
Dual crosslinking polymer networks: correlation between polymer topologies and self-healing efficiency†
Soonyoung
Choi
ab,
Junhee
Kim
a,
Eunyong
Seo
 ac,
Hyocheol
Jung
a,
Ji-Eun
Jeong
a,
Young Il
Park
a,
Jin Chul
Kim
a,
Dong Woog
Lee
*b,
Byeong-Su
Kim
*d and
Sang-Ho
Lee
*ae
ac,
Hyocheol
Jung
a,
Ji-Eun
Jeong
a,
Young Il
Park
a,
Jin Chul
Kim
a,
Dong Woog
Lee
*b,
Byeong-Su
Kim
*d and
Sang-Ho
Lee
*ae
aCenter for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea. E-mail: slee@krict.re.kr
bSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: dongwoog.lee@unist.ac.kr
cDepartment of Chemical Engineering, Ulsan College, Ulsan 44610, Republic of Korea
dDepartment of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: bskim19@yonsei.ac.kr
eAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
First published on 2nd February 2023
Abstract
The topologies of polymers can impact the performance of polymeric materials, including their chemical and physical properties. In this work, a dynamic covalent bond of boronate ester was introduced by the addition of 4-vinylphenylboronic acid to the rich hydroxyl groups of polyglycidols (PGs) with different topologies, including branched cyclic, hyperbranched, and linear PGs. The formation of the dual crosslinked polymer networks, which consisted of dynamic covalent bonds (B–O) and static covalent bonds, was confirmed by thermogravimetric analysis and a swelling test. In addition, the mechanical properties of the cured materials were evaluated using a rheometer, dynamic mechanical analysis, and nanoindentation. Scratch tests and tensile tests were used to determine the self-healing effectiveness of polymer topologies. Intriguingly, based on the polymer topologies, the crosslinked network with a branched cyclic structure (bc-cPGB) exhibited a greater self-healing efficiency and modulus than hyperbranched networks (hb-cPGB). These findings indicate that the physical properties of polymer networks are influenced by the network mesh space and preferred intermolecular crosslinking of the branched cyclic structure. In addition, to maximize the benefits of the dual crosslinking system, the dynamic B–O bonds were utilized for recycling cured materials, and the PG prepolymer was successfully recovered from cPGB by adding pinacol to THF with a yield of 99.5%. These findings demonstrate the significance of topology control in highly adaptable advanced functional materials.
Introduction
In recent years, polymeric self-healing materials have attracted a great deal of academic and industrial interest as a potential technology to preserve material properties and functions throughout their service life.1,2 Generally, self-healing of polymeric materials can be achieved by chemical, physical, and physicochemical approaches,3 enabling the repetitive association and/or dissociation of polymer networks in response to stimuli, such as heat,4 light,5 redox response,6 and pH changes.7–9 With their environmental and economic benefits, these self-healing materials can be utilized in numerous industrial fields, including automotive coatings, adhesives, solar cells, electronic skins, sensors, and supercapacitors.10Over the past decade, efforts have been focused on the design of dynamic polymer networks with self-healing properties11 utilizing Diels–Alder chemistry,12 disulfide bonds,13 hindered urea bonds,14 and boron-based bonds.15–20 In these studies, the stability of reactive groups resulting from chain cleavage is the key factor for enhanced self-healing performance.21 The cleaved polymeric segments generated from damage may undergo segmental rearrangements resulting in conformational changes or diffusion leading to polymeric network rearrangements. In the rearrangement process, a self-healing reaction will not occur if the reactive group is converted to an inactive group by undesired side reactions or is not spatially synchronized with the opposing reactive groups. To induce an effective self-healing process, requiring a delicate interplay between chemical reactions and physical network rearrangement, the stability of reactive groups is crucial.
