DOI:
10.1039/D2PY01563C
(Paper)
Polym. Chem., 2023,
14, 1639-1645
Cu(0)-RDRP of acrylates using an alkyl iodide initiator†
Received
14th December 2022
, Accepted 13th March 2023
First published on 17th March 2023
Abstract
In the vast majority of atom transfer radical polymerizations, alkyl bromides or alkyl chlorides are commonly employed as initiators, and minimal attention has been given to alkyl iodides. Herein, we report the room temperature Cu(0)-mediated reversible deactivation radical polymerization of acrylates utilizing alkyl iodide as an initiator. Kinetic experiments were conducted showing a linear dependence of Mn with conversion, good agreement between theoretical and experimental molecular weights, and low dispersity values (Đ ∼ 1.05), even at high monomer conversions. The high-end group fidelity of the iodide-terminated polymer was confirmed via MALDI-ToF-MS analysis and successful in situ chain extensions at near-quantitative conversions. Polymerization of methyl acrylate with various targeted degrees of polymerizations (DPn = 25–2400), resulted in the synthesis of well-defined polymers with low dispersities (Đ < 1.15), even at higher molecular weights (e.g. Mn = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Đ ∼ 1.13). The compatibility of the methodology with various solvents, including acetonitrile (MeCN), dimethylformamide (DMF), acetone, and isopropanol (IPA) as well as a range of acrylic monomers was also investigated yielding polymers with excellent control over the molar mass distributions. A series of block copolymers consisting of different block segments were also synthesized in one pot without any intermediate purification steps, thus highlighting the potential of an alkyl iodide initiator in a controlled polymerization.
000, Đ ∼ 1.13). The compatibility of the methodology with various solvents, including acetonitrile (MeCN), dimethylformamide (DMF), acetone, and isopropanol (IPA) as well as a range of acrylic monomers was also investigated yielding polymers with excellent control over the molar mass distributions. A series of block copolymers consisting of different block segments were also synthesized in one pot without any intermediate purification steps, thus highlighting the potential of an alkyl iodide initiator in a controlled polymerization.
Introduction
During the last century, there have been extraordinary developments in polymer science. Starting from living ionic polymerization methodologies, a plethora of fundamentally different approaches for polymer synthesis, have been discovered.1 Among those, reversible deactivation radical polymerization (RDRP), also referred to as controlled radical polymerization (CRP), has flourished in the last three decades.2,3 Starting with the discovery of nitroxide-mediated polymerization (NMP) in 1986,4 other RDRP methodologies were subsequently discovered including atom transfer radical polymerization (ATRP) in 1995,5,6 reversible addition–fragmentation chain-transfer (RAFT) polymerization in 1998,7 and, more recently, iodide-mediated polymerizations.8,9
Arguably, ATRP remains to date the most studied controlled radical polymerization method due to its simplicity and versatility.10 ATRP operates by utilizing a transition metal (usually copper) to first trigger and then mediate the polymerization. In conventional ATRP, the metal in its low oxidation state (e.g. CuI) coordinates to an amine ligand (e.g. CuI/L) forming a complex which acts as a catalyst. The catalyst activates the carbon–halogen bond of an alkyl halide initiator generating a radical and a metal complex in a higher oxidation state (e.g. CuII/L), known as the deactivator. The generated radical, also referred to as propagating radical, can be either immediately deactivated by the catalyst in the higher oxidation state, propagate by reacting with one more vinyl monomers followed by deactivation, or undergo an irreversible reaction with another radical. The latter leads to the accumulation of CuII during the polymerization and is responsible for the control over the polymerization through the persistent radical effect mechanism. As a result, conventional ATRP necessitates a certain extent of termination prior to gaining control over the molar mass distributions. In addition, high amounts of catalyst are typically needed for a successful polymerization. To overcome both barriers, a number of alternative ATRP approaches