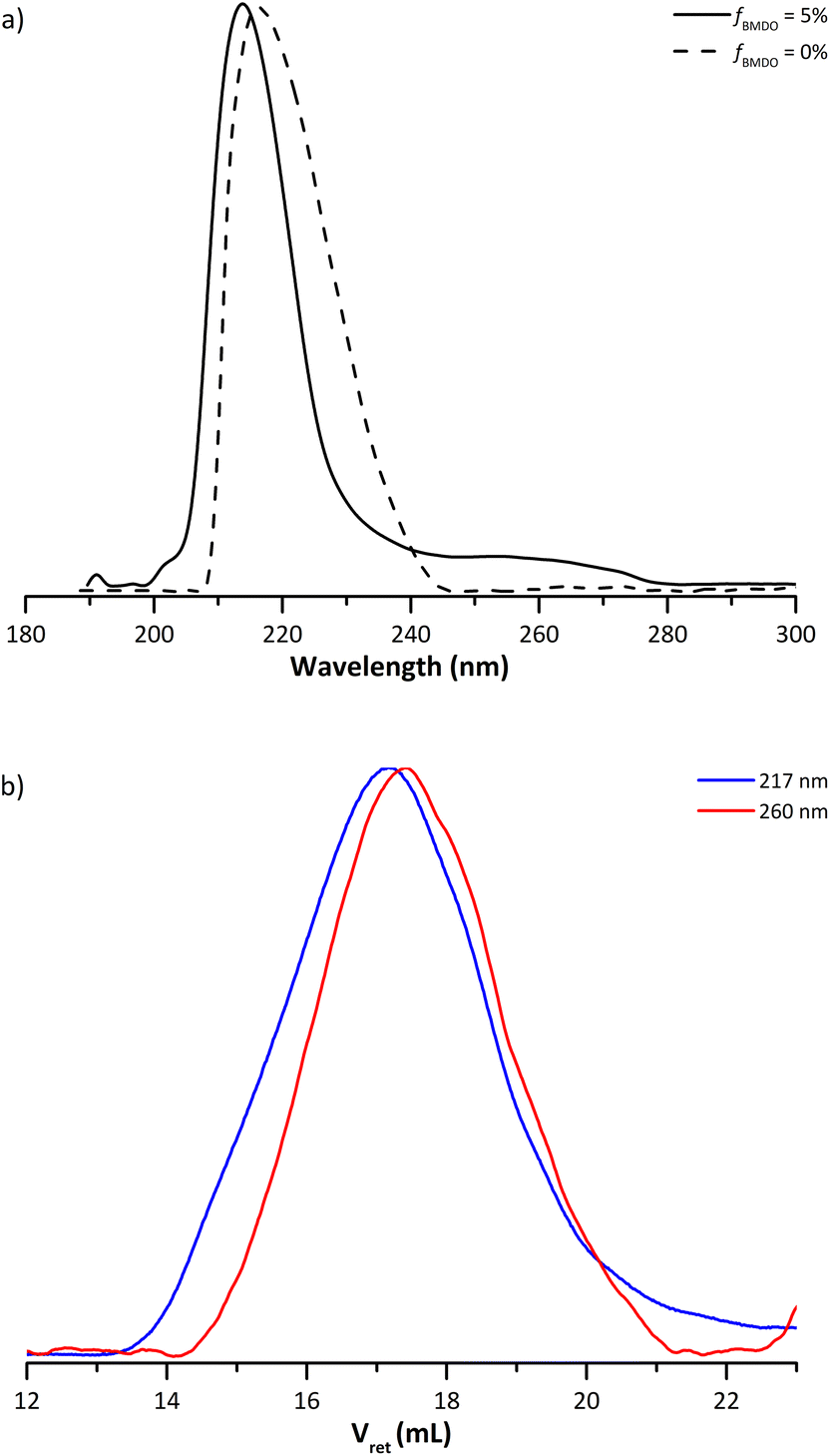 | ||
| Fig. 3 (a) SEC traces from the UV detector obtained for the dried extract from experiments E1MMA (fBMDO,0 = 0%) and E1BMDO (fBMDO,0 = 5%, Table 1), normalized on the maximum of absorption. (b) SEC traces from the UV detector set at 217 nm (blue) and 260 nm (red) obtained for experiment E1BMDO (Table 1). | ||
Eventually, the curve obtained from DSC analysis performed on the dried extract showed one thermal transition at 86 °C (Fig. 6) that is likely attributed to the Tg of P(MMA-co-BMDO) copolymer chains as the Tg of PBMDO10 and of a PMMA sample prepared by emulsion polymerization22 have been reported at 16 °C and 127 °C, respectively.