As a representative example of self-healing materials, boronic ester-based polymeric networks have received considerable attention due to their high self-healing efficiency and biocompatibility.17,19,22–27 In addition, boron–oxygen (B–O) bonds within boronic ester possess a unique combination of high thermodynamic stability and kinetic tunability. For example, Sumerlin et al. reported photo-initiated radical thiol–ene click chemistry crosslinked polymer networks carrying dynamic covalent boronic esters.16 The self-healing was conducted at room temperature and under ambient conditions without solvents. Using telechelic diboronic ester, Guan et al., also reported boronic ester transesterification to improve self-healing efficiencies.22 Intriguingly, Li et al. demonstrated that a phenol compound containing three hydroxyl groups could facilitate the dynamic exchange of B–O bonds with a phenol compound containing two hydroxyl groups.18 Although successful self-healing can be achieved through efficient boronic ester transesterification, numerous studies continue to focus on enhancing the thermodynamic stability and kinetic tunability of boronic ester transesterification by controlling pH or alcohol structures.
Meanwhile, controlling the topology of a polymer is essential to determine its physical properties and functions in the bulk and solution state.28 Due to the absence of polymeric chain ends, macrocyclic polymers have a higher glass transition temperature (Tg) than their linear counterparts in the bulk state.29 In addition, the hydrodynamic volume of macrocyclic polymers in solution is smaller than that of linear polymers due to their reduced conformational degree of freedom.30 Due to their branching structure, hyperbranched polymers with intermediate structures between linear polymers and dendrimers have a lower Tg value than linear polymers.31 As a representative example, hyperbranched polyglycidols (hb-PGs) possess a number of remarkable characteristics, including a highly flexible aliphatic polyether backbone, multiple hydroxyl groups, and excellent biocompatibility.32,33 In addition, the degree of branching (DB) affects the microstructures and physical properties of hb-PGs.28,34–37 We have recently reported a recyclable metal-free catalytic system for the cationic ring-opening polymerization (CROP) of glycidol under ambient conditions38 and the metal-free ring-opening polymerization of glycidol to produce a diverse array of hb-PGs and branched cyclic PGs (bc-PGs) with precise control of their topologies (Scheme 1a).28 There was a significant correlation between the topologies and physical properties of the topology-controlled PGs. For instance, topology-controlled PGs with a similar degree of polymerization (DP) and similar DB values (DBbc-PG = 0.48 and DBhb-PG = 0.51) exhibited distinct Tg values, attributed to intermolecular and/or intramolecular hydrogen bonding (H-bonding) between the polymer chains (Tg,hb-PG > Tg,bc-PG). Moreover, their unique solution properties, such as hydrodynamic volumes and diffusion coefficients (D), were found to be dependent on the polymer topologies (Dhb-PG < Dbc-PG). Despite the abundance of literature on polymeric topologies, studies to improve the self-healing efficiency as a function of polymeric topologies remain scarce.
 | ||
| Scheme 1 (a) Previous study on controlling the topologies of polyglycidols (PGs). (b) Self-healable polymer network based on various polymer topologies using dynamic boronic ester bonds. | ||
Experimental
Materials and methods
Controlled experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500; Mw/Mn = 1.03–1.11). A 300 MHz Bruker Ultrashield spectrometer was used to perform 1H NMR. All spectra were recorded in ppm units using DMSO-d6 and methanol-d4 as deuterated solvents at room temperature. On a Q500 calorimeter, polymer networks were subjected to thermogravimetric analysis (TGA) under airflow at a heating rate of 10 °C min−1 from 25 to 800 °C (TA Instruments). A rheometer oscillatory frequency sweep was performed to evaluate the mechanical properties of the circular film (Anton Paar, MCR102). At room temperature, the modulus of the parallel plates (diameter, 25 mm) was measured over the frequency range of 0.1 to 1000 rad s−1 with a constant strain of 0.1%. tanδ was determined by dynamic mechanical analysis (DMA) with a constant strain of 0.1% at a heating rate of 3 °C min−1 from −50 to 100 °C using a Q800 calorimeter (TA Instruments). DMA was subjected to a stress relaxation test with a constant strain of 3% at a range of temperatures (50–90 °C). For the tanδ and stress relaxation experiments, rectangular bars were prepared with dimensions of 30 mm × 5 mm × 0.5 mm and 60 mm × 10 mm × 2 mm, respectively. Using a nanoindentation tester, hardness and modulus indentation of coatings were determined (Anton Paar TriTec, NHT3). Loading (0 to 2 mN, 60 mN min−1), holding (2 mN), and unloading (reversed the loading step) measurements were performed. The reported data represent the mean of three separate measurements.