have recently been developed, including eATRP,11,12 photoATRP,13–16 ARGET-ATRP,17,18 and, (P)ICAR-ATRP.19,20 A common feature of these methodologies is that they require a stimulus or an additive to in situ re-generate the active catalyst and as a result they can operate at significantly lower catalyst concentrations.21–25 A copper-mediated polymerization approach that has received considerable attention during the last fifteen years is Cu(0)-mediated RDRP in which a zero-valent metal, usually copper wire, is used to provide the initial source of copper in the reaction.26 Although the mechanism of this approach has been heavily debated in the literature, it is not the subject of this investigation.27–34 Irrespective of the mechanism, it can be commonly agreed that Cu(0)-mediated RDRP is a versatile and robust polymerization methodology that can polymerize various monomers with extremely high end-group fidelity.35–38 The increased popularity of Cu(0)-RDRP can be attributed to its’ simplicity as it can be conducted at mild polymerization conditions, it requires very low catalyst concentrations, and it allows for the vast majority of the catalyst to be removed by simply lifting the stirring bar at the end of the reaction. In addition, in many occasions the copper wire can be re-used several times upon polymerization.39
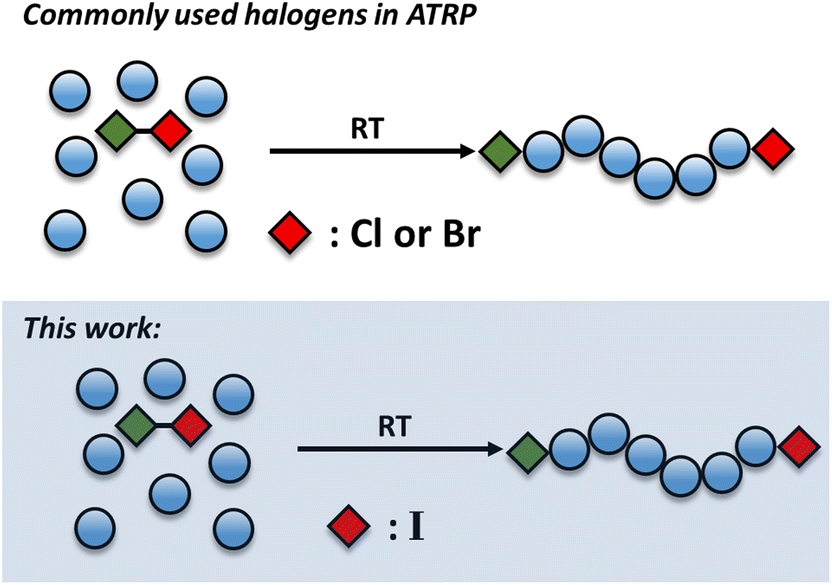
Regardless of the catalytic system employed and the way by which the active catalyst is being regenerated, all copper-mediated polymerizations employ alkyl halides initiators. The halogen atom affects the thermodynamic properties of the catalyst/initiator system and has a significant impact on the ATRP reaction.40–43 However, in the vast majority of studies, the common halogens employed are either bromine or chlorine. While the bromine-containing initiators are typically used in organic media, the chlorine-ones are often preferred in aqueous polymerizations due to the higher stability of the carbon chlorine bond against dissociation.44 Although theoretical calculations of iodine-containing initiators have been conducted, they have been rarely employed in ATRP. For example, Matyjaszewski and co-workers showed that alkyl iodide presents a faster activation rate when compared to its alkyl bromide analogue, while the KATRP had the opposite trend.40,41 This suggests that the kdeact in the case of alkyl iodide should be higher than the alkyl bromide. However, during the ATRP reaction of methyl acrylate using methyl 2-iodo-propionate as an initiator at 90 °C, very limited conversion (20%) was detected after 20 hours.45 An iodide haloform initiator has also been emoloyed by Percec and co-workers in Cu(0)-RDRP. Although the primary focus of this paper was the synthesis of ultra high molecular weight polymers, the authors also conducted an experiment whereby iodomethane was employed as a bi-functional initiator yielding the preparation of telechelic polymers at 80% of conversion (Đ ∼ 1.18).33 This paper suggests that Cu(0)-RDRP may be particularly advantageous to afford a controlled polymerization when conventional mono-functional iodide-containing initiators are employed and inspired our current study.
In this work we utilize an alkyl iodide initiator to trigger the Cu(0)-RDRP of various acrylates at room temperature (Scheme 1). This is in contrast to iodine-transfer polymerization (ITP), controlled radical polymerization catalysed via different transition metals (Ruthenium, Rhenium, Iron, etc.) or reversible complexation mediated polymerization (RCMP) whereby high temperatures and/or free radical initiators are typically required to catalyze a successful polymerization.46–56 Under relatively mild conditions, we seek to employ a mono-functional initiator to target a wide range of molecular weights at near-quantiative convesions and without compromising the control over the molar mass distributions. Indeed, poly(methyl acrylate) with excellent control over the molar mass distributions (Đ as low as 1.05) could be obtained. Notably, a series of polymers with various molecular weights could be targeted (DPn = 25–2400) at high monomer conversions and the resulting polymers possess high end-group fidelity as demonstrated by in situ chain extensions and block copolymer formation. The effect of the solvent was also investigated and a range of solvents were found to be compatible with our polymerization approach.
