
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers of intrinsic microporosity containing aryl-phthalimide moieties: synthesis, modeling, and membrane gas transport properties†
Fidel E.
Rodríguez-González
 a,
Cenit
Soto
b,
Laura
Palacio
b,
Ana L.
Montero-Alejo
*c,
Néstor
Escalona
de,
Eduardo
Schott
f,
Bibiana
Comesaña-Gándara
*g,
Claudio A.
Terraza
*ah and
Alain
Tundidor-Camba
*ah
a,
Cenit
Soto
b,
Laura
Palacio
b,
Ana L.
Montero-Alejo
*c,
Néstor
Escalona
de,
Eduardo
Schott
f,
Bibiana
Comesaña-Gándara
*g,
Claudio A.
Terraza
*ah and
Alain
Tundidor-Camba
*ah
aResearch Laboratory for Organic Polymers (RLOP), Department of Organic Chemistry, Pontificia Universidad Católica de Chile, P.O. Box 306, Post 22, Santiago, Chile
bInstitute of Sustainable Processes, University of Valladolid, Dr Mergelina, s/n, 47011, Valladolid, Spain
cDepartamento de Física, Facultad de Ciencias Naturales, Matemática y del Medio Ambiente (FCNMM), Universidad Tecnológica Metropolitana, José Pedro Alessandri 1242, Ñuñoa, Santiago, Chile. E-mail: amonteroa@utem.cl
dDepartamento de Ingeniería Química y Bioprocesos Escuela de Ingeniería Pontificia Universidad Católica de Chile, Av. Vicuña Mackenna 4860, Macul, Santiago, Chile
eANID – Millennium Science Initiative Program-Millennium Nuclei on Catalytic Process towards Sustainable Chemistry (CSC), Chile
fDepartamento de Química Inorgánica, Facultad de Química y de Farmacia, Centro de Investigación en Nanotecnología y Materiales Avanzados CIEN-UC, Pontificia Universidad Católica de Chile, Santiago, Chile
gIU CINQUIMA, University of Valladolid, Paseo Belén 5, 47011, Valladolid, Spain. E-mail: bibiana.comesana@uva.es
hUC Energy Research Center, Pontificia Universidad Católica de Chile, Santiago, Chile. E-mail: cterraza@uc.cl; atundido@uc.cl
First published on 26th April 2023
Abstract
High-performance polymers for membrane gas separation require the careful design of the structure-porous relationship. In this work, five phthalimide-based polymers of intrinsic microporosity (PIMs) were obtained via the double nucleophilic aromatic substitution with the commercially available 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethylspirobisindane (TTSBI) monomer. The phthalimide monomers were synthesized considering different sizes and positions of the alkyl-substituents to evaluate their influence on the physical properties of the polymers and their potential use as gas separation membranes. Four polymers were soluble in the low-boiling solvents chloroform and tetrahydrofuran, facilitating the casting of self-standing films to evaluate their gas separation properties. The thermally stable membranes showed 5% weight lost between 537 °C and 549 °C. As powders, these four polymers showed apparent BET surface areas ranging from 434 to 661 m2 g−1. The experimental BET surface areas correlated with those obtained by molecular simulation models of the synthesized polymers. A linear function is proposed as a tool to predict, with a known uncertainty, the surface area values of this type of polymer from the corresponding computational models. As a trend, increasing the volume of the ortho-substituent in the aryl-phthalimide group increases the permeability of the membranes, reaching generally better performances than Matrimid® and close to those of PIM-1, considering their place on the Robeson diagrams of the O2/N2, CO2/CH4 and CO2/N2 gas pairs. Aging studies between 63 and 122 days showed a decrease in permeability, accompanied by the typical increase in selectivity that tends to move the data parallel to the upper Robeson limits.
Introduction
Microporous materials have attracted attention from both the academic and industrial world. There are microporous inorganic materials, including zeolites, activated carbons, or silica, as well as microporous inorganic–organic materials like metal–organic framework (MOFs)1 and microporous organic materials including hypercrosslinked polymers (HCPs),2 covalent triazine frameworks (CTFs),3 conjugated microporous polymers (CMPs),4 porous aromatic frameworks (PAFs),5 covalent organic frameworks (COFs)6 and polymers of intrinsic microporosity (PIMs).7–9 In general, microporous materials exhibit high surface areas and pore sizes comparable to a small molecule, showing potential applications in catalysis,10–13 water treatment,14 adsorbents for organic compounds,15 energy and gas storage16,17 and gas separation membrane.18–21PIMs are a class of solution-processable polymers, having the ability to form robust films. The solubility of these materials is due to their contorted structure, which facilitates the interaction between solvent and highly rigid chains. In addition, the rigidity and contortion of the molecular structure cause an inefficient packing of the chains, generating microporosity.22
From a synthetic point of view, the first PIM synthesized (PIM-1) was obtained from the double nucleophilic aromatic substitution reaction between a tetraol (5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethylspirobisindane (TTSBI) and 2,3,5,6-tetrafluoroterephthalonitrile (TFTPN) in the presence of potassium carbonate, N,N-dimethylformamide as a solvent at low temperature (60 °C).23 However, Guiver et al. prefer to perform the reaction at higher temperatures (120 °C).24PIM-1 showed excellent performance in the gas separation field for a wide range of commercial gas pairs.25
Other polymerization reactions could be used to prepare PIMs based on forming fused ring structures or free-rotating restricted fragments by steric hindrance. These include CANAL-type polymerization,26 Tröger's base formation,27 and catalyzed cross-coupling reactions.28 Nevertheless, polymerization using dibenzodioxins formation is the most popular and versatile procedure due to the number of monomers (biscatechol and tetrahalide derivatives) available that organic synthesis offers. In this sense, aromatic tetrahalide-containing monomers must have electro-withdrawing groups on strategic positions of the aromatic ring, favoring the success of the reaction. Some of these groups are nitrile, sulfone, phenazine, fluorine, trifluoromethyl, carbonyl, and phthalimide (Fig. 1).