500; Mw/Mn = 1.03–1.11). A 300 MHz Bruker Ultrashield spectrometer was used to perform 1H NMR. All spectra were recorded in ppm units using DMSO-d6 and methanol-d4 as deuterated solvents at room temperature. On a Q500 calorimeter, polymer networks were subjected to thermogravimetric analysis (TGA) under airflow at a heating rate of 10 °C min−1 from 25 to 800 °C (TA Instruments). A rheometer oscillatory frequency sweep was performed to evaluate the mechanical properties of the circular film (Anton Paar, MCR102). At room temperature, the modulus of the parallel plates (diameter, 25 mm) was measured over the frequency range of 0.1 to 1000 rad s−1 with a constant strain of 0.1%. tanδ was determined by dynamic mechanical analysis (DMA) with a constant strain of 0.1% at a heating rate of 3 °C min−1 from −50 to 100 °C using a Q800 calorimeter (TA Instruments). DMA was subjected to a stress relaxation test with a constant strain of 3% at a range of temperatures (50–90 °C). For the tanδ and stress relaxation experiments, rectangular bars were prepared with dimensions of 30 mm × 5 mm × 0.5 mm and 60 mm × 10 mm × 2 mm, respectively. Using a nanoindentation tester, hardness and modulus indentation of coatings were determined (Anton Paar TriTec, NHT3). Loading (0 to 2 mN, 60 mN min−1), holding (2 mN), and unloading (reversed the loading step) measurements were performed. The reported data represent the mean of three separate measurements.
Results and discussion
Design of poly(glycidol)s carrying 4-vinylphenylboronic ester as a crosslinking building blocks
A B–O dynamic covalent bond can generally be formed through transesterification and re-esterification of alcohols with boronic acids. In addition, the vinyl group in VPA can be used to crosslink polymer networks using a photocuring system. To confirm the reactivity between boronic acid and various alcohol moieties within the PG backbone (i.e., L1,3 = primary alcohol, L1,4 = secondary alcohol, and T1 = diol), control experiments with small molecule analogs using ethanol as a primary alcohol, 2-propanol as secondary alcohol, and 1,2-propanediol as diol with VPA were conducted (Fig. S1†). As anticipated, the reactivity of diol with boronic acid was significantly higher than that of mono alcohols at varying ratios ([alcohol]/[VPA] = 1, 2, 4, and 8 molar ratio), whereas 1H NMR analyses confirmed the formation of boronic ester from mono alcohols to be around 20% in excess of the alcohols.According to our previous research, the diol units in topology-controlled PGs comprised approximately 40 mol%; therefore, 15 and 30 mol% of the crosslinking agent was added at room temperature for 3 h.28 As shown in Fig. 1, 1H NMR spectra indicated the successful synthesis of PGs containing 4-vinylphenylboronic ester, including bc-PGB, hb-PGB, and lin-PGB. For instance, the peaks originating from the 4-vinylphenyl group in bc-PGB could be clearly assigned, including those of the polyether backbone protons (3.95–3.38 ppm), vinyl protons (6.84–5.17 ppm), and phenyl protons (7.79–7.32 ppm). The fraction of boronic ester was calculated by 30 mol% in PGB backbone based on the peak integration between polyether backbone protons and vinyl group on VPA. In addition, hb-PG and lin-PG containing 30 mol% of boronic ester (hb-PGB and lin-PGB) were synthesized using the same protocol (Fig. 1b and c).
