
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential dual-curing of electron-deficient olefins and alcohols relying on oxa-Michael addition and anionic polymerization†
Susanne M.
Fischer
 ab,
Viktor
Schallert
ab,
Johanna M.
Uher
ab and
Christian
Slugovc
*ab
ab,
Viktor
Schallert
ab,
Johanna M.
Uher
ab and
Christian
Slugovc
*ab
aChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, 8010 Graz, Austria. E-mail: slugovc@tugraz.at
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
First published on 20th February 2023
Abstract
Herein we propose the preparation of crosslinked polymers from off-stoichiometric oxa-Michael formulations proceeding via a self-limiting base catalyzed reaction between difunctional Michael acceptors and substoichiometric amounts of diols followed by anionic polymerization of the remaining vinyl groups. The properties of the resulting polymers can easily be tuned by varying the amount of diols.
The combination of two polymerization mechanisms taking place simultaneously or sequentially is defined as dual-curing. The resulting polymer network exhibits different and often superior properties than the individual polymers. Additionally, dual-curing facilitates processing, especially when low-viscosity formulations are required and allows for the preparation of customized intermediate and final materials by changing the composition or curing conditions.1 A variety of reactions can be used in dual-curing formulations. However, the Michael reaction is of special interest due to its “click” nature featuring high selectivity, high yield and mild reaction conditions.2 In this reaction, an electron-deficient olefin (Michael acceptor) reacts with a nucleophile (Michael donor) which is typically an acetoacetate (carba-Michael), amine (aza-Michael), thiol (thia-Michael), or alcohol (oxa-Michael). Typical Michael acceptors are maleimides, acrylamides or acrylates. The latter have been used in polymer formulations for decades mainly in radical (photo)polymerization or anionic polymerization. A somewhat unusual way to initiate anionic polymerization is via Lewis base catalysis, although phosphine-initiated polymerization of acrylonitrile has been known since the 1950s.3 However, the polymerization of less electron-deficient monomers such as acrylates4 or methacrylates5 with Lewis base catalysis has been described recently.
This polymerization can be exploited in the preparation of two-stage materials from off-stoichiometric Michael formulations in which the monomer in excess, the vinyl compound, undergoes radical homopolymerization after the first self-limiting reaction. The second reaction can then be triggered by different stimuli such as light or temperature or has a sufficiently different reaction rate compared to the first reaction.6 Two-stage curing enhances the thermo-mechanical properties of the polymer, which is important in applications that require high glass transition temperatures (Tg) and hardness.7 Two-stage curing of off-stoichiometric formulations has been described for carba-Michael,8 thia-Michael8a,9 and aza-Michael polymerization.10 However, to the best of our knowledge dual-curing of oxa-Michael formulations has not yet been reported.
This might be due to the lower reactivity of alcohols compared to other Michael donors complicating the synthesis of oxa-Michael polymers. With highly reactive Michael acceptors such as divinyl sulfone, the lack of reactivity of the alcohol can be compensated and satisfying conversions are obtained.11 However, weaker Michael acceptors such as diacrylates can only be converted if strong Lewis bases such as tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP) or strong Brønsted bases such as phosphazene bases are employed as catalysts.12 Nevertheless, only the preparation of hyperbranched13 but not cross-linked polymers has been reported so far. Recently, Yang and co-workers reported the phosphazene base catalyzed oxa-Michael polymerization of 2-hydroxyethyl acrylate and introduced diolefins and another phosphazene base at the last stage of the reaction resulting in a combination of oxa-Michael and Rauhut–Currier reactions. With this approach, high molecular weight polymers with promising mechanical properties are obtained after UV-curing of the remaining double bonds.14
Herein we present the preparation of crosslinked polymers by sequential dual-curing of off-stoichiometric formulations of difunctional Michael acceptors and diols. In the first stage polyaddition of alcohols and vinyl groups occurs and in the second stage remaining vinyl groups are anionically polymerized (Fig. 1). The second stage reaction is promoted by the same catalyst and occurs after full consumption of the alcohol.