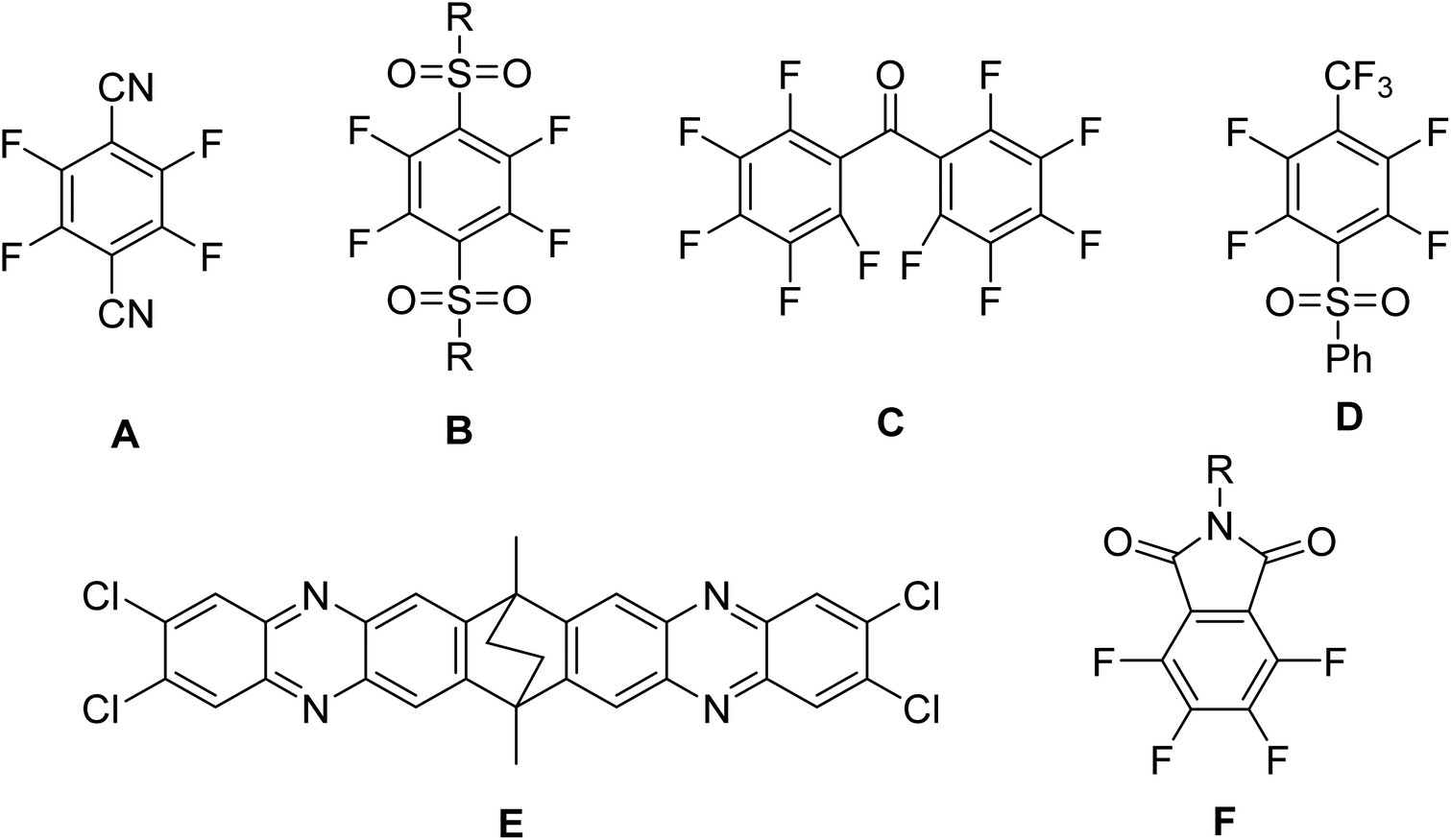 | ||
| Fig. 1 Aromatic tetrahalide-containing monomers bearing electro-withdrawing groups. | ||
Makhseed et al. prepared a series of phthalimide-based microporous polymers from the commercial TTBSI and several synthesized 3,4,5,6-tetrafluoro-N-R-phthalimides (structure F, Fig. 1), where the R substituent was phenyl, hexyl, 4-methoxyphenyl, 3,4,5-trimethoxyphenyl, 4-tert-butylphenyl, 2,6-diisopropylphenyl and 1-adamantyl.29 Those polymers provided surface areas between 595–889 m2 g−1 and were soluble in CHCl3, making them good candidates for gas adsorption and gas separation. Moreover, the authors prepared a transparent self-standing film obtained from 3,4,5,6-tetrafluoro-N-2,6-diisopropylphenylphthalimide, which could be tested as a gas separation membrane; however, they do not perform gas permeation measurements.
Due to the complexity of determining the three-dimensional structure of PIMs, computational chemistry simulations have been widely used to understand their molecular structures and to probe the pore morphology.10,30–44 Despite the challenge of creating amorphous models, it is a great advantage to predict relevant properties related to gas separation processes at a minimal cost. With the help of extensive exploration using molecular models, it has been possible to suggest adjustments to the structures of the monomeric units such that they confer greater rigidity to the polymer.30,42 Nowadays, macromolecular packing models can be obtained systematically and at a relatively low cost using the Polymatic simulated polymerization algorithm, followed by a compression/decompression scheme over the amorphous periodic unit cell.33,40,42–44 The models are then used for estimating properties such as surface area, the fractional free volume (FFV) and the distribution of cavity sizes.
Inspired by the Makhseed et al. work, we propose to deepen the understanding of the structure-microporosity relationship of the phthalimide-based polymers as the direct route to design better polymers for gas separation by membranes. We aim to answer whether gas transport depends on the size and position of the alkyl-substituent groups on the aryl-phthalimide. One question came to us: does gas transport depend on the size and position of the substituent groups on the phthalimide?
To respond to above question, we prepared three new 3,4,5,6-tetrafluoro-N-R-phthalimides, where the R substituent was 2,6-dimethylphenyl, 2,4,6-trimethylphenyl and 2,6-diethylphenyl (Fig. 2; IM2, IM3 and IM5, respectively), and synthesized two of the tetrafluorinated monomers (R: phenyl; 2,6-diisopropylphenyl) reported by Makhseed et al. (Fig. 2; IM1 and IM4, respectively). Each tetrafluorinated monomer was reacted with TTBSI to obtain five PIMs. Solubility, thermal properties, BET surface area, ability to form free-standing film, gas transport properties and physical aging were established and discussed.
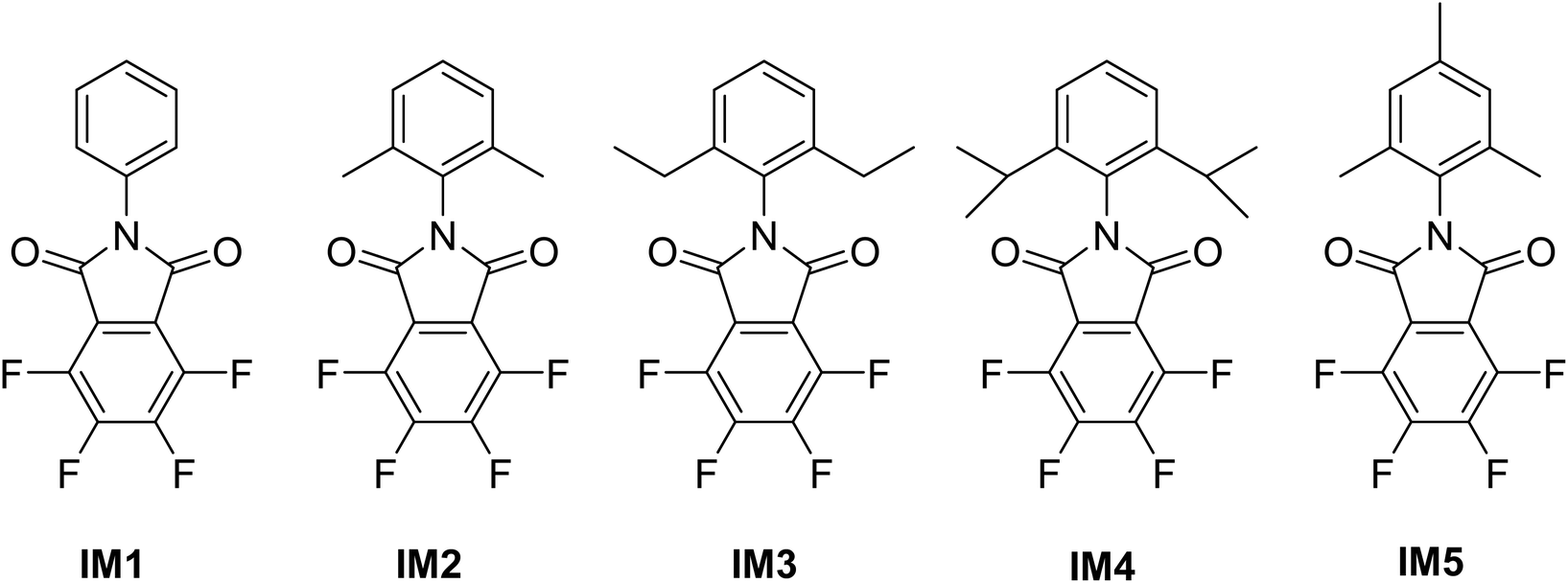 | ||
| Fig. 2 Tetrafluorophthalimide monomers synthesized for this study. | ||
We also took the advance of the Polymatic algorithm to simulate amorphous polymeric structures and made molecular models of the synthesized PIMs. Bulk density, surface area, and fractional free volume were calculated for the amorphous periodic cell models and the resulting data were compared to those obtained from the experiments.