 | ||
| Fig. 1 1H NMR spectra of (a) bc-PGB, (b) hb-PGB, and (c) lin-PGB carrying 30 mol% of 4-vinylphenylboronic ester (MeOH-d4, room temperature). | ||
Preparation of crosslinked polymer networks via photo-radical curing process
The synthesized PGBs were then employed in the photo-radical curing process with PBPO as the initiator under 365 nm UV light for 6 h (Fig. 2). To determine the degree of crosslinking on the obtained films, a swelling test in ethanol was conducted for 5 h. All cured PGB samples (cPGBs) exhibited nearly constant masses before and after the swelling test, indicating the quantitative conversion of vinyl groups in PGBs during curing. Interestingly, the degree of swelling of bc-cPGB was greater than that of hb-cPGB and lin-cPGB. It is attributable to the preferred intermolecular crosslinked polymer network and larger network mesh space by the cyclic structure of bc-PGB, resulting in a more stable polymer network and a greater swelling degree in ethanol compared to other polymer structures.40 On the other hand, the de-swelling behavior of hb-cPGB occurred after 1 h in ethanol. It is likely due to the gradual formation of intramolecular bonds in the hyperbranched structure, which would lead to the disruption of the intermolecular crosslinked polymer network.41 In contrast, lin-cPGB exhibited no swelling due to the dense crosslinking of its polymer networks. | ||
| Fig. 2 (a) Photocuring system for PGBs, (b) swelling tests of cPGBs in ethanol as a function of time, and (c) photos of cPGBs before and after swelling tests. All images are in the same scale. | ||
Furthermore, the swelling behavior of these crosslinked polymer networks in toluene or water was evaluated (Fig. S2†). Due to the hydrophilic polymer backbone, no swelling behavior was observed in toluene. However, the dissociation of boronic ester in water resulted in the degradation of polymer networks.
Thermal and mechanical properties of crosslinked polymer networks
The glass transition temperature (Tg) of bc-cPGB and hb-cPGB was determined by DMA using the tanδ peak (Fig. S3†). Intriguingly, similar Tg values were observed even though the crosslinked polymer networks may differ due to competing crosslinking mechanisms between intermolecular and intramolecular crosslinking depending on the prepolymer structure. Generally, fully crosslinked polymer networks with a cyclic structure have a lower Tg value than those with a linear structure due to the increased chain mobility resulting from the mesh structure of the cyclic topology.42 In this study, however, the crosslinking densities of the samples were restricted to 30%, and hyperbranched prepolymer (hb-PGB) had a higher probability of forming intramolecular crosslinking than cyclic prepolymer (bc-PGB), resulting in a lower Tg value than expected.38
TGA was also used to determine the thermal stability of bc-cPGB and hb-cPGB (Fig. S4†). From 376 °C to 388 °C, the weight of both cPGBs decreased gradually, indicating the thermal decomposition of the boronic ester linkage. Even though the crosslinking densities of bc-cPGB and hb-cPGB were comparable, the decomposition temperature (Td5) of bc-cPGB was higher than that of hb-cPGB, indicating that bc-cPGB is more thermally stable and that the intramolecular crosslinking pathway of hb-PGB was enhanced (Td5,bc-cPGB = 254 °C and Td5,hb-cPGB = 218 °C).
To investigate the mechanical property of cured materials based on the polymer topologies, the viscoelastic property was analyzed further with a rheometer (Fig. 3a). At all frequencies, the storage modulus (G′) of both networks is greater than the loss modulus (G′′), indicating that elasticity predominates than viscosity. G′ and G′′ of bc-cPGB are surprisingly higher than those of hb-cPGB, indicating a higher mechanical property of bc-cPGB because of the unique ring in cyclic polymer networks contributing to elasticity of the network.39 Similarly, tensile tests revealed that the tensile strength and Young's modulus of bc-cPGB are greater than those of hb-cPGB (Fig. 3b). In addition, nanoindentation of the surface mechanical properties of the samples revealed greater indentation hardness (HIT) and indentation modulus (EIT) due to the unique topology of cyclic structure (Fig. 3c). These results demonstrated that polymer networks with branched cyclic polymer matrices possessed superior overall mechanical properties than those with hyperbranched polymer matrices.