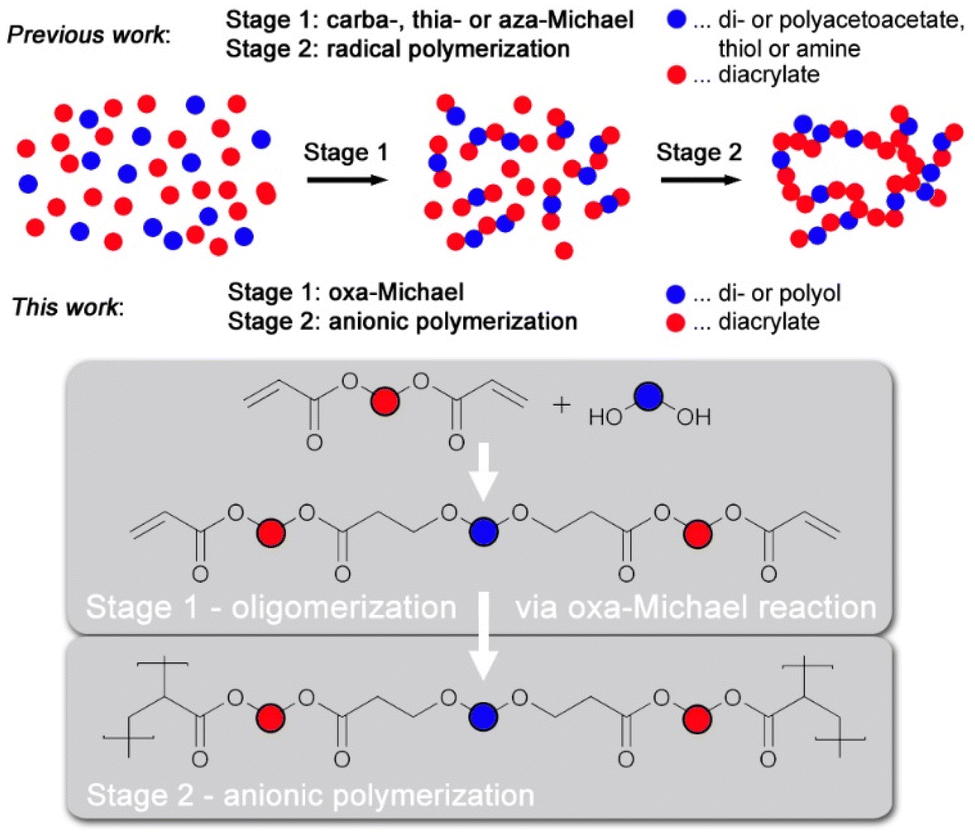 | ||
| Fig. 1 Off-stoichiometric Michael formulations with carba, thia- or aza-Michael donors undergoing 2nd stage curing by radical polymerization (previous work) and with oxa-Michael donors with anionic curing in the 2nd stage using the same base for both stages. | ||
To test which organocatalysts are active in both oxa-Michael and anionic polymerization of acrylates, the performance of Lewis and Brønsted bases active in catalyzing oxa-Michael reactions11,12 were evaluated in anionic polymerization. For this purpose, methyl acrylate (Fig. 2) was mixed with 5 mol% of catalyst and stirred at 60 °C for 24 h. No solvent was used in all reactions. Only the strong Lewis base TTMPP and the strong Brønsted base 1-tert-butyl-2,2,4,4,4-pentakis-(dimethylamino)-2λ5,4λ5-catenadi-(phosphazen) (P2-tBu) were effective in oligomerisation of the monomer under these conditions (Fig. 2 and S1–S3†). Weaker Lewis or Brønsted bases failed and no reaction occurred (Fig. S4†). The only exception was the reasonable strong Lewis base tris(4-methoxyphenyl)phosphine (TMPP)15 which promoted about 30% of dimerization in a Rauhut–Currier reaction (Fig. S5†). Subsequently, the substrate scope was enlarged to other mono- and multifunctional acrylates (Fig. 2) as well as divinyl sulfone as representative example of a strong Michael acceptor. Methyl acrylate, ethyl acrylate and benzyl acrylate were oligomerised using the aforementioned conditions resulting in average number molecular masses (Mn) of 2000–4000 Da as determined by size exclusion chromatography (Table S1 and Fig. S6, S9†). t-Butyl acrylate could not be converted indicating a strong influence of the steric demand of the monomer. Using di- or triacrylates, namely butane-1,4-diol diacrylate (BDDA) or trimethylolpropane triacrylate as the monomers resulted in mostly insoluble polymer networks with a sol fraction smaller than 10 wt% (Fig. S13†). The reaction can also be performed at room temperature, albeit lower conversions were obtained within the same time. In case of the more reactive divinylsulfone, polymerization can also be realized with weaker Lewis bases such as 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-en (DBU) or triphenylphosphine (TPP) (Fig. S15†).