Overall, this first experiment conducted with BMDO indicated that very classical emulsion conditions could lead to the formation of a stable latex made of P(MMA-co-BMDO) copolymers. This copolymerization system was then investigated in more detail.
Influence of the pH on the copolymerization
As the stability of BMDO is conditioned to the absence of acidic species, we anticipated above that successful emulsion copolymerizations should take place at basic pH. In the following, we conducted emulsion copolymerizations using a different base while keeping the other experimental parameters constant (i.e., temperature, initiator, solid content). The use of K2CO3 (E2BMDO, Table 1) instead of NaHCO3 (E1BMDO, Table 1) had no significant influence on the conversion (94% and 99%, respectively) and particle size. In both experiments the conversion was high, the particles rather small and the final pH similar (∼8). Nevertheless, the incorporation of BMDO was slightly increased as experiment E2BMDO yielded a copolymer with an average of 4.6 mol% of BMDO units in the chains. A third experiment was performed without adding any base to the medium (E3BMDO, Table 1). Only 73% conversion of MMA was achieved after 90 min and barely any BMDO was incorporated in the copolymers. The final pH dropped to 2.3. It is very likely that, in this case, BMDO was hydrolyzed at a much faster rate compared to the experiments conducted in the presence of either NaHCO3 or K2CO3. The use of a base is thus of paramount importance to achieve a successful copolymerization of MMA with BMDO under aqueous emulsion conditions. NaHCO3 was selected for the following experiments, as it represented the better option to achieve full conversion of MMA with high incorporation of BMDO units.Influence of the initial fraction of BMDO in the monomer feed
In the next series of experiments the initial fraction of BMDO in the feed of monomers was varied between 2 and 10 mol% (Table 1). The syntheses all led to nearly full conversion of MMA (>98%) in less than 90 min. Stable latexes were formed in every case (Fig. 4). According to the DLS analysis, the hydrodynamic diameter of the particles decreased with the increase in BMDO (from 67 nm to 45 nm, for fBMDO,0 = 0 and 2%, respectively, Table 1), which translates in the aspect of latex that was less turbid (Fig. 4). The hydrodynamic diameter did not vary significantly when the BMDO content further increased (45 nm to 47 nm, for fBMDO,0 = 2 and 10%, Table 1). | ||
| Fig. 4 Photos of the latexes prepared with different initial molar fraction of BMDO. From left to right: experiments E1MMA, fBMDO,0 = 0%; E4BMDO, fBMDO,0 = 2%; E1BMDO, fBMDO,0 = 5%; E5BMDO, fBMDO,0 = 10% (Table 1). | ||
The BMDO content in the copolymer (FBMDO), determined by 1H NMR, increased with the initial fraction of BMDO. However, the more BMDO in the initial feed, the less is proportionally incorporated into the chains. While an initial fraction of BMDO of 2 mol% (E4BMDO, Table 1) led to 100% of the BMDO being incorporated in the copolymer (FBMDO = 2%), the synthesis starting with 10 mol% of BMDO (E5BMDO, Table 1) led to the incorporation of 64% of the BMDO in the copolymer (FBMDO = 6.4%). Yet, thanks to the emulsion polymerization process, an incorporation of 64% into the copolymer after only 2 h is still in stark contrast with what can be obtained for the bulk copolymerization of BMDO with MMA. Indeed, as mentioned above, the bulk copolymerization of MMA with BMDO (50/50 mol/mol) conducted at 50 °C using AIBN led to a copolymer containing only 13 mol% of BMDO units after 45 h of reaction.5,13
The molar mass of the copolymer significantly decreased as the amount of BMDO increased. The presence of secondary backbiting reactions due to the insertion of BMDO units in the chains could explain this phenomenon.10 It is nevertheless difficult to conclude on the evolution of the dispersity. It tends to increase with the addition of BMDO, except for the copolymer formed in the presence of 10 mol% BMDO (Table 1). Indeed, the SEC chromatogram of this copolymer does not exhibit a shoulder towards the high molar masses, unlike copolymers formed with 2 and 5 mol% of BMDO in the initial feed (Fig. 5a). Furthermore, the UV chromatograms at 217 nm and 260 nm are perfectly superimposed for this copolymer, attesting that all the chains would contain an even fraction of BMDO (Fig. 5b) contrary to what was shown for the copolymer formed with 5 mol% of BMDO (Fig. 3b).