 | ||
| Fig. 3 (a) Storage (G′) and loss (G′′) modulus of obtained cPGB films, (b) tensile strength, Young's modulus, and elongation at break of cPGB films, and (c) indentation hardness (HIT) and indentation modulus (EIT) of cPGB surface. | ||
Effect of polymer topologies on self-healing ability
The boronic ester-based self-healing material functions by exchanging boronic ester through transesterification and re-esterification as a dynamic covalent bond.43 Simple tensile tests confirmed the self-healing property of polymer networks in this study. A razor blade was used to cut and separate cPGB samples, and a small amount of water was added to the cut surface. Then, the cut surfaces were physically placed into contact by hand. As shown in Fig. 4a, bc-cPGB and hb-cPGB samples were completely healed within approximately three days at room temperature while lin-cPGB was not healed during the identical time period (Fig. S5†). It is possibly due to the compact crosslinked bonds with boronic acid and low chain mobility of lin-cPGB. The healed bc-cPGB and hb-cPGB were then utilized in a 500 g (1000 times the mass of the sample) tensile test to confirm their self-healing efficiencies. Under the boronic ester exchange reaction, both bc-cPGB and hb-cPGB films could withstand the 500 g weight. In addition, to determine the quantitative self-healing efficiency, the healed cPGB samples were utilized for the UTM tensile test based on the healing time (Fig. 4b). Intriguingly, bc-cPGB exhibited a relatively higher self-healing efficiency (95% after 1 h), as defined by the ratio of the maximum stress after self-healing to that of pristine, than that of hb-cPGB (59% after 3 days) because of its unique cyclic structure and increased chain mobility.44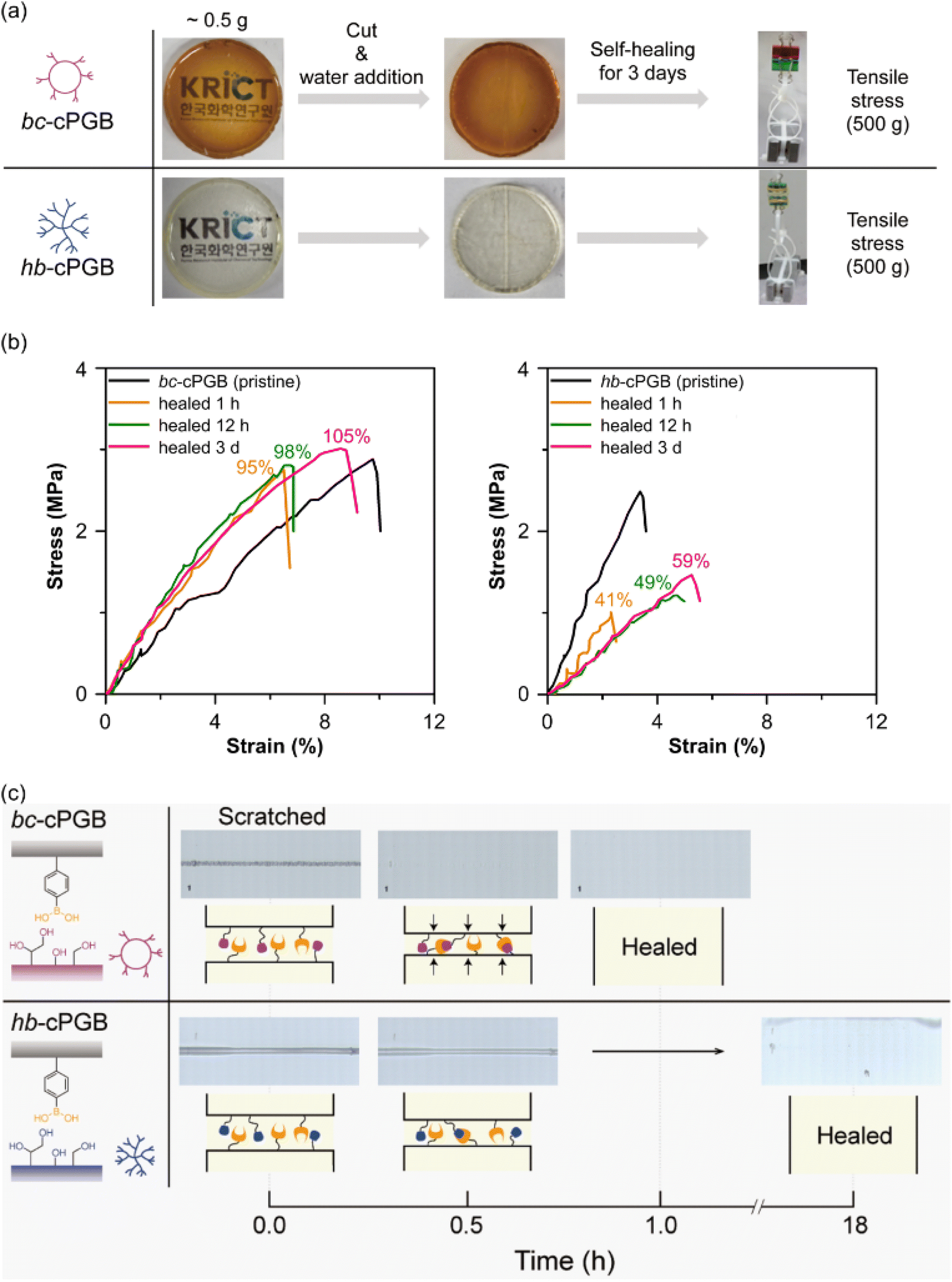 | ||