 | ||
| Fig. 2 Reaction of different Lewis and Brønsted bases with alkyl acrylates (R = Me, Et or Bz). | ||
Having identified suitable catalysts for the anionic polymerization of acrylates, the dual curing approach was first investigated using a model formulation made up from methanol (0.5 equiv.), methyl acrylate (1.0 equiv.) and TTMPP (0.025 equiv.). Upon heating the formulation to 60 °C, the reaction progress was monitored using 1H-NMR spectroscopy (Fig. S16†). Within the first 10 min methanol was fully consumed and the formation of other vinyl-coupling products could only be observed distinctly later. After 4 h, 13% of the remaining vinyl groups were consumed and after 24 h still about 10% vinyl groups are present in the reaction mixture. Anionic polymerization and Rauhut–Currier derived products were identified (Fig. S17–19†).
Using difunctional acrylates and sub-stoichiometric amounts of difunctional alcohols results in the formation of polymer networks. The reaction of BDDA with butane-1,4-diol (BA) employed in different ratios (0.2–0.5 equiv., in respect to BDDA) and 5 mol% TTMPP was investigated at 60 °C. Within the first minutes, BA is fully consumed (Fig. S20†) by the corresponding oxa-Michael reaction forming acrylate-terminated oligomers. Afterwards, a slow increase of the viscosity is noted resulting in a largely insoluble material after 24 h. Alternatively, the reaction can also be performed sequentially by using room temperature for the first stage (oxa-Michael) and an elevated temperature of 100 °C for the second stage. In this case, a network with similar characteristics could be obtained after only 6 h compared to the 24 h reaction time in case of curing at 60 °C. For the characterization of the networks, sol–gel analysis was performed. The network poly(BDDAcoBA0.5) made of 1 equiv. BDDA and 0.5 equiv. BA gave 11 wt% soluble fraction (CH2Cl2) or 18 wt% soluble fraction (acetone). The soluble fraction was analyzed by NMR spectroscopy revealing the presence of the expected vinyl-terminated oxa-Michael derived oligomers (Fig. S23–25†), TTMPP and its oxidized analogue. However, the catalyst could not be fully recovered, indicating that most of it is covalently bound to the polymer. The Infrared-spectroscopy (IR) of the sol- and the gel-fraction revealed very similar spectra only distinguished by a reduced relative intensity of the bands corresponding to acrylate groups (1597, 1460, 1408, 809 cm−1, Fig. S26†) in the gel fraction. In all samples, the characteristic ether band at 1107 cm−1 indicative for the presence of oxa-Michael derived structures was observed. Thermal properties were studied with differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Both, the Tg (Table S3†) as well as the thermal stability increase with decreasing BA share. The polymer obtained from the acrylate alone, poly(BDDA), shows a quick mass loss at temperatures above 350 °C (Fig. 3). Copolymerization of BA causes a mass loss starting at about 180 °C. The amount of the mass loss occurring at this temperature is proportional to the BA content in the polymer. Interestingly, poly(BDDA) exhibits a minor mass loss starting at 180 °C probably indicative for the presence of a low share of oxa-Michael derived repeat units in the sample. On the other hand, poly(BDDAcoBA1) which is ideally exclusively build up by oxa-Michael derived repeat units seems to feature a small share of anionically derived repeat units. Size exclusion chromatography supports this speculation (cf. ESI, Fig. S30†).