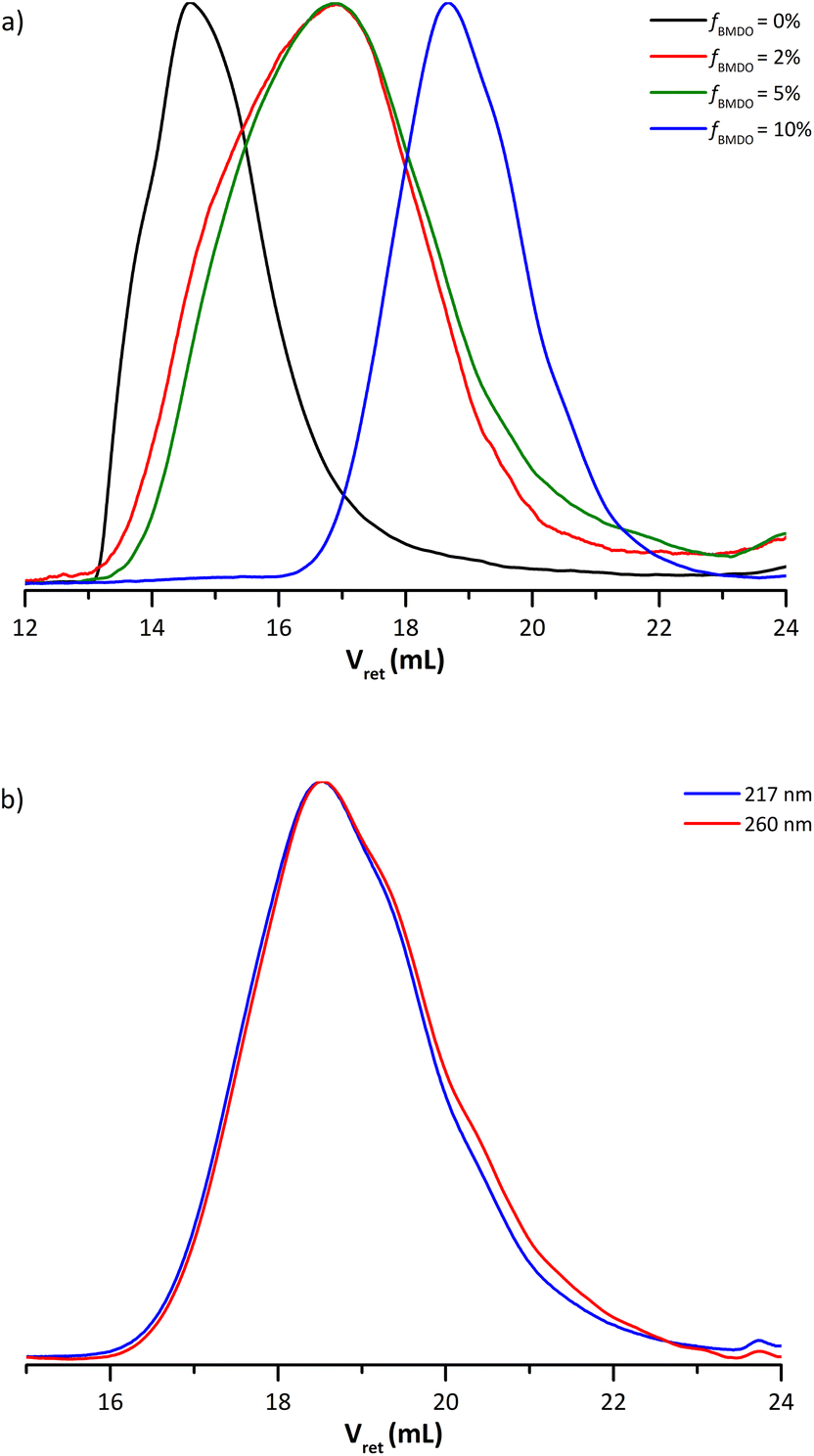 | ||
| Fig. 5 (a) Molar mass distributions of dry extracts for latexes E4BMDO (red), E5BMDO (blue), E1BMDO (green) and EMMA1 (black) (Table 1). (b) UV chromatograms from SEC-THF analysis at 217 nm (blue) and 260 nm (red) for the dried latex E5BMDO. | ||
The DSC curves also showed that for all the samples containing BMDO units, a single Tg could be detected (Fig. 6). This Tg decreases when the BMDO content increases (Table 1). Again, these results are in good agreement with what was expected from the Tg of the respective homopolymers.10,22 One can note that the width of this transition is increasing as the number of incorporated BMDO units increases, which seems to