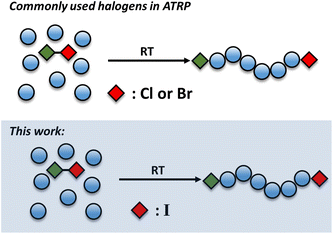 |
| | Scheme 1 Conceptual scheme illustrating the previous literature and the current approach. | |
Results and discussion
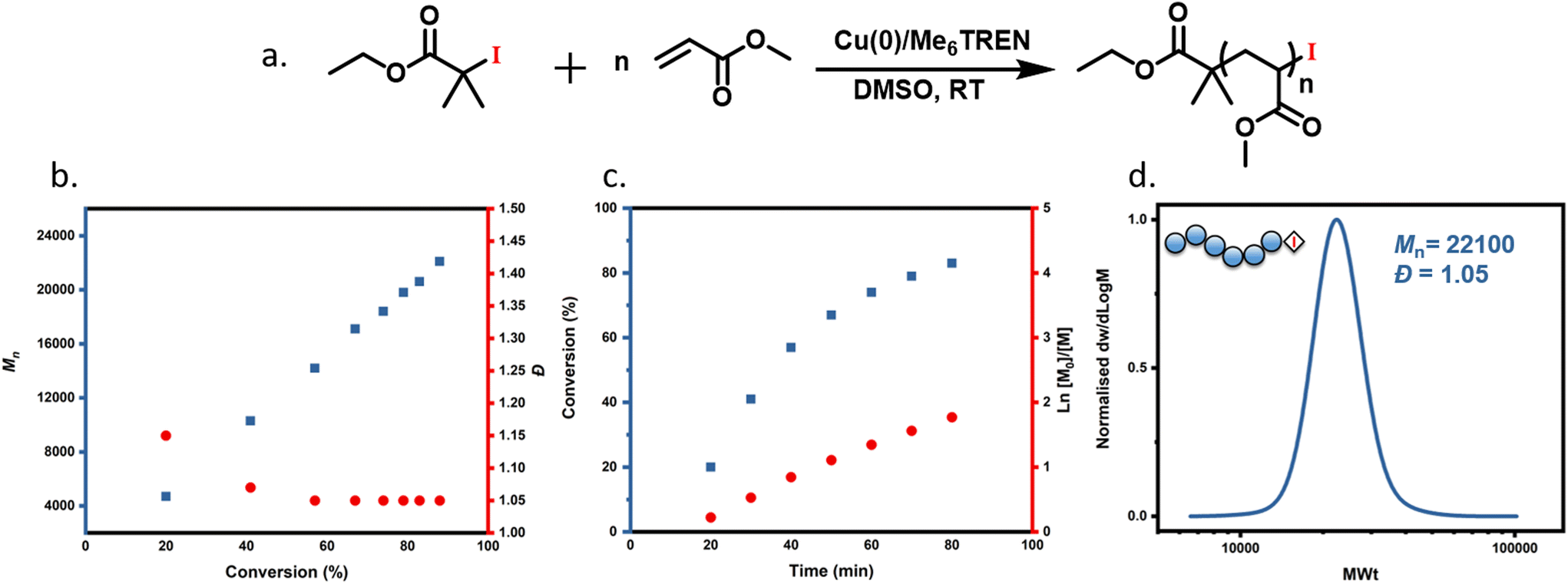
To begin exploring the Cu(0)-RDRP of acrylates using an alkyl iodide initiator, we chose a model system where dimethylsulfoxide (DMSO) was used as a solvent, methyl acrylate (MA) as a monomer, Me6TREN as a ligand and ethyl 2-iodo-2-methylpropionate (EIP) as the initiator. The molar ratio of components used was set as follows: [MA]:[EIP]:[Cu(0)]:[Me6TREN] equals 200:1:5 cm:0.18 (Fig. 1a). It is noted that in contrast to previous reports with alkyl bromide initiators, no external deactivator was employed as CuI2 is not a commercially available reagent. A detailed kinetic experiment showed 88% monomer conversion achieved in less than 2 h, as detected by 1H NMR (Fig. S1†), while near quantitative monomer conversion (96%) was atchieved in 3 h. A linear evolution of Mn with conversion was observed with the dispersity gradually decreasing throughout the polymerization (Fig. 1b and Table S1†), thus indicating a controlled polymerization. In addition, a linear dependence of ln([Mo]/[M]) on the time was also obtained as shown in Fig. 1c. Furthermore, SEC analysis revealed a continued shift to higher molecular weights with narrow molar mass distributions (Đ = 1.05) (Fig. 1d). It is noted that this is the lowest Đ and highest conversion ever reported with an iodide-containing initiator in copper-catalyzed RDRP. For instance, Percec and co-workers reported a dispersity of 1.18 at 80% monomer conversion while Matyjaszewski's group reached only 20% of conversion when iodide-containing initiators were employed.33,45 Taken altogether, this data suggest that alkyl iodide can be successfully used in Cu(0)-RDRP as a mono-functional alkyl halide initiator, with improved monomer conversions, reaction times and Đ when compared to previously reported conventional ATRP of MA using alkyl iodide initiator.33,45
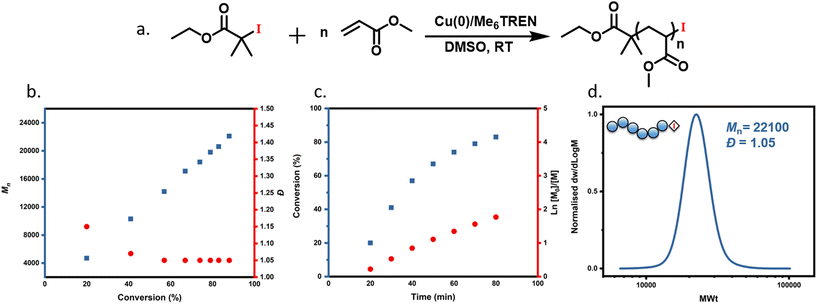 |
| | Fig. 1 (a) Reaction scheme showing Cu(0)-RDRP of MA with alkyl iodide initiator and the kinetic analysis of MA polymerization with molar ratio of [MA]:[EIP]:[Me6TREN] = [100]:[1]:[0.18] using 5 cm of Cu(0) wire and 1:1 monomer to solvent volume ratio. (b) Evolution of Mn and Đ with respect to monomer conversion, (c) evolution of conversion and ln([M0]/[M]) over time, and (d) SEC trace of the last time-point of the kinetic experiment. | |