| Fig. 4 (a) Photos of the healing process for different cured polymer networks. (b) Stress–strain curves of bc-cPGB (left) and hb-cPGB (right); pristine (black line) and healed samples. (c) Optical microscopy images of the scratch test of bc-cPGB (top) and (b) hb-cPGB (bottom). | ||
To further investigate the self-healing performance based on polymer topologies, the scratch healing property of bc-cPGB and hb-cPGB was analyzed using a scratch tester. Optical microscopy was used to observe cPGB samples that had self-healed under conditions of 80% relative humidity and room temperature. Surprisingly, as shown in Fig. 4c, the scratched surface of bc-cPGB was completely healed in 1 h, whereas the scratched surface of hb-cPGB required 18 h to heal under the same conditions, indicating that the tendency of the self-healing effect is consistent with the tensile test. Thus, a stress relaxation experiment was conducted to investigate the dynamic exchange of B–O bonds in polymer networks based on the various polymer topologies. DMA was used to conduct the stress relaxation experiment within the linear viscoelastic region of polymer networks (Fig. S6†). Clearly, both bc-cPGB and hb-cPGB samples were perfectly relaxed between 50 °C and 90 °C. Particularly, a shorter relaxation time was observed for bc-cPGB than hb-cPGB at all temperatures, indicating that bc-cPGB has faster exchange dynamics due to its unique cyclic topology. Based on the Arrhenius equation, the activation energy (Ea) for the self-healing of polymer networks was determined by plotting the graph of ln(τ*) versus 1000/T (Fig. 5a). τ* (s) is the relaxation time at G/G0 = 1/e, τ0 is τ* at an infinite temperature, R is the gas constant (8.314 J mol−1 K), and T is the temperature (K).