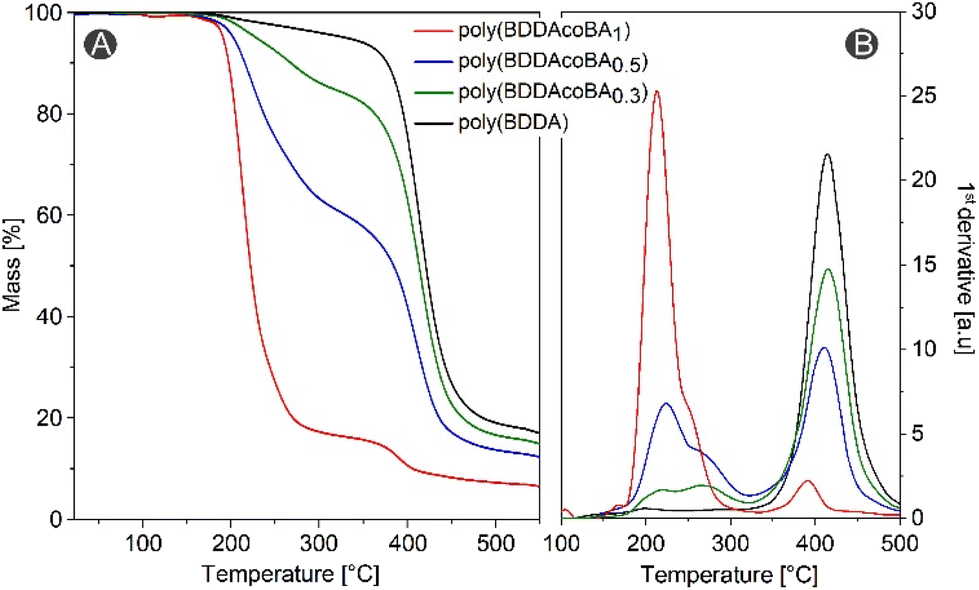 | ||
| Fig. 3 Thermogravimetric analysis of the polymer-networks made from BDDA with and without copolymerizing different amounts of BA using a heating rate of 10 °C min−1; (A) mass vs temperature and (B) 1st derivative of curves shown in (A). | ||
In case of the stronger difunctional Michael acceptor divinyl sulfone (DVS) also weaker Lewis bases such as DBU cause anionic polymerization (Fig. S15†). Therefore, dual curing of formulations of DVS (1 equiv.) and varying amounts of the differently acidic diols butane-1,4-diol (BA), (Z)-butene-1,4-diol (BE) or butyne-1,4-diol (BY) was catalyzed with DBU (0.05 equiv.) at 60 °C for 24 h or at 25 °C for 15 min (stage 1) followed by 120 °C for 1 h (stage 2). The reaction of DVS and 0.5 equiv. BE was monitored by 1H NMR spectroscopy. After 15 min at 60 °C, BE was completely consumed. Vinyl groups were converted to about 54%, i.e. vinyl groups have almost exclusively reacted in oxa-Michael additions. After 1 h, the vinyl group amount decreased to 20% (Fig. S31†). After 24 h, solubility tests were performed with CH2Cl2 (17 wt% soluble fraction) or acetone (9 wt%). The soluble fraction consists of the catalyst DBU and oxa-Michael products with a very low share of vinyl sulfone groups according to NMR and IR spectroscopy (Fig. S31–S33†).
The resulting polymeric materials are characterized by an increasing hardness as exemplified for the series poly(DVScoBE) with various BE shares. While poly(DVScoBE1) and poly(DVScoBA0.75) are viscous liquids, poly(DVScoBA0.5) and poly(DVScoBA0.25) are solids characterized with ShoreD hardness of 46.8 (±2.6) and 82.0 (±2.2), respectively.‡ Thermal analysis revealed the increase of the Tg with decreasing diol share (Fig. 4). Remarkably, a variation of the Tg within the range of about 100 °C could be obtained by simply changing the share of the diol. For poly(DVS) and poly(DVScoBY0.25) no Tg could be detected in the DSC runs conducted up to a temperature of 150 °C. At higher temperatures thermal decomposition starts as determined by TGA. For DVS based polymers, the oxa-Michael and the vinyl polymerization derived parts degrade at similar temperatures (Fig. S39–S41†).16,17
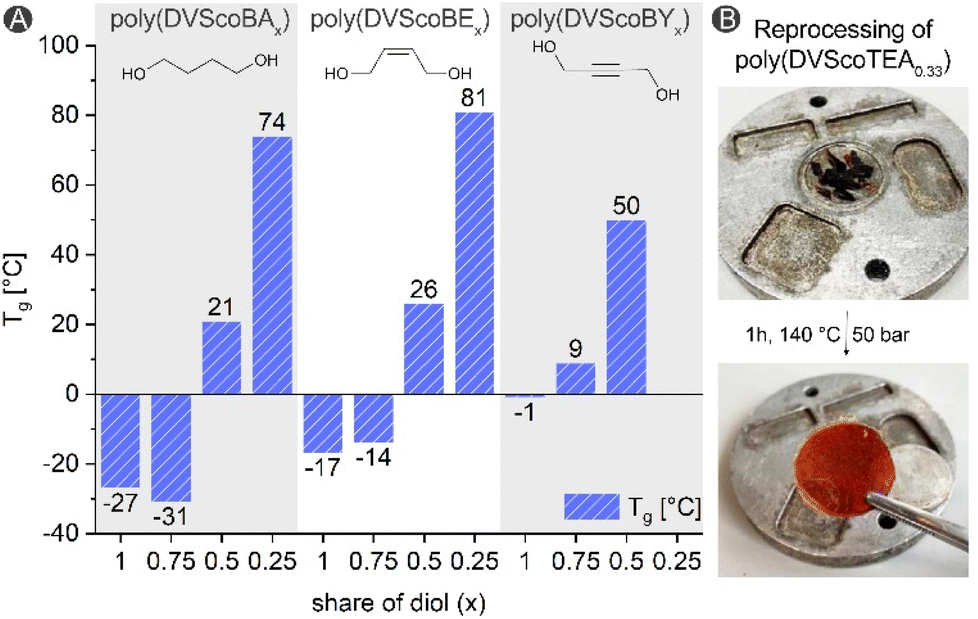 | ||
| Fig. 4 (A) Glass transition temperatures determined with DSC (3rd heating run, heating rate 40 °C min−1); (B) reprocessing of poly(DVScoTEA0.33) using a platen press (1 h, 140 °C, 50 bar). | ||