Considering the use of iodo-compounds in the presence of vinyl monomers, the possibility of a potential degenerative transfer reaction cannot be excluded. In fact, degenerative transfer is often cited as the main reason that iodine-containing initiators have been avoided in ATRP.42,57 To explore this, we conducted a series of control experiments, as shown in Table 1. In the first control experiment, polymerization of MA was attempted at room temperature and in the absence of copper wire. However, no monomer conversion could be observed even after 24 h, thus strongly suggesting that copper wire is necessary to abstract the Iodine and generate radicals (Table 1, row 1). In a similar fashion, no polymerization took place when either the ATRP initiator or the ligand were excluded (Table 1, row 2 and 3), and as such it was concluded that all the reagents are essential for a successful RDRP at room temperature. However, the low dispersity obtained (Fig. 1) can be attributed to either sufficient ATRP deactivation, degenerative chain-transfer or a combination thereof. To investigate the possibility of degenerative chain transfer, we sought for an alternative radical source (i.e. to replace copper wire). As a room temperature free radical initiator was not available in our laboratory, we first repeated our key experiment at 80 °C and narrow molar mass distributions were also obtained (Đ ∼ 1.06) (Table S2†). In fact, regardless of the polymerization temperature (60 °C, 80 °C or 110 °C), very low dispersities (<1.09) were achieved accompanied with an increased polymerization rate at higher temperatures (Table S2†). This highlights sufficient deactivation and minimal side reactions under the studied ATRP conditions. Instead, when the identical experiment was repeated using AIBN as a radical source at 80 °C (i.e. without copper wire/ligand), significantly broader molar mass distributions were obtained (Đ ∼ 1.7), albeit with a good control over the molecular weight (Table S2†). These experiments highlight that (i) degenerative chain transfer can occur in the presence of Iodide-containing initiators (when a radical source such as AIBN is employed) leading to good control over the molecular weight and high dispersity, in line with the literature and (ii) the low dispersity obtained is primarily attributed to the ATRP deactivation rather than degenerative chain transfer. Taking into account that the synthesis of polyacrylates through RCMP typically occurs at higher temperatures (i.e. 110 °C), rather than room temperature, we also performed a control experiment at 110 °C and in the absence of any copper catalyst (but in the presence of ligand that is responsible for the halogen abstraction/radical generation). Within 1.5 hours the polymerization mixture turned viscous, with 1H NMR showing 80% monomer conversion. The SEC analysis of the sample showed a monomodal molecular weight distribution with controlled molecular weight, albeit with a very high dispersity (Table 1, entry 4), thus confirming an RCMP mechanism at higher temperatures with poor control over the molecular weight distributions. Taken altogether, these control experiments indicate that the low dispersity obtained is due to the Cu(0)-RDRP mechanism although the possibility of degenerative chain transfer cannot be excluded.