Experimental
Instrumentation and measurement
FT-IR spectra were obtained using a PerkinElmer Spectrum-Two spectrometer (PerkinElmer Inc., Waltham, MA, USA) with a coupled Universal Attenuated Total Reflection (UATR) unit over the range of 4000–450 cm−1. 1H NMR (400 MHz) and 13C NMR (101 MHz) spectra were recorded on a Varian AV Agilent (Agilent Tech., Santa Clara, CA, USA) instrument operated at 400 MHz for 1H and 75 MHz for 13C or a Bruker Avance III HD (Bruker Corporation, Karlsruhe, Germany) spectrometer using DMSO-d6 or CDCl3 as solvents. Viscosimetric measurements at single point (inherent viscosity) were made in a Desreux–Bischoff type viscosimeter at 30 °C (c = 0.5 g dL−1) in chloroform. An average of five measurements was reported. A Fisons EA 1108-CHNS-O equipment (Thermo Scientific, Waltham/MA, USA) was used to make the elemental analysis. Thermogravimetric analysis (TGA) was performed to powder samples using a Shimadzu TGA-50 (Columbia, United States) instrument under a nitrogen atmosphere with a heating rate of 10 °C min−1 from room temperature to 850 °C. Wide-angle X-ray diffraction (WAXD) was conducted to powder samples on a Bruker D8 ADVANCE A25 advanced diffractometer equipped with a Goebel mirror and using CuKα radiation (λ = 1.542 Å) as the radiation source and a LYNXEYE detector (Bruker, Billerica, MA, USA). The diffractograms were recorded in the 2θ range of 5° to 40°, with a measurement time of 0.5 s per each step of 0.02°. The average d-spacing was calculated using Bragg's Law equation: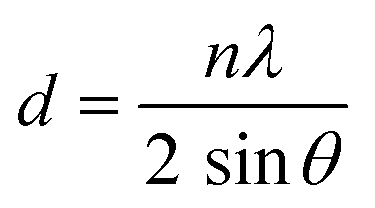 | (1) |
Gas permeation measurements
Uniform film samples with a thickness in the range of 75–130 micrometres were affixed on brass drilled discs using epoxy adhesive and protected with fiberglass filter paper. Pure gas permeability coefficients were determined using a constant volume/variable pressure apparatus based on the “time lag” method, at 35 °C and an upstream pressure of 3 bar, as described elsewhere.46 Before starting the permeability tests, the membrane samples were left overnight under a vacuum inside the permeation cell. First, helium permeability measurements were carried out at three different pressures (1, 2 and 3 bar) to verify the absence of pinholes in the membrane before performing the measurements with the following gases: N2, O2, CH4 and CO2. All membranes were stored under ambient conditions during the aging process and thoroughly degassed before measuring them again. The gas permeability coefficient (P) was calculated from the increase in the permeate pressure (pt) as a function of time (t) in the pseudo-steady state, according to the expression: | (2) |
thus,
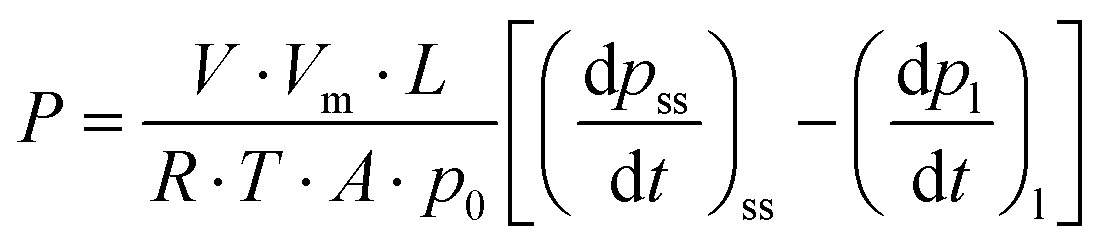 | (3) |
 | (4) |
According to the solution-diffusion model, the solubility coefficient (S) was calculated from the expression:
| P = D × S | (5) |
Then, the ideal selectivity α(i,j) of a membrane for the separation of a mixture of two gases was calculated as the ratio of the membrane permeabilities to each pure gas:
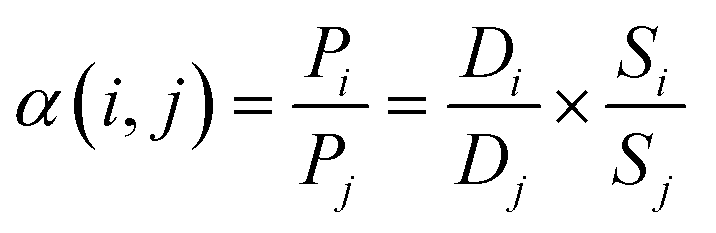 | (6) |
Computational method
According to the experimentally evaluated polymers, we set out to obtain representative models of the PIM-SBI-IM(2–5) polymers, including the model of the PIM-1 as a reference. The polymerization of the respective repeating units, including all possible stereoisomers, was performed with the Polymatic algorithm. To sample the structures, eleven independent models of each polymer were generated by randomly packing forty-eight repeating units in a cubic periodic box, whose side was defined to provide an initial packing density of 0.3 g cm−3. According to previous reports, this initial density was required to obtain a higher percentage of polymerization.40 Within the simulation box, four configurations with different stereochemistry were considered for PIM-SBI-IM(2–5) models, twelve repeating units of each one. In the PIM-1, twenty-four repeating units of each of the two configurations with different stereochemistry that are present in the polymer were packed. Forces on atoms were calculated by considering the general AMBER (GAFF)47 and the Transferable Potentials for Phase Equilibria (TraPPE-UA) force fields48–53 to describe bonding and non-bonding interactions, respectively. Atomic charges were estimated from the electrostatic potential using a grid-based method for a model with two units of each monomer optimized at the HF/6-31G* level of theory by the Gaussian package.54 For PIM-1 models, previously reported atomic charges were used.40During the simulated polymerization, bonds were formed between the reactive atoms at a distance less than or equal to 6 Å. The reactive sites were the carbon atoms initially bonded to the fluorine atoms and the oxygen of the hydroxyl groups. In addition, geometric restrictions were used for forming the bonds, as proposed by Polymatic for this type of simulated reaction.40 The structure was minimized after a new bond was formed, and for every five new bonds, intermediate molecular dynamics (MD) steps were performed. MD steps were performed with the LAMMPS package,55 at 1000 K temperature, during 10 ps using 1 fs of time step. The Ewald sum was used to calculate the long-range electrostatic interactions, described by the Coulomb potential. The Lennard–Jones (LJ) potential was used to represent the short-range van der Waals interactions. The cut-off distance for the LJ interactions and the real part of the Ewald sum was set to 15 Å. The repeating units used for PIM-1 and the PIM-SBI-IM(2–5) polymers are shown in ESI Fig. 1–5,† and the atom types and the partial charge of the atoms are shown in ESI Tables 1–5.†
Through 21 compression/decompression steps performed by MD, each polymer was obtained with a density that simulates the experimental value. This is an established protocol for generating structures of microporous polymers. In this protocol, MD steps alternate between canonical ensemble (NVT) and isobaric-isothermal ensemble (NPT), using a maximum temperature and pressure of 1000 K and 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bar to reach the balanced structure at a final temperature and pressure of 300 K and 1 bar, respectively. Constant temperature and pressure were maintained using a Nosé–Hoover thermostat and barostat during MD steps as implemented in LAMMPS.40