 | ||
| Fig. 5 (a) Arrhenius plots and activation energy of polymer networks and (b) scheme for the structure of cured sample depending on the polymer topologies. | ||
Recyclable property of crosslinked polymer networks
We then prompted to take advantages of the dual cross-linking system, in which the dynamic bonds can be further exploited to recycle the resulting polymers. As a heterogeneous system, bc-cPGB was reacted with pinacol in THF at 60 °C in an effort to investigate the recyclable property of polymer networks. Due to the replacement of the boronic ester bonds in the polymer main backbone by pinacol and the subsequent precipitation of bc-PG in cold diethyl ether, the sample transformed into a cloudy solution after 6 h (Fig. 6a). Notable is the confirmation of recycled bc-PG and poly(4-vinylphenylboronic ester) (PVPBE) by 1H NMR and SEC analysis (Fig. 6 and Fig. S7†). Particularly, recycled bc-PG exhibited identical molecular weight and molecular weight distribution with a high yield (99.5% for recycled bc-PG), attributed to heterogeneous system. We highlight that this study unveils the effect of polymer topologies on the self-healing efficiency. Moreover, the remarkable recyclability of bc-PG is demonstrated due to dual cross-linking system in this study. | ||
| Fig. 6 (a) Mechanism for de-crosslinking of polymer networks using pinacol; (b) 1H NMR spectrum for recycled polymers (c) SEC traces of bc-PG of before and after recycling. | ||
Conclusions
Dual crosslinking polymer networks consisting of dynamic covalent bonds as a function of self-healing polymer networks and covalent bonds as a function of crosslinking polymer networks were formed by a simple photocuring reaction involving topology-controlled PGBs carrying 4-vinylphenylboronic ester and photo-radical initiator. The storage modulus (G′), loss modulus (G′′), tensile strength, Young's modulus, indentation hardness (HIT), and indentation modulus (EIT) of the obtained cPGBs were evaluated. Due to the intramolecular crosslinking pathway of hb-PGB, bc-cPGB with its cyclic structure, exhibited superior physical properties to hb-cPGB with its hyperbranched structure. The bc-cPGB and hb-cPGB exhibited self-healing performance by dynamic exchange of B–O bond, with the bc-cPGB exhibiting faster self-healing due to a lower Ea value than the hb-cPGB, because of enhanced chain mobility resulting from its unique cyclic structure. In contrast, lin-cPGB exhibited no self-healing properties due to its dense crosslinking with boronic acid and low chain mobility. These results suggest that topology control of polymer materials is essential for the development of highly tunable, advanced functional materials.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. S. C.: experimentation, analysis, and writing; J. K. and E. S.: experimentation and analysis; H. J., J.-E. J., Y. I. P., and J. C. K.: analysis; D. W. L. and B.-S. K: conceptualization and supervision; S.-H. L.: conceptualization, supervision, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Ministry of Trade, Industry & Energy (MOTIE, Korea) under the Industrial Technology Innovation Program (no. 20011123) and the Korea Research Institute of Chemical Technology (KRICT) (no. KS2041-00). This work was also supported by the National Research Foundation of Korea (NRF-2022R1F1A1074416 and NRF-2021R1A2C3004978).References
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- B. S. Sumerlin, Science, 2018, 362, 150–151 CrossRef CAS PubMed.
- A. J. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- Y. Fang, J. Li, X. Du, Z. Du, X. Cheng and H. Wang, Polymer, 2018, 158, 166–175 CrossRef CAS.
- M. S. de Luna, V. Marturano, M. Manganelli, C. Santillo, V. Ambrogi, G. Filippone and P. Cerruti, J. Colloid Interface Sci., 2020, 568, 16–24 CrossRef PubMed.
- Y. Hong, J.-M. Kim, H. Jung, K. Park, J. Hong, S.-H. Choi and B.-S. Kim, Biomacromolecules, 2020, 21, 4913–4922 CrossRef CAS PubMed.
- D. H. Son, H. E. Bae, M. J. Bae, S.-H. Lee, I. W. Cheong, Y. I. Park, J.-E. Jeong and J. C. Kim, ACS Appl. Polym. Mater, 2022, 4, 3802–3810 CrossRef CAS.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- J. Baek, S. Kim, I. Son, S.-H. Choi and B.-S. Kim, ACS Macro Lett., 2021, 10, 1080–1087 CrossRef CAS PubMed.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- G. Y. Kim, S. Sung, M. P. Kim, S. C. Kim, S.-H. Lee, Y. I. Park, S. M. Noh, I. W. Cheong and J. C. Kim, Appl. Surf. Sci., 2020, 505, 144546 CrossRef CAS.
- M. Röttger, T. Domenech, R. van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 256, 62–65 CrossRef PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- Z.-H. Zhao, D.-P. Wang, J.-L. Zuo and C.-H. Li, ACS Mater. Lett., 2021, 3, 1328–1338 CrossRef CAS.