Table 1
1H NMR and SEC analysis of control experiments
| [MA]:[EIP]:[Cu(0)]:[Me6TREN] |
Temp. (°C) |
Time |
Conv. (%) |
M
n (SEC) (Da) |
Đ
|
| 50:1:—:0.18 |
RT |
24 h |
— |
— |
— |
| 50:—:5 cm:0.18 |
RT |
24 h |
— |
— |
— |
| 50:1:5 cm:— |
RT |
24 h |
— |
— |
— |
| 50:1:—:0.18 |
110 |
1.5 h |
80 |
7600 |
1.66 |
| 50:1:5 cm:0.18 |
110 |
5 min |
82 |
4700 |
1.09 |
Since we validated the controlled behaviour of Cu(0)-RDRP of MA utilising alkyl iodide as an initiator, we then sought to further characterize the synthesized polymers and examine their end-group fidelity. To do so, low molecular weight PMA (targeted DP = 25) was initially synthesized and then analyzed via matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF-MS). Fig. 2a shows the MALDI spectrum whereby it can be confirmed that every polymer chain is initiated by the expected initiator and has a living iodide at the ω-end. Notably, no other end-groups were observed, highlighting that very high end-group fidelity can be maintained. To further highlight the excellent end-group functionality of the polymers, as well as the stability of the iodine end-group, we subsequently conducted an in situ chain extension experiment. PMA with DP 200 was first synthesized and upon near-quantitative monomer conversion (i.e. 96%), a second aliquot of a deoxygenated MA and DMSO solution was added, and the polymerization was allowed to proceed for 3 further hours. 