000 bar to reach the balanced structure at a final temperature and pressure of 300 K and 1 bar, respectively. Constant temperature and pressure were maintained using a Nosé–Hoover thermostat and barostat during MD steps as implemented in LAMMPS.40
The five optimized models of the polymers with the highest density were used to analyze the structure and estimate properties. The fractional free volume and the surface area were calculated using the “Atom Volumes and Surfaces” tool of the Materials Studio computational package.54 The procedure is based on measuring the Connolly surface using a spherical probe, where the probe is imaginarily rolled over the entire structure. Guided by the experimental measurement of the BET surface area, the probe for the calculation of this magnitude was defined with a radius of 1.82 Å, corresponding to the kinetic radius for N2, and empirically, the van der Waals radii of the atoms in the structure were scaled by a factor of 1.35 (vdW scale factor).56 A 1.65 Å radio probe was used to calculate the free volume, according to a previous report.57 The fractional free volume was estimated by the ratio between the free volume and the volume of the simulation cell. In addition, the structural analysis of polymers was supplemented by computing the radial distribution function between pairs of atoms using the Visual Molecular Dynamics (VMD) program.58
Reagents, intermediates, monomers and solvents
Aniline (99%); 2,6-dimethylaniline (98%); 2,6-diethylaniline (98%) 2,6-diisopropylaniline (97%); 2,4,6-trimethylaniline (98%) and 3,4,5,6-tetrafluorophthalic acid (98%) were obtained from AK Scientific, Inc. (San Francisco, USA). Anhydrous N,N-dimethylformamide (DMF, 99.8%); anhydrous potassium carbonate (99%); anhydrous tetrahydrofuran (THF, 99.9%); chloroform (for analysis); isopropanol (for analysis); methanol (for analysis); 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethylspirobisindane (TTSBI, 96%) and thionyl chloride (99.5%) were purchased from Merck-Sigma Aldrich Chile. TTSBI was washed in chloroform, recrystallized in isopropanol and dried in a vacuum oven at 80 °C for 24 h before use. All other reagents and solvents purchased were used without further purification. 3,4,5,6-Tetrafluorophatalic anhydride was synthesized according to a published procedure.59 Details of the synthesis are given in the ESI.†Synthesis of monomers and polymers: general procedure
Membrane preparation
Thin flat films were prepared from a 2–4% w/v solution of the polymers in THF (typically 500 mg in 20 mL of solvent). Each solution was filtered through a 5 μm nylon syringe filter and poured into a 7 cm diameter Petri dish. The membranes were formed by slow evaporation of the solvent (usually for four days) at ambient conditions. The dense membranes were peeled off and immersed in methanol for 24 h just before measuring the gas transport properties. The thickness of the membranes ranged from 75–130 μm.Results and discussion
Synthesis and characterization of tetrafluorophthalimide monomers IM(1–5)
Monomers IM(1–5) were synthesized by modifying the procedure reported by Makhseed et al.29 First, the commercial 3,4,5,6-tetrafluorophthalic acid was dehydrated to 3,4,5,6-tetrafluorophatalic anhydride by using thionyl chloride. Then, each aniline derivative was put to react with a slight excess of 3,4,5,6-tetrafluorophthalic anhydride in glacial acetic acid resulting in a high purity grade of each phthalimide derivative. In this type of reaction, it is usual to use an excess of amine; however, this reagent was not removed quantitatively during the isolation process, even after many washes with acidic water. When an excess of the anhydride was used, the presence of amine was not detected by NMR, while the anhydride was removed by washing with acidic water and then with water, transforming it into carboxylic acid, which was soluble in water at room temperature. Finally, the purity of each monomer was verified by elemental analysis.The successful reaction of amines A(1–5) to tetrafluorophthalimide monomers IM(1–5) was confirmed by IR and NMR spectroscopies. In the IR spectra, the characteristic imide group absorption bands were observed in the range of 1790 cm−1 and 1710 cm−1 due to asymmetrical and symmetrical carbonyl stretching vibrations, respectively. Additionally, the successful conversion was corroborated by the absence of any broad band in the 3600–3000 cm−1 zone. In this region, the N–H and O–H stretching vibrations associated to amic acid structures could be observed if the imide group was not obtained. The chemical structures of the monomers, as well as the complete conversion from amine to imide, were also consistently demonstrated by NMR spectroscopy. Details of the spectroscopic characterization, including the complete assignment of all signals in the 1H NMR spectra, are given in ESI Fig. 6.†
Synthesis and characterization of polymers
Polymers, labeled as PIM-SBI-IM1, PIM-SBI-IM2, PIM-SBI-IM3, PIM-SBI-IM4, and PIM-SBI-IM5, were synthesized from a combination of tetrafluorophthalimide monomers with commercial bis-catechol TTSBI, according to a procedure reported by Neil Mckeown et al.23 (Scheme 1). Briefly, both monomers reacted in the presence of anhydrous potassium carbonate and DMF as a solvent at 65 °C for 72 h under a nitrogen atmosphere. Chain growth occurs via double nucleophile aromatic substitution forming dioxin rings. | ||
| Scheme 1 Synthesis of tetrafluorophthalimide-containing monomers IM(1–5) and microporous polymers PIM-SBI-IM(1–5). Reagents and conditions: (i) Glacial acetic acid, reflux 16 h. (ii) Anhydrous DMF, anhydrous K2CO3, 65 °C, N2 (g), 72 h. | ||
The chemical structure of the polymers was firstly established by IR and 1H NMR spectroscopies. The polymers all retained their two characteristic bands related to the asymmetrical and symmetrical carbonyl stretching vibrations of the imide groups around 1770 cm−1 and 1710 cm−1, respectively. Additionally, the absence of the O–H stretching absorption bands in the range of 3600–3000 cm−1 provides clear evidence that the reaction of the tetrafluorophtalimides with TTSBI was significantly efficient. PIM-SBI-IM1 demonstrated poor solubility in all common deuterated solvents tested, preventing characterization by solution NMR; therefore, it was only spectroscopically characterized by IR technique.