indicate a stronger heterogeneity in the polymer chains, consistent with the reactivity ratios of BMDO and MMA.
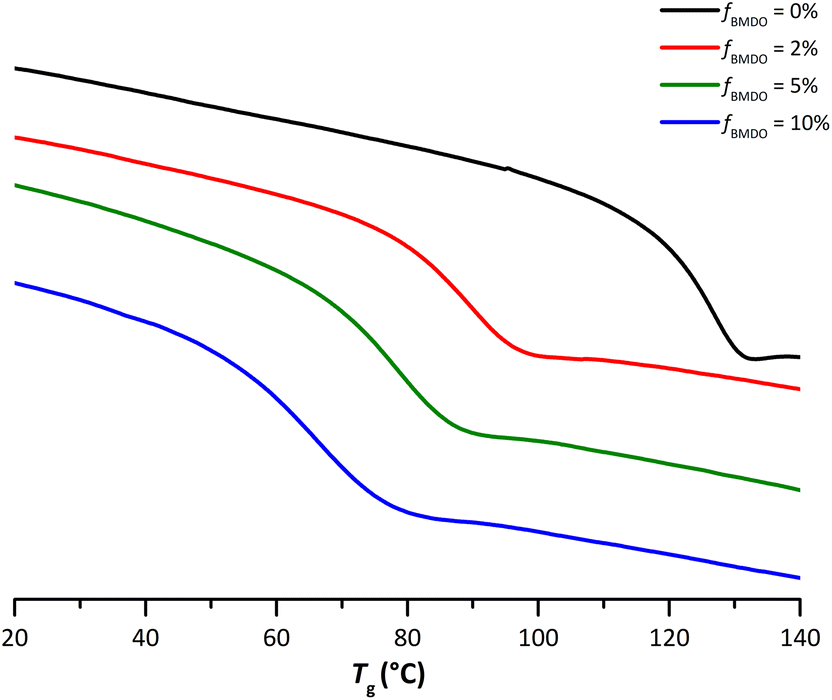 | ||
| Fig. 6 Thermograms from DSC analyses performed on the dried extract of latexes of PMMA (E1MMA) and P(MMA-co-BMDO) with increasing amounts of BMDO: fBMDO,0 = 2% (E4BMDO), fBMDO,0 = 5% (E1BMDO), fBMDO,0 = 10% (E5BMDO) (Table 1). | ||
To better comprehend the evolution of Tg with BMDO content in the copolymer, the experimental values were compared to those obtained by calculation according to Fox equation (eqn (1)).23 Although the Tg of the copolymers was systematically lower than predicted (Fig. 7), the experimental trend follows the theoretical one.