1H NMR and SEC analysis indicated a successful chain extension, with the SEC traces shifting to higher molecular weights and a final dispersity of 1.09 (Fig. 2b).
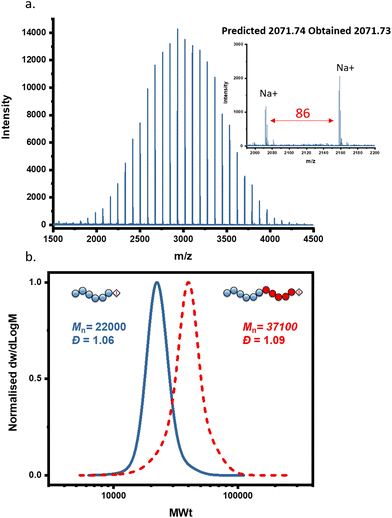 |
| | Fig. 2 (a) MALDI-ToF-MS analysis of PMA synthesized via Cu(0)-RDRP with molar ratio of [MA]:[EIP]:[Me6TREN] = [25]:[1]:[0.18] using 5 cm of Cu(0) wire and 1:1 monomer to solvent volume ratio. (b) SEC traces of the PMA200 (blue, solid) and its in situ chain extension with MA (red, dashed). | |
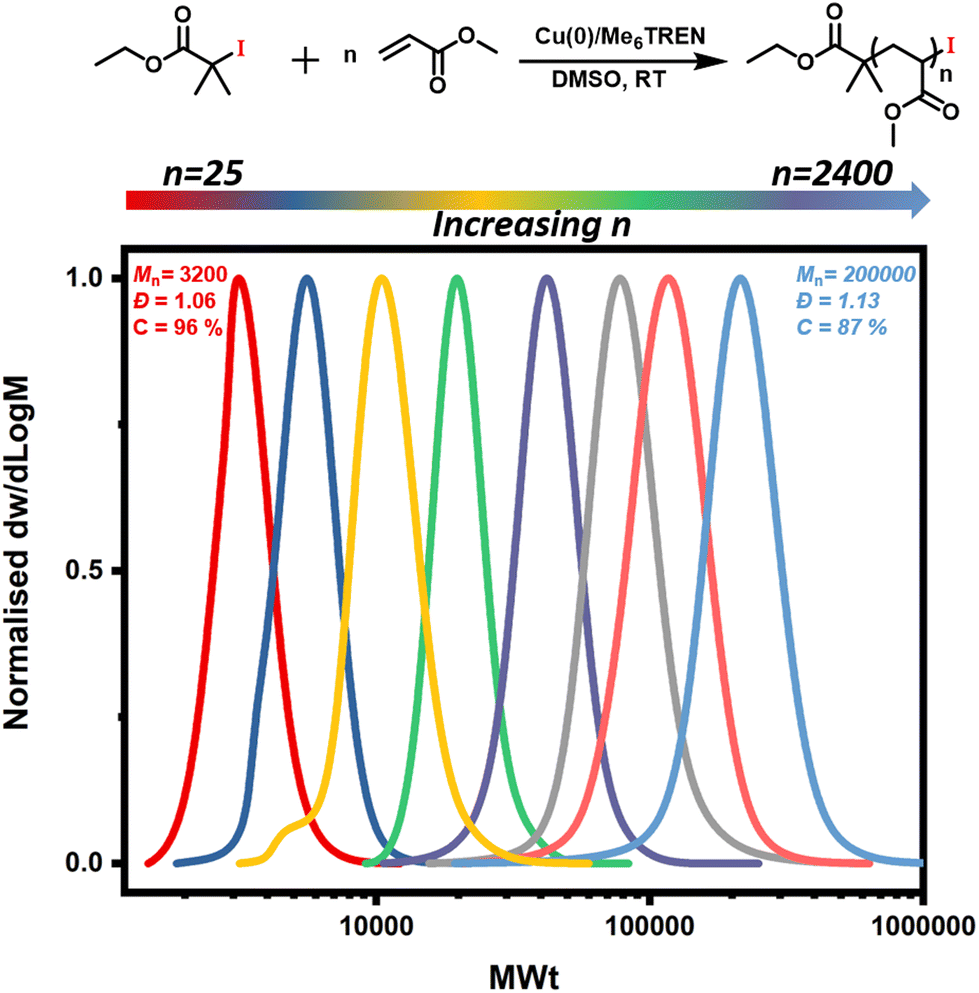
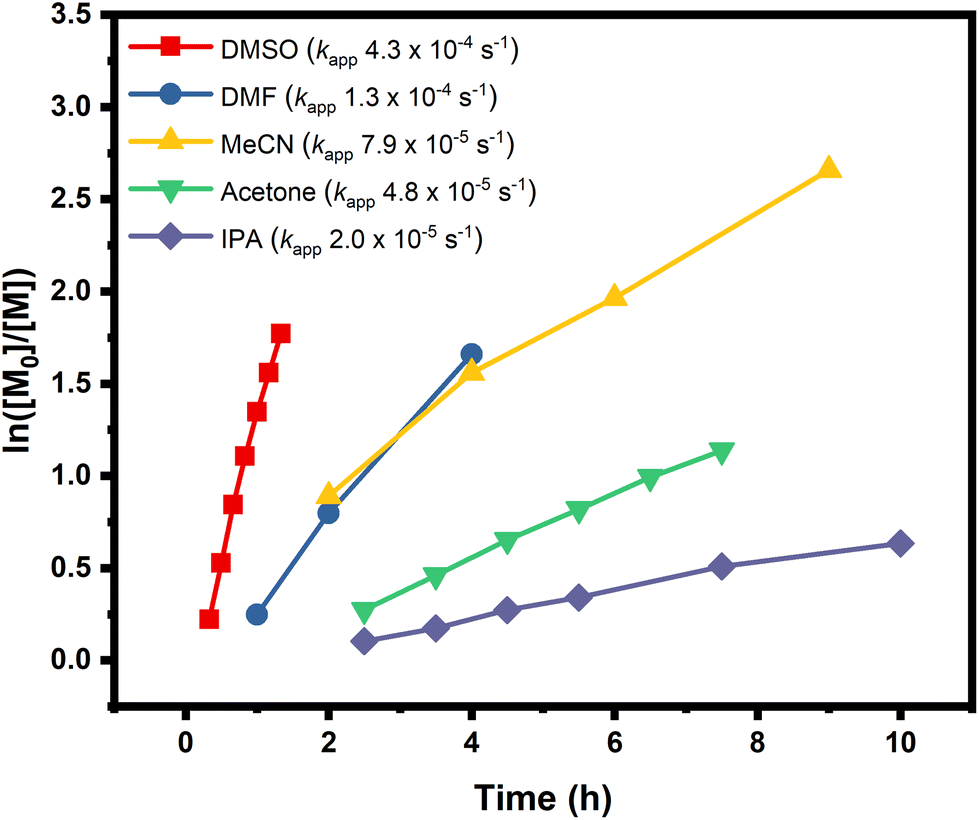
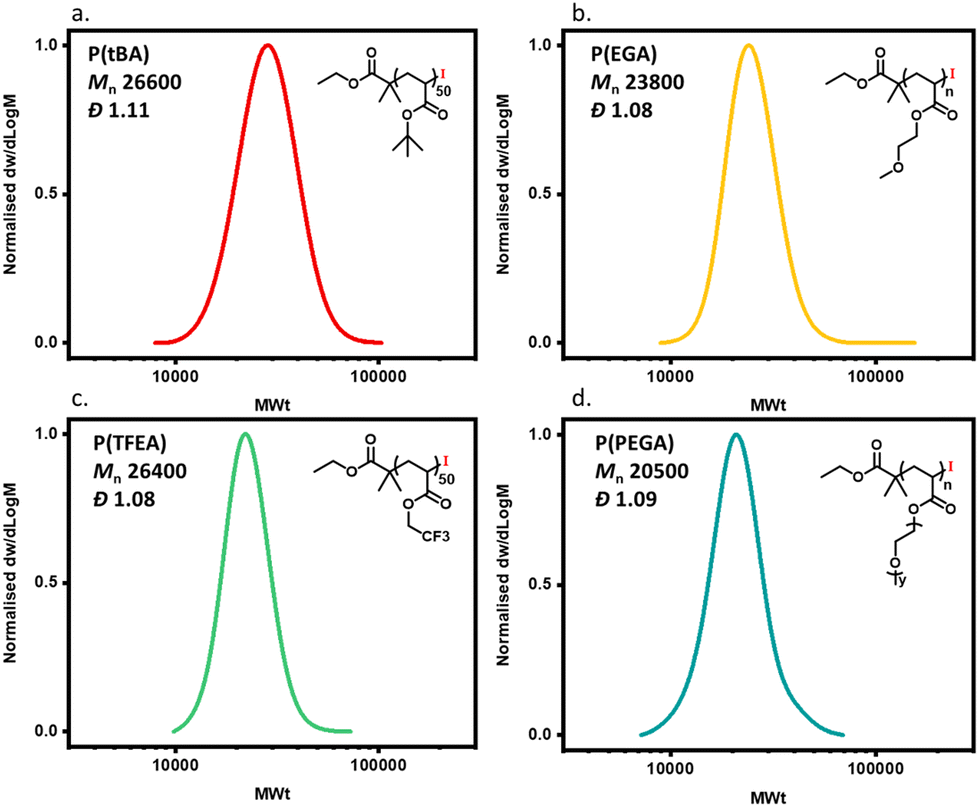
In order to expand the scope, we also conducted the synthesis of a series of PMAs with various targeted DPs (Fig. 3 and Table S3). Starting with DP 25, the synthesis resulted in a well-defined PMA with Mn of 3200 and Đ of 1.06. Increasing the targeted DP up to 2400 did not influence the control over the polymerization yielding PMA with Mn of 200000 and Đ of 1.13. Overall, very good control was maintained for all the targeted DPs, with high monomer conversions and low dispersities in all cases (Fig. 3 and Table S3). Next, a range of solvents was investigated. For this purpose, five different solvents were employed for the polymerization of PMA, under the