The other four polymers (PIM-SBI-IM2, PIM-SBI-IM3, PIM-SBI-IM4 and PIM-SBI-IM5) were soluble in deuterated chloroform enabling characterization by solution 1H NMR. The precise assignment of the signals in the 1H NMR spectra was carried out with the help of the 1H NMR spectra of corresponding monomers. The broad aromatic and aliphatic signals in the 1H NMR spectra indicate the formation of long polymeric chains, where the chemical shifts of the protons occupying the same position in the repeating unit may have small differences due to magnetic anisotropy effects, causing a broadening of the signals. Details of the spectroscopic characterization are given in ESI Fig. 7 and 8.†
PIM-SBI-IM2, PIM-SBI-IM3, PIM-SBI-IM4 and PIM-SBI-IM5 were soluble in chloroform and THF, while PIM-SBI-IM1 was not soluble in any commonly used solvent, not even in quinoline, which is often used as a last resort to prepare PIMs films. As a result, it was not possible obtaining a film of PIM-SBI-IM1, while the other four polymers were able to form self-standing films by slow evaporation of THF. The PIM-SBI-IM(2–5) solutions exhibited high inherent viscosity values (1.21–1.41 dL g−1), which is indicative of high molecular weights. ESI Fig. 9† shows a robust self-supporting film indicating that molecular weights of these polymers were adequate to obtain good quality films.
Properties of polymers
Thermogravimetric analysis (Fig. 3) proved the complete removal of residual solvents and humidity from the membranes. Mass decreases were not detected for any polymer below 450 °C, and the 5% weight loss due to thermal decomposition was in the range of 537–549 °C. In addition, all four polymers were more resistant to thermal degradation than PIM-1, as shown in Table 1. This higher thermal stability is due probably to the high content of fused aromatic rings and the presence of the imide group in the repeating units of the polymers.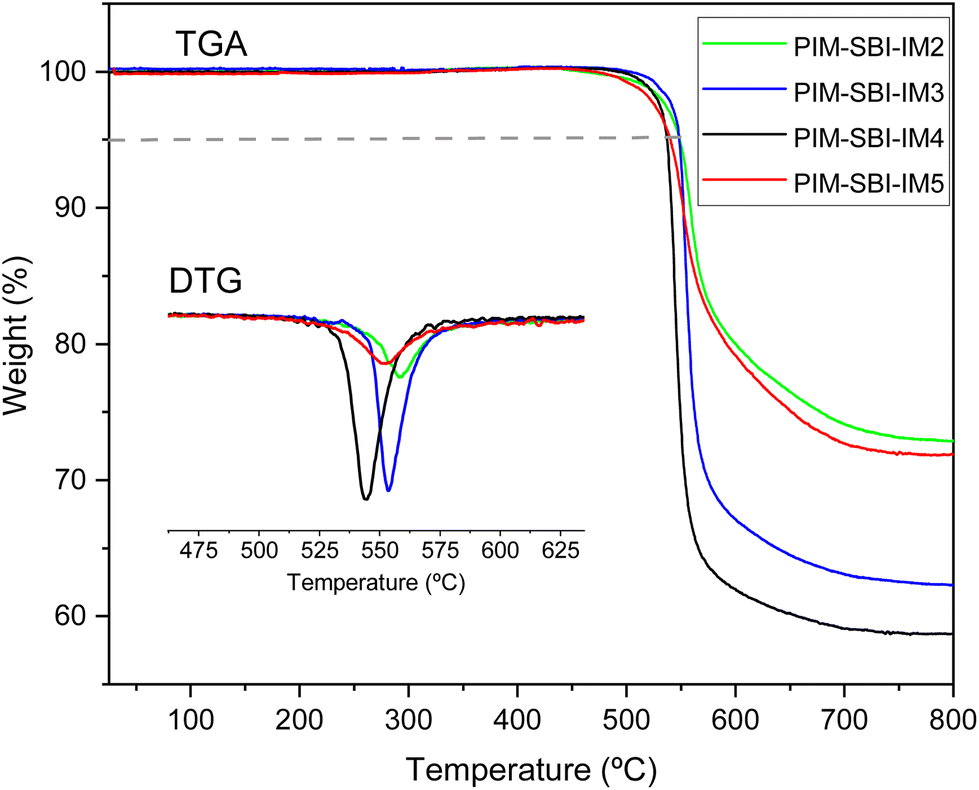 | ||
| Fig. 3 Thermogravimetric analysis of the PIM-SBI-IM(2–5) membranes. | ||
| Polymer | Experimental | Computational simulation | |||||
|---|---|---|---|---|---|---|---|
| SABET (m2 g−1) | Densitya (g cm−3) | T d,5% (°C) | Amorphous unit cellb | SAsim (m2 g−1) | Density (g cm−3) | FFV | |
| a Density of the PIM-SBI-IM(2–5) membranes were calculated geometrically from the thickness, area and mass of a piece of each film. b Amorphous unit cell of each polymer shows the outer (blue) and inner (gray) Connolly surfaces of the cavities. | |||||||
| PIM-SBI-IM2 | 471 | 1.202 | 549 |

|
721 ± 89 | 0.963 ± 0.017 | 0.253 ± 0.015 |
| PIM-SBI-IM3 | 434 | 1.166 | 549 |

|
767 ± 64 | 0.935 ± 0.009 | 0.264 ± 0.012 |
| PIM-SBI-IM4 | 661 | 1.056 | 537 |

|
935 ± 113 | 0.898 ± 0.022 | 0.289 ± 0.018 |
| PIM-SBI-IM5 | 495 | 1.158 | 541 |

|
820 ± 65 | 0.935 ± 0.005 | 0.271 ± 0.008 |
| PIM-1 | 760 [ref. 61] | 1.114 [ref. 62] | 493 [ref. 63] |

|
967 ± 83 | 0.936 ± 0.011 | 0.299 ± 0.015 |
Wide angle X-ray diffraction (WAXD) analysis (Fig. 4) evidenced the amorphous structure of these materials since very broad signals were observed in the diffractograms. Three well-defined peaks were observed, corresponding to different interchain distances estimated from the Bragg's Law. However, the WAXD results do not allow to distinguish structural differences between polymers. The smallest distance, at approximately 5 Å, is due to the more compactly packed regions of the structure. The signal near to 7 Å can be attributed to regions with inefficient packing of the polymeric chains, due to their rigid and contorted structure and bulky side groups. Finally, the signal near to 10 Å should be mainly related to spaces generated between the spiro-centers.60 This last idea is in agreement with the spirocenters-spirocenters radial distribution functions (ESI Fig. 15†) obtained for the computational polymeric models of PIM-SBI-IM(2–5).
 | ||
| Fig. 4 Wide angle X-ray diffraction (WAXD) analysis of the PIM-SBI-IM(2–5) membranes. | ||
The nitrogen adsorption/desorption isotherms at 77 K for all polymers in powder form was carried out. (Fig. 5A). The polymers showed the characteristic shape of microporous materials with an isotherm type I. Apparent BET surface areas were calculated, ranging from 434 to 661 m2 g−1 (Table 1). PIM-SBI-IM4 showed the highest surface area. Apparently, the two isopropyl groups close to imide ring increase the fractional free volume in the solid state, which may result in a less efficient packing of the polymer. Pore size distribution was calculated according to the Horvath and Kawazoe (HK) model (Fig. 5B). The values were around 0.7 nm indicating a great contribution of ultramicropores.