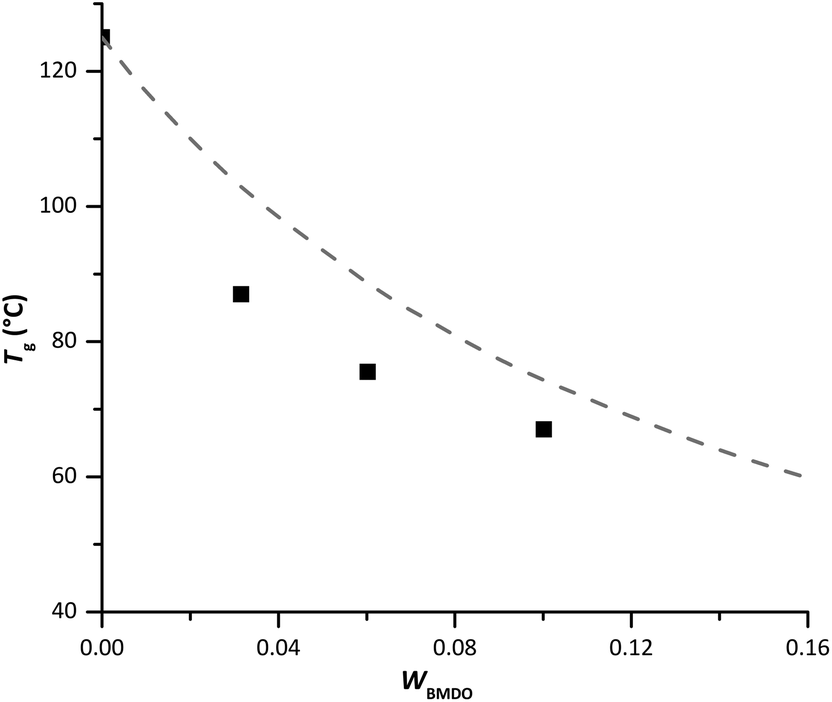 | ||
| Fig. 7 Evolution of experimental (square) and theoretical (dashed line) Tg with the mass fraction of BMDO. Theoretical values were calculated with the Fox equation. | ||
Degradation of the P(MMA-co-BMDO) copolymers
The degradation of the P(MMA-co-BMDO) copolymers was probed on the dry extract of the latexes. Several methods can be used to degrade CKA-containing vinyl copolymers. Yet, the most convenient and broadly used technique is the degradation under accelerated hydrolytic conditions, i.e. in basic conditions using a strong base such as KOH.11,24,25 Dry extracts were thus dissolved in THF and degraded by adding a solution of potassium hydroxide in methanol (Scheme 2).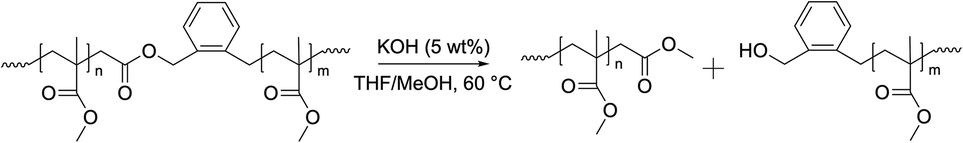 | ||
| Scheme 2 Degradation of P(MMA-co-BMDO) copolymers under accelerated conditions in the presence of KOH. | ||
This protocol was first applied to the PMMA sample. As expected, its molar mass did not change significantly after treatment of the dry extract by KOH (from 660 kg mol−1 initially, to 632 kg mol−1 after degradation, Table 2). On the other hand, SEC traces corresponding to experiments performed in the presence of BMDO shifted toward the lower molar masses after treatment of the dried extracts with KOH in THF/MeOH (Fig. 8). All copolymers exhibited a loss of molar mass of more than 80% (Table 2).