following conditions [MA]:[EIP]:[Me6TREN] = [200]:[1]:[0.18] using 5 cm of Cu(0) wire and 1:1 monomer to solvent volume ratio. Irrespective of the chosen solvent, well-controlled polymers were obtained with good control over the molecular weight and low dispersity values (Đ < 1.2). However, the polymerization rate was significantly affected (Fig. 4), in agreement with the literature.58 In particular, the fastest polymerization took place in DMSO (kapp = 4.3 × 10−4 s−1), whereby 90% monomer conversion was reached within 2 hours. The next solvent with the fastest polymerization rate was DMF (kapp = 1.3 × 10−4 s−1) followed by MeCN, acetone and finally IPA.59–61 A range of different acrylic monomers were also polymerized including the hydrophilic (EGA), the functional (tBA), the semi-fluorinated (TFEA) and the macromonomer (PEGA480). All the polymerizations proceeded efficiently resulting in low dispersity polymers (Đ < 1.1) (Fig. 5 and Table S4†). Finally, the in situ synthesis of diblock copolymers with different block segments was attempted (Fig. 6). A PMA200 macro-initiator was first synthesized and upon reaching near-quantitative monomer conversion, the second monomer was added in one pot. In all the cases, the successful formation of block copolymers was confirmed by both 1H NMR and SEC analysis yielding low dispersity final materials.