Recently, we described covalent adaptable networks relying on the reversibility of oxa-Michael derived bonds.17 To evaluate if polymers obtained from off-stoichiometric formulations still have self-healing characteristics, poly(DVScoBE0.5) was cut into small pieces (3–4 mm), post-cured at 100 °C for 1 h and pressed at 130 °C for 1 h with 50 bars. Thereafter, a uniform material was obtained (Fig. S45†). The IR spectra of the reprocessed polymer is virtually identical with its virgin counterpart. Decreasing the alcohol content to 0.25 equiv. does not result in proper healing of the material indicating that a sufficient amount of reversible bonds is necessary to obtain a uniform material (Fig. S45†). Moreover, the synthesis of the polymer network made of 1 equiv. of DVS and 0.66 equiv. triethanolamine (TEA) described by Ratzenböck et al.,17 was repeated with a halved amount of TEA to obtain poly(DVScoTEA0.33) (Scheme S2†). Using sub-stoichiometric amounts of the alcohol resulted in an increase of the Tg to 52 °C compared to 3 °C when stoichiometric amounts are used. The self-healing properties of this material were then tested as shown in Fig. 4B and are retained.
Conclusion
In conclusion, we demonstrated the preparation of dual-cured polymers via the combination oxa-Michael and anionic polymerization. The polymerization tendency increases with increasing Lewis (or Brønsted) basicity of the initiator and with increasing electron-deficiency of the vinyl groups of the monomer. For acrylates highly active catalysts such as P2-tBu or the electron rich phosphine TTMPP must be employed whereas for DVS also less active catalysts such as DBU or DMAP can be used. Polymers from off-stoichiometric oxa-Michael formulations cure in a two stage process. The oxa-Michael reaction is finished within minutes at room temperature, while the anionic polymerization of the remaining vinyl groups needs higher temperatures to proceed at reasonable pace. The second stage is typically performed by heating to 100–120 °C for 1 h. The resulting alcohol-deficient copolymers exhibit a higher thermal stability, higher hardness and higher Tg values compared to their stoichiometric oxa-Michael polymer congeners. These properties of the polymers and as would seem natural other properties can therefore easily be tuned upon changing the amount and the nature of the alcohol component.Author contributions
S. M. F. and J. M. U. set up and performed the experiments and analysed the results. V. S. and J. M. U. performed thermoanalytic characterizations and reprocessing experiments. S. M. F and C. S. wrote the manuscript. C. S. conceived and directed the research, analysed the results and reviewed the manuscript. All authors contributed to the discussion and interpretation of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development, and the Christian Doppler Research Association (Christian Doppler Laboratory for Organocatalysis in Polymerization) is gratefully acknowledged.References
- O. Konuray, X. Fernández-Francos, X. Ramis and À. Serra, Polymers, 2018, 10, 178 CrossRef PubMed.