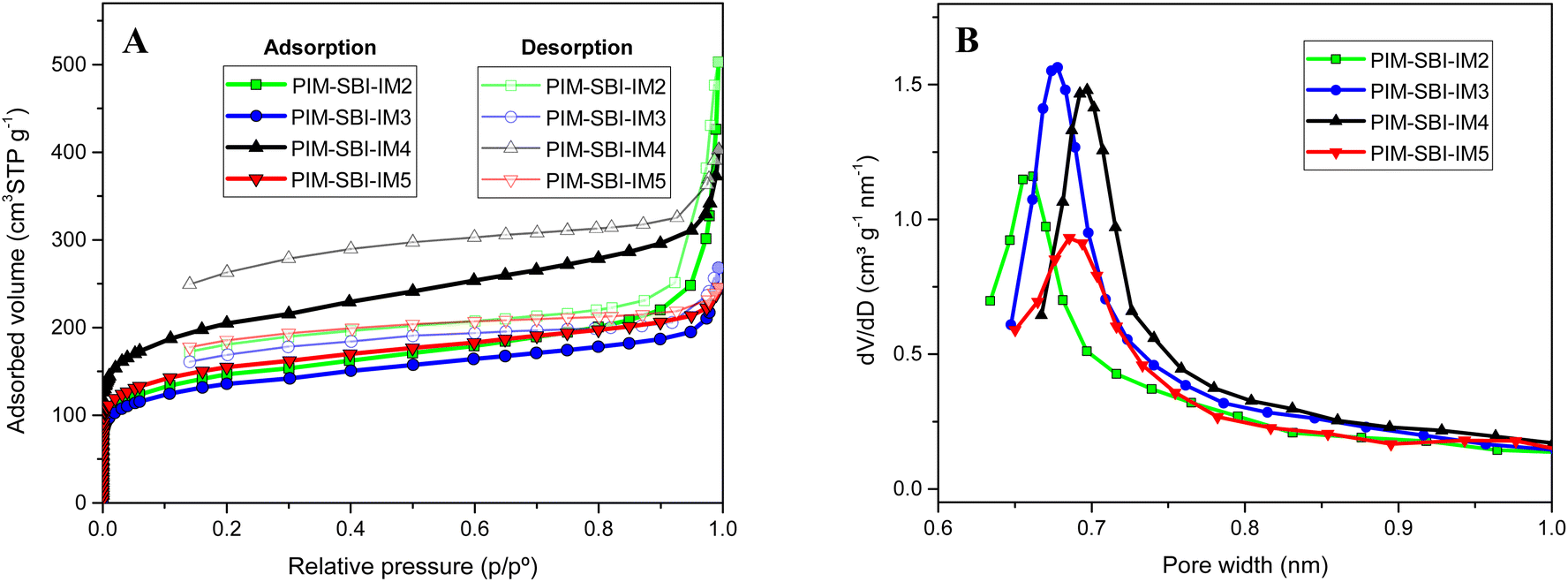 | ||
| Fig. 5 (A) Nitrogen adsorption and desorption isotherms and (B) pore size distribution of PIM-SBI-IM(2–5) polymers, calculated according to the Horvath and Kawazoe (HK) method. | ||
Computational simulation of polymers
The distributions of the dihedral angles involving the spiro-centers and those associated with the planarity of the dibenzodioxin rings in PIM-1 and PIM-SBI-IM(2–5) proved the final models fulfilled the standard geometrical features for this type of structure (ESI Fig. 10†). On validating the models, we set out to perform the simulation of the properties. The simulated properties of the PIM-SBI-IM(2–5) and PIM-1 are reported in Table 1, along with the corresponding experimental results for better comparison. Table 1 also shows one of the amorphous unit cells representing each polymer, where the inner and outer Connolly surfaces of the cavities are displayed in gray and blue, respectively, as geometrically were delineated by a cylindrical probe with a radius of 1.65 Å.The simulated density of PIM-1 corresponded with the value reported in similar simulations.40 Likewise, the simulated packed of PIM-SBI-IM(2–5) tended to have a lower density than the corresponding prepared polymeric membranes, and consequently, the simulated surface area (SAsim) showed the opposite trend, as does the calculated fractional free volume (FFV). Still, experimental and simulated surface area values evidenced a clear correlation with a regression coefficient (R2) of 0.89 and a standard deviation (σ) of 46 m2 g−1 (Fig. 6). Based on these results, we propose the linear regression function obtained for estimating the surface area from polymeric models with new structural modifications. The data showed that polymers containing aryl-phthalimide groups can achieve properties similar to PIM-1. Thus, PIM-SBI-IM4, whose fluorinated monomer contains the largest groups in the ortho position of the aryl-phthalimide, had the largest surface area within the series. In contrast, PIM-SBI-IM2 and PIM-SBI-IM3 showed the lowest surface area, indicating the possibility of generating more compact structures. Furthermore, by including a methyl group in the aryl-phthalimide para position (PIM-SBI-IM5), the surface area tended to increase even with small volume substituents in the ortho position.
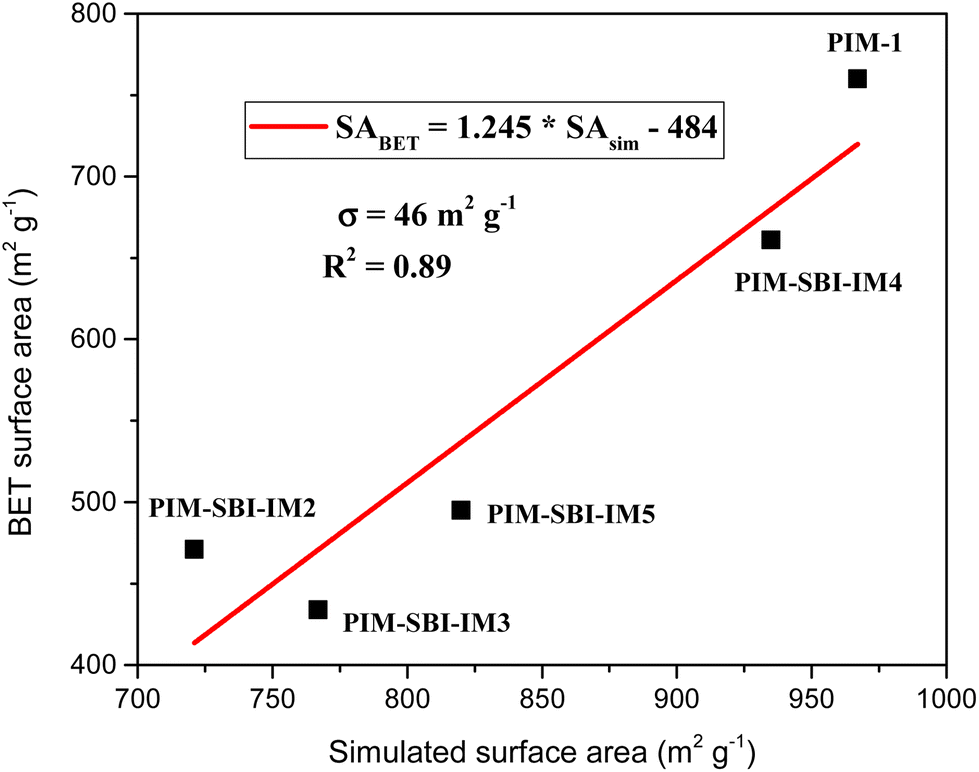 | ||
| Fig. 6 Linear regression of experimental (SABET) and simulated (SAsim) surface area from the PIM-SBI-IM(2–5) and PIM-1. | ||
Gas permeation properties
The aging time expressed in days is indicated in parenthesis. PIM-1 and Matrimid® are reported for comparison (1 Barrer = 10−10 cm3 (STP) cm s−1 cm s−2 cmHg−1).Table 2 shows the results of N2, O2, CH4, and CO2 permeability measurements for PIM-SBI-IM2, PIM-SBI-IM3, PIM-SBI-IM4 and PIM-SBI-IM5 freshly methanol treated and aged membranes, and ideal selectivity calculations for the corresponding gas pairs of most significant interest. The table also shows values reported for Matrimid® and PIM-1 since these polymers are frequently used as a reference in studies focused on membrane gas separation.