- N. Fujita, S. Shinkai and T. D. James, Chem. – Asian J., 2008, 3, 1076–1091 CrossRef CAS PubMed.
- S. Cho, S. Y. Hwang, D. X. Oh and J. Park, J. Mater. Chem. A, 2021, 9, 14630–14655 RSC.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- C. C. Deng, W. L. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef CAS PubMed.
- S. Tajbakhsh, F. Hajiali, K. Guinan and M. Marić, React. Funct. Polym., 2021, 158, 104794 CrossRef CAS.
- S. Cho, S. Y. Hwang, D. X. Oh and J. Park, J. Mater. Chem. A, 2021, 9, 14630 RSC.
- A. Zych, J. Tellers, L. Bertolacci, L. Ceseracciu, L. Marini, G. Mancini and A. Athanassiou, ACS Appl. Polym. Mater., 2021, 3, 1135–1144 CrossRef CAS.
- L. L. Robinson, J. L. Self, A. D. Fusi, M. W. Bates, J. Read de Alaniz, C. J. Hawker, C. M. Bates and C. S. Sample, ACS Macro Lett., 2021, 10, 857–863 CrossRef CAS PubMed.
- S. E. Kim, Y.-R. Lee, M. Kim, E. Seo, H.-J. Paik, J. C. Kim, J.-E. Jeong, Y. I. Park, B.-S. Kim and S.-H. Lee, Polym. Chem., 2022, 13, 1243–1252 RSC.
- L. Gao, J. Oh, Y. Tu, T. Chang and C. Y. Li, Polymer, 2019, 170, 198–203 CrossRef CAS.
- M. D. Hossain, J. C. Reid, D. Lu, Z. Jia, D. J. Searles and M. J. Monteiro, Biomacromolecules, 2018, 19, 616–625 CrossRef CAS PubMed.
- A. Khalyavina, L. Häußler and A. Lederer, Polymer, 2012, 53, 1049–1053 CrossRef CAS.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- A. Dworak, S. Slomkowski, T. Basinska, M. Gosecka, W. Walach and B. Trzebicka, Polimery, 2013, 58, 641–649 CrossRef CAS.
- Y. Segawa, T. Higashihara and M. Ueda, J. Am. Chem. Soc., 2010, 132, 11000–11001 CrossRef CAS PubMed.
- T. Higashihara, Y. Segawa, W. Sinananwanich and M. Ueda, Polym. J., 2012, 44, 14–29 CrossRef CAS.
- C. Schubert, M. Schömer, M. Steube, S. Decker, C. Friedrich and H. Frey, Macromol. Chem. Phys., 2018, 219, 1700376 CrossRef.
- S. Y. Lee, M. Kim, T. K. Won, S. H. Back, Y. Hong, B.-S. Kim and D. J. Ahn, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- S. E. Kim, H. J. Yang, S. Choi, E. Hwang, M. Kim, H.-J. Paik, J.-E. Jeong, Y. I. Park, J. C. Kim, B.-S. Kim and S.-H. Lee, Green Chem., 2022, 24, 251–258 RSC.
- J. Song, L. Palanikumar, Y. Choi, I. Kim, T.-y. Heo, E. Ahn, S.-H. Choi, E. Lee, Y. Shibasaki, J.-H. Ryu and B.-S. Kim, Polym. Chem., 2017, 8, 7119–7132 RSC.
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 CrossRef CAS PubMed.
- S. Sharifi, I. Asenjo-Sanz, J. A. Pomposo and A. Alegria, Macromolecules, 2022, 55, 3627–3636 CrossRef CAS PubMed.
- J. Yu, K. Li, L. Li, L. Liu, Z. Zhang, M. Guo, N. Zhou and X. Zhu, Polym. Chem., 2019, 10, 2872–2880 RSC.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 RSC.
- C. Kim, H. Ejima and N. Yoshie, J. Mater. Chem. A, 2018, 6, 19643–19652 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00025g |
| This journal is © The Royal Society of Chemistry 2023 |