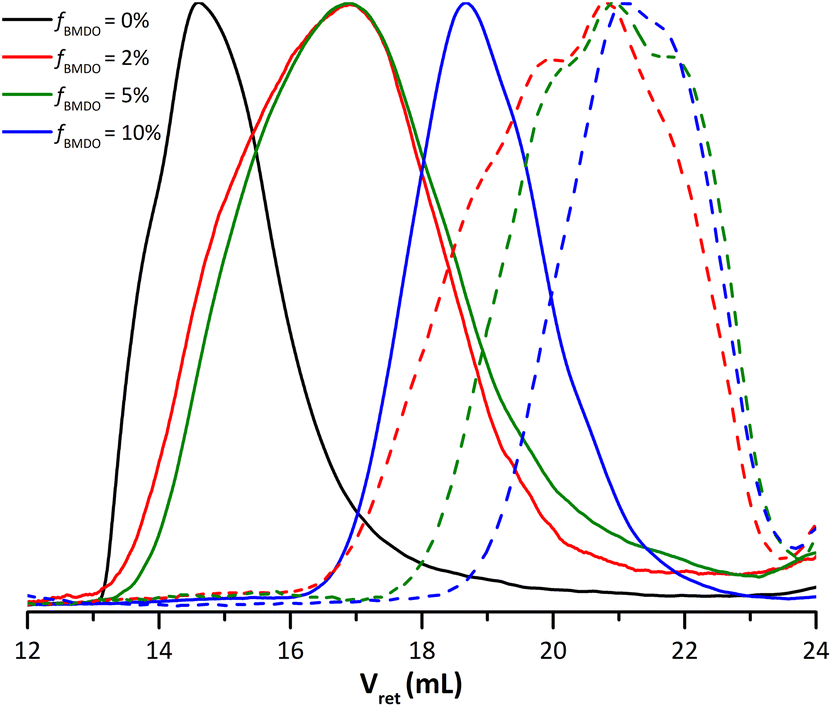 | ||
| Fig. 8 Molar mass distribution of dry extracts (plain lines) and degradation products (dashed lines) obtained by degradation of dry extracts using KOH in THF/MeOH for latexes synthesized with varying fraction of BMDO in the feed of monomers (black: E1MMA; red: E4BMDO; green: E1BMDO; blue: E5BMDO, Table 2). | ||
| Run |
F
BMDOa (%) |
M
nb (kg mol−1) |
Đ |
M
thn,degc (kg mol−1) |
M
expn,degb (kg mol−1) |
Đ | % Mn loss |
|---|---|---|---|---|---|---|---|
| a Average molar fraction of BMDO in the copolymer. b Experimental number-average molar mass determined by SEC-THF using a calibration based on PMMA standards. c Theoretical number-average molar mass calculated according to the following equation: Mthn,deg = MMMA × (1/FBMDO − 1) + MBMDO. | |||||||
| E1MMA | 0 | 660 | 2.6 | 660 | 632 | 2.9 | 4 |
| E4BMDO | 1.9 | 98.2 | 5.9 | 5.1 | 8.6 | 3.1 | 91 |
| E1BMDO | 3.8 | 61.5 | 5.8 | 2.7 | 5.9 | 2.5 | 90 |
| E5BMDO | 6.4 | 28.5 | 1.9 | 1.6 | 5.2 | 1.9 | 82 |
Those results proved the degradability of the copolymers and confirmed the incorporation of BMDO units. It is worth mentioning that molar mass values obtained remain higher than the calculated theoretical ones. Several factors may explain this observation. The rROP of CKAs is known to lead to some ring retention.8 The inserted ring-closed BMDO units would not degrade. The presence of such units can usually be assessed by 13C NMR analysis. However, in our case, the relatively low BMDO content did not allow us obtaining exploitable data by this technique. It is also possible that the mole fraction of BMDO in the copolymer, as determined by NMR, is overestimated due to the inaccuracy of the 1H NMR signal integration. Eventually, it is very likely that the distribution of the degradable ester functions along the chain is uneven resulting in asymmetrical degradation products.26 Nevertheless, the dispersity tends toward 2 when increasing the BMDO content (Table 2), which is indeed what one would expect for a statistical polymer degradation. In any case, the results of this degradation proved that we did obtain stable latexes made of degradable PMMA-based polymers.