- X. Ramis, X. Fernández-Francos, S. De la Flor, F. Ferrando and À. Serra, Thermosets Struct. Prop. Appl. Second Ed, 2018, 511 Search PubMed.
- L. Horner, W. Jurgeleit and K. Klüpfel, Liebigs Ann. Chem., 1955, 591, 108 CrossRef CAS.
- G. Rui, Q. Lv, J. Lu, T. Wu, S. Zhao, R. Huang, B. Han and W. Yang, Polymer, 2021, 214, 123245 CrossRef CAS.
- Y. Zhang, M. Schmitt, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2013, 135, 17925 CrossRef CAS PubMed.
- O. Konuray, X. Fernández-Francos, S. De la Flor, X. Ramis and À. Serra, Polymers, 2020, 12, 1084 CrossRef CAS PubMed.
- (a) D. P. Nair, N. B. Cramer, M. K. McBride, J. C. Gaipa, R. Shandas and C. N. Bowman, Polymer, 2012, 53, 2429 CrossRef CAS PubMed; (b) C. L. Sherman, R. C. Zeigler, N. E. Verghese and M. J. Marks, Polymer, 2008, 49, 1164 CrossRef CAS.
- (a) N. Moszner and V. Rheinberger, Macromol. Rapid Commun., 1995, 16, 135 CrossRef CAS; (b) J. Pavlinec and N. Moszner, J. Appl. Polym. Sci., 1997, 65, 165 CrossRef CAS; (c) O. Konuray, X. Fernández-Francos, X. Ramis and Á. Serra, Polymers, 2019, 11, 1408 CrossRef CAS PubMed.
- D. P. Nair, N. B. Cramer, M. K. McBride, J. C. Gaipa, N. C. Lee, R. Shandas and C. N. Bowman, Macromol. Symp., 2013, 329, 101 CrossRef CAS.
- (a) G. González, X. Fernández-Francos, À. Serra, M. Sangermano and X. Ramis, Polym. Chem., 2015, 6, 6987 RSC; (b) M. Retailleau, A. Ibrahim, C. Croutxé-Barghorn, X. Allonas, C. Ley and D. Le Nouen, ACS Macro Lett., 2015, 4, 1327 CrossRef CAS PubMed; (c) T. Gencoglu, T. N. Eren, J. Lalevee and D. Avci, J. Photochem. Photobiol., A, 2021, 404, 112959 CrossRef CAS; (d) M. Retailleau, A. Ibrahim, C. Croutxé-Barghorn and X. Allonas, Prog. Org. Coat., 2016, 100, 51 CrossRef CAS.
- (a) S. Strasser and C. Slugovc, Catal. Sci. Technol., 2015, 5, 5091 RSC; (b) S. Strasser, C. Wappl and C. Slugovc, Polym. Chem., 2017, 8, 1797 RSC; (c) K. Ratzenböck, M. Mehraj, U. Din, S. M. Fischer, E. Zagar, D. Pahovnik, A. D. Boese, D. Rettenwander and C. Slugovc, Chem. Sci., 2022, 13, 6920 RSC.
- (a) S. M. Fischer, P. Kaschnitz and C. Slugovc, Catal. Sci. Technol., 2022, 12, 6204 RSC; (b) H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Polym. Chem., 2018, 9, 4685 RSC.
- Q. Jiang, Y. L. Zhang, Y. Du, M. Tang, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, Polym. Chem., 2020, 11, 1298 RSC.
- H. Yao, Y. Song, W. Huang, L. Jiang, Q. Jiang, X. Xue, B. Jiang and H. Yang, Macromolecules, 2022, 55, 8283 CrossRef CAS.
- S. M. Fischer, S. Renner, A. D. Boese and C. Slugovc, Beilstein J. Org. Chem., 2021, 17, 1689 CrossRef CAS PubMed.
- O. P. Lee, H. Lopez Hernandez and J. S. Moore, ACS Macro Lett., 2015, 4, 665 CrossRef CAS PubMed.
- K. Ratzenböck, J. M. Uher, S. M. Fischer, D. Edinger, V. Schallert, E. Žagar, D. Pahovnik and C. Slugovc, Polym. Chem., 2023, 14, 651 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00035d |
| ‡ The hardness of poly(DVS) could not be determined via a ShoreD measurement because of its brittleness. |
| This journal is © The Royal Society of Chemistry 2023 |