| Polymer | Permeability Pi [Barrer] | Ideal selectivity α(Pi/Pj) | |||||
|---|---|---|---|---|---|---|---|
| N2 | O2 | CH4 | CO2 | O2/N2 | CO2/CH4 | CO2/N2 | |
| PIM-SBI-IM2 (122 days) | 81.3 (8.1) | 300 (42.0) | 122 (11.1) | 1362 (174) | 3.69 (5.16) | 11.2 (15.7) | 16.8 (21.3) |
| PIM-SBI-IM3 (121 days) | 80.6 (53.4) | 279 (199) | 140 (87.6) | 1393 (941) | 3.45 (3.73) | 9.95 (10.8) | 17.3 (17.6) |
| PIM-SBI-IM4 (63 days) | 309 (259) | 954 (867) | 536 (413) | 4705 (4204) | 3.09 (3.35) | 8.78 (10.2) | 15.2 (16.2) |
| PIM-SBI-IM5 (92 days) | 133 (79.6) | 446 (279) | 200 (122) | 2165 (1330) | 3.36 (3.51) | 10.8 (10.9) | 16.3 (16.7) |
| Matrimid® [ref. 64] | 0.320 | 2.10 | 0.280 | 10.0 | 6.60 | 35.7 | 31.3 |
| PIM-1 [ref. 65] | 238 | 786 | 360 | 3496 | 3.30 | 9.71 | 14.7 |
The diffusion and solubility coefficients calculated from the time-lag measurements are reported in ESI Table 6.† All freshly methanol treated and aged membranes presented in this study were significantly more permeable to all the tested gases than Matrimid®. The data plotted on the Robeson diagrams (Fig. 7) show that PIM-SBI-IM2, PIM-SBI-IM4 and PIM-SBI-IM5 exceed the 1991 upper bound for O2/N2 separation, while only PIM-SBI-IM4 exceeds the 1991 upper bound for CO2/CH4 gas pair. On the other hand, the performance of all membranes is below the 2008 upper bound for CO2/N2 separation. The fresh PIM-SBI-IM4 membrane showed the highest permeability for all gases tested, which is in accordance with the results obtained from the BET measurements and computational calculations of surface area and FFV of the polymers (Table 1). According to the permeability/selectivity trade-off, this polymer showed the lowest selectivity for all gas pairs. However, the fresh PIM-SBI-IM4 membrane exhibited the best performance for gas separation within the freshly methanol treated membranes series. The results were close to those showed by PIM-1 for the O2/N2 and CO2/CH4 gas pairs, clearly surpassing it for the CO2/N2 pair. Comparing the results reported in Tables 1 and 2, it is evident that generally bulkier alkyl groups in the ortho positions of the aryl-phthalimide increase the surface area and FFV values of the polymers, resulting in higher values of gas permeability.
 | ||
Fig. 7 Robeson diagrams for the O2/N2, CO2/CH4 and CO2/N2 gas pairs, showing the position of the data for fresh films of PIM-SBI-IM2 ( ), PIM-SBI-IM3 ( ), PIM-SBI-IM3 ( ), PIM-SBI-IM4(■), PIM-SBI-IM5 ( ), PIM-SBI-IM4(■), PIM-SBI-IM5 ( ) and PIM-1 ( ) and PIM-1 ( ); and aged films of PIM-SBI-IM2(122 days, ); and aged films of PIM-SBI-IM2(122 days,  ), PIM-SBI-IM3 (121 days, ), PIM-SBI-IM3 (121 days,  ), PIM-SBI-IM4 (63 days, ), PIM-SBI-IM4 (63 days,  ), PIM-SBI-IM5 (92 days, ), PIM-SBI-IM5 (92 days,  ). ). | ||
The PIM-SBI-IM(2–5) membranes were aged for a short time ranging from 63 to 122 days. Despite the difference in aging time, it is possible to analyze these data for trends, considering that the largest conformational changes in PIMs occur during the first few months of aging.65–67 As is often the case for PIMs, the gas permeability of all PIM-SBI-IM(2–5) decreased due to films aging and, in concordance with the permeability/selectivity trade-off, the selectivity tended to increase (Fig. 7). In particular, PIM-SBI-IM4 demonstrated the lowest physical aging within the series, even improving its gas separation performance for all considered gas pairs over time. The increase in selectivity of PIM-SBI-IM4 membrane, for the separation of the three gas pairs (O2/N2, CO2/CH4 and CO2/N2), accompanied by a less drastic decrease in permeability with respect to the other membranes, suggests that the rearrangement of its polymeric chains occurs such that the interconnection regions between the free volume elements are tightened without a substantial reduction of the total FFV of the polymer. In contrast, the decrease of FFV showing by PIM-SBI-IM2, PIM-SBI-IM3 and PIM-SBI-IM5 due to aging is apparently not accompanied by a significant tightening of the interconnection sites between the free volume elements, resulting in very poor improvements of the selectivity. The latter is also in agreement with the diffusion and solubility data shown in ESI Tables 6 and 7.† In all cases, the results indicate that the decrease in permeability upon aging is due to a decrease in the corresponding diffusion coefficients, while the increase in selectivity is essentially due to an increase in diffusivity-selectivity, since the corresponding solubility coefficients and solubility-selectivity almost do not change upon aging.
Conclusions
In conclusion, five microporous polymers for membrane gas separation were obtained via the double nucleophilic aromatic substitution from the commercially available TTSBI monomer and five synthesized 3,4,5,6-tetrafluoro-N-R-phthalimides, three of which are reported for the first time. Four of the polymers formed self-standing films from which measurements of N2, O2, CH4 and CO2 permeability data were carried out. They exhibited excellent thermal stability with 5% weight loss values below 450 °C, and relatively high BET surface areas (434–661 m2 g−1) that are comparable to those of PIM-1. Also, wide angle X-ray diffraction measurements showed the amorphous and microporous nature of the four prepared membranes. Computational models of the studied polymers were obtained, from which it was possible to predict morphological properties such as surface area and free volume fraction, and then compare them with the experimental values. Additionally, a linear function was proposed as a tool to predict the surface area values of future PIMs from the corresponding computational models. In relation to the gas permeation properties, it was found that the larger the alkyl substituent in the ortho positions of the aryl-phthalimide fragments, gas permeability of the polymeric membranes tends to be higher. On the other hand, a substitution at the para position of aryl-phthalimide moieties also seems to be favorable for achieving higher permeability values.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was funded by the Agencia Nacional de Investigación y Desarrollo (ANID) through the Fondecyt project number 1190772 and the Ph.D. fellowship number 21180253. In addition, this work was also supported by Grant PROYEMER-2021-05 funded by the University of Valladolid (Spain) and Grant TED2021-131170A-I00 funded by MCIN/AEI/10.13039/501100011033 and by “European Union NextGenerationEU/PRTR”. Fidel. E. Rodríguez-González thanks to Professor Ángel Lozano from ICTP-CSIC, Spain, for accepting him in the research internship. L. Palacio would like to thank the “Ministerio de Ciencia e Innovación” for the financial support through the research project PID2019-109403RB-C21/AEI/10.13039/501100011033.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- K. Konstas, J. W. Taylor, A. W. Thornton, C. M. Doherty, W. X. Lim, T. J. Bastow, D. F. Kennedy, C. D. Wood, B. J. Cox, J. M. Hill, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2012, 51, 6639–6642 CrossRef CAS PubMed.