Conclusion
Ab initio aqueous emulsion copolymerization of BMDO with MMA was performed for the first time and provided a straightforward route for the synthesis of nanoparticles made of degradable vinyl polymers. Indeed, very conventional conditions allowed yielding stable latexes made of P(MMA-co-BMDO) copolymers in a simple and fast process in water compared to the already published studies published so far on solution or bulk copolymerization. Emulsion copolymerization proved to be successful only in basic media, which is probably due to an increase rate of hydrolysis of BMDO under acidic conditions. By adjusting the pH to basic conditions with NaHCO3, the incorporation rates of BMDO varied from 2 to 6.4 mol% depending on the initial BMDO fraction. The incorporation of BMDO units was however higher than what is obtained in the case of homogeneous copolymerization. Eventually, all copolymers could be degraded under accelerated conditions with a loss of molar mass of at least 80% for every BMDO-containing copolymer.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Agence Nationale de la Recherche (grant number ANR-18-CE06-0014 CKAPART).References
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- P. Eyerer and A. Franck, in Polymers – Opportunities and Risks I: General and Environmental Aspects, ed. P. Eyerer, Springer, Berlin, Heidelberg, 2010, pp. 391–427 Search PubMed.
- Nature, 2021, 590, 363–364 Search PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef CAS.
- W. J. Bailey, S.-R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913–1920 CrossRef CAS.
- W. J. Bailey, Makromol. Chem., 1985, 13, 171–190 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- A. Tardy, N. Gil, C. M. Plummer, C. Zhu, S. Harrisson, D. Siri, J. Nicolas, D. Gigmes, Y. Guillaneuf and C. Lefay, Polym. Chem., 2020, 11, 7159–7169 RSC.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152–6159 CrossRef CAS.
- G. G. d'Ayala, M. Malinconico, P. Laurienzo, A. Tardy, Y. Guillaneuf, M. Lansalot, F. D'Agosto and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 104–111 CrossRef.
- C. Riachi, N. Schüwer and H.-A. Klok, Macromolecules, 2009, 42, 8076–8081 CrossRef CAS.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397–2403 CrossRef CAS.
- W. J. Bailey, Z. Ni and S.-R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef CAS.
- M. C. D. Carter, A. Hejl, S. Woodfin, B. Einsla, M. Janco, J. DeFelippis, R. J. Cooper and R. C. Even, ACS Macro Lett., 2021, 10, 591–597 CrossRef CAS PubMed.
- M. C. D. Carter, J. DeFelippis, R. C. Even and A. Hejl, United States, US11111328B2, 2021.
- J. M. Siebert, D. Baumann, A. Zeller, V. Mailänder and K. Landfester, Macromol. Biosci., 2012, 12, 165–175 CrossRef CAS PubMed.
- A. J. Kresge and T. S. Straub, J. Am. Chem. Soc., 1983, 105, 3957–3961 CrossRef CAS.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS.
- S. Agarwal, R. Kumar, T. Kissel and R. Reul, Polym. J., 2009, 41, 650–660 CrossRef CAS.
- A. Tardy, J.-C. Honoré, D. Siri, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Polym. Chem., 2017, 8, 5139–5147 RSC.
- J. G. Tsavalas and D. C. Sundberg, Langmuir, 2010, 26, 6960–6966 CrossRef CAS PubMed.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- C. Zhu and J. Nicolas, Polym. Chem., 2021, 12, 594–607 RSC.
- D. Gigmes, P. H. M. V. Steenberge, D. Siri, D. R. D′hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 39, 1800193 CrossRef PubMed.
- B. R. Kordes, L. Ascherl, C. Rüdinger, T. Melchin and S. Agarwal, Macromolecules, 2023, 56, 1033–1044 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00040k |
| ‡ These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2023 |