- A. P. Cote, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- C. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed.
- Q. Sun, Z. Dai, X. Liu, N. Sheng, F. Deng, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204–5209 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- F. E. Rodríguez-González, V. Niebla, M. V. Velázquez-Tundidor, L. H. Tagle, R. Martin-Trasanco, D. Coll, P. A. Ortiz, N. Escalona, E. Pérez, I. A. Jessop, C. A. Terraza and A. Tundidor-Camba, React. Funct. Polym., 2021, 167, 104998 CrossRef.
- S. Bhowmik, R. G. Jadhav and A. K. Das, J. Phys. Chem. C, 2018, 122, 274–284 CrossRef CAS.
- E. Al-Hetlani, M. O. Amin, C. G. Bezzu and M. Carta, R. Soc. Open Sci., 2020, 7, 200741 CrossRef CAS PubMed.
- B. S. Ghanem, M. Hashem, K. D. M. Harris, K. J. Msayib, M. Xu, P. M. Budd, N. Chaukura, D. Book, S. Tedds, A. Walton and N. B. McKeown, Macromolecules, 2010, 43, 5287–5294 CrossRef CAS.
- P. Bhanja, S. K. Das, K. Bhunia, D. Pradhan, T. Hayashi, Y. Hijikata, S. Irle and A. Bhaumik, ACS Sustainable Chem. Eng., 2018, 6, 202–209 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau, S. Thomas and M. D. Guiver, Macromolecules, 2008, 41, 9656–9662 CrossRef CAS.
- N. Du, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromolecules, 2010, 43, 8580–8587 CrossRef CAS.
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669–2673 CrossRef CAS PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. van Puyvelde and B. van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- S. A. Felemban, C. G. Bezzu, B. Comesaña-Gándara, J. C. Jansen, A. Fuoco, E. Esposito, M. Carta and N. B. McKeown, J. Mater. Chem. A, 2021, 9, 2840–2849 RSC.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem. – Eur. J., 2005, 11, 2610–2620 CrossRef CAS PubMed.
- N. Du, J. Song, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromol. Rapid Commun., 2008, 29, 783–788 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- S. Liu, Z. Jin, Y. C. Teo and Y. Xia, J. Am. Chem. Soc., 2014, 136, 17434–17437 CrossRef CAS PubMed.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 12727–12731 CrossRef CAS PubMed.
- S. Makhseed, F. Ibrahim and J. Samuel, Polymer, 2012, 53, 2964–2972 CrossRef CAS.
- R. Williams, L. A. Burt, E. Esposito, J. C. Jansen, E. Tocci, C. Rizzuto, M. Lanč, M. Carta and N. B. McKeown, J. Mater. Chem. A, 2018, 6, 5661–5667 RSC.
- H. Zhou, W. Jin, H. Zhou and W. Jin, Membranes, 2019, 9, 3 CrossRef PubMed.
- C. Zhang, L. Fu, Z. Tian, B. Cao and P. Li, J. Membr. Sci., 2018, 556, 277–284 CrossRef CAS.
- S. J. Rukmani, T. P. Liyana-Arachchi, K. E. Hart and C. M. Colina, Langmuir, 2018, 34, 3949–3960 CrossRef CAS PubMed.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- W. Fang, L. Zhang and J. Jiang, Mol. Simul., 2010, 36, 992–1003 CrossRef CAS.
- M. Heuchel, D. Fritsch, P. M. Budd, N. B. McKeown and D. Hofmann, J. Membr. Sci., 2008, 318, 84–99 CrossRef CAS.
- S. Yi, B. Ghanem, Y. Liu, I. Pinnau and W. J. Koros, Sci. Adv., 2019, 5, eaaw5459 CrossRef PubMed.
- J. Zhou, X. Zhu, J. Hu, H. Liu, Y. Hu and J. Jiang, Phys. Chem. Chem. Phys., 2014, 16, 6075 RSC.
- W. Fang, L. Zhang and J. Jiang, J. Phys. Chem. C, 2011, 115, 14123–14130 CrossRef CAS.
- L. J. Abbott, K. E. Hart and C. M. Colina, Theor. Chem. Acc., 2013, 132, 1334 Search PubMed.
- K. E. Hart, L. J. Abbott, N. B. McKeown and C. M. Colina, Macromolecules, 2013, 46, 5371–5380 CrossRef CAS.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesaña-Gándara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 CrossRef CAS PubMed.
- C. Song, F. Hu, Z. Meng, S. Li, W. Shao, T. Zhang, S. Liu and X. Jian, RSC Adv., 2020, 10, 4258–4263 RSC.
- K. A. Thompson, R. Mathias, D. Kim, J. Kim, N. Rangnekar, J. R. Johnson, S. J. Hoy, I. Bechis, A. Tarzia, K. E. Jelfs, B. A. McCool, A. G. Livingston, R. P. Lively and M. G. Finn, Science, 2020, 369, 310–315 CrossRef CAS PubMed.
- N. S. Murthy, in Polymer Morphology: Principles, Characterization, and Processing, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016, pp. 14–36 Search PubMed.
- R. Recio, L. Palacio, P. Prádanos, A. Hernández, Á. E. Lozano, Á. Marcos, J. G. de la Campa and J. de Abajo, J. Membr. Sci., 2007, 293, 22–28 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1999, 103, 4508–4517 CrossRef CAS.
- C. D. Wick, M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 2000, 104, 8008–8016 CrossRef CAS.
- C. D. Wick, J. M. Stubbs, N. Rai and J. I. Siepmann, J. Phys. Chem. B, 2005, 109, 18974–18982 CrossRef CAS PubMed.
- N. Rai and J. I. Siepmann, J. Phys. Chem. B, 2007, 111, 10790–10799 CrossRef CAS PubMed.
- J. M. Stubbs, J. J. Potoff and J. I. Siepmann, J. Phys. Chem. B, 2004, 108, 17596–17605 CrossRef CAS.
- S. D. D. S. Biovia, Dassault Systèmes, Materials Studio, [22.1.0.34462], 2022 Search PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ‘t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- L. Gong, W. Wu, D. Lin and K. Yang, J. Mater. Chem. A, 2021, 9, 254–258 RSC.
- G. Genduso, B. S. Ghanem, Y. Wang and I. Pinnau, Polymers, 2019, 11, 361 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- N. Savjani, S. J. Lancaster, S. Bew, D. L. Hughes and M. Bochmann, Dalton Trans., 2011, 40, 1079–1090 RSC.
- T. J. Corrado, Z. Huang, D. Huang, N. Wamble, T. Luo and R. Guo, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022204118 CrossRef CAS PubMed.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS PubMed.
- C. Scholes and S. Kanehashi, Membranes, 2019, 9, 41 CrossRef PubMed.
- S. He, X. Jiang, S. Li, F. Ran, J. Long and L. Shao, AIChE J., 2020, 66, e16543 CrossRef CAS.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- C. L. Staiger, S. J. Pas, A. J. Hill and C. J. Cornelius, Chem. Mater., 2008, 20, 2606–2608 CrossRef CAS.
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- R. R. Tiwari, J. Jin, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2017, 537, 362–371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01584f |
| This journal is © The Royal Society of Chemistry 2023 |