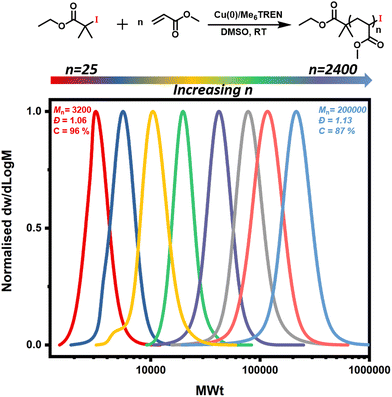 |
| | Fig. 3 Reaction scheme showing Cu(0)-RDRP of MA with alkyl iodide initiator and SEC traces for the Cu(0)-RDRP of MA with targeted DPs 25–2400 and molar ratio of [MA]:[EIP]:[Me6TREN] = [n]:[1]:[0.18] using 5 cm of Cu(0) wire and 1:1 monomer to solvent volume ratio. | |
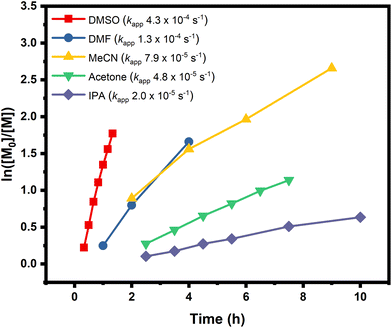 |
| | Fig. 4 Polymerization kinetics of MA in different solvents, using molar ratio of [MA]:[EIP]:[Me6TREN] = [200]:[1]:[0.18], 5 cm of Cu(0) wire and 1:1 monomer to solvent volume ratio. The graph shows the evolution of ln([M0]/[M]) over time and the corresponding kapp for different solvents. | |
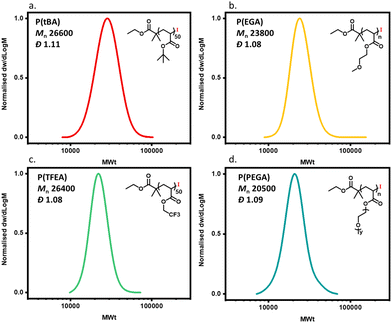 |
| | Fig. 5 SEC traces for different polymers obtained through Cu(0)-RDRP using alkyl iodide initiator: (a) poly(tert butyl acrylate), (b) poly(ethylene glycol methyl ether acrylate), (c) poly(trifluoroethyl acrylate) and (d) poly(poly ethylene glycol methyl ether acrylate). | |
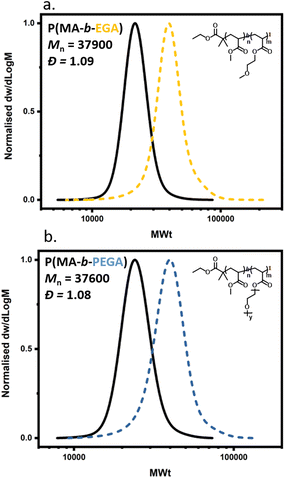 |
| | Fig. 6 SEC traces for different diblock copolymers obtained through Cu(0)-RDRP using alkyl iodide initiator via in situ chain extension of PMA (dashed trace) with: (a) ethylene glycol methyl ether acrylate (yellow/dashed), (b) poly ethylene glycol methyl ether acrylate(blue/dashed). | |
Conclusions
To conclude, in this work we show that alkyl iodides can be successfully used as alky halide initiators in Cu(0)-RDRP at room temperature. The obtained polymers showed exceptional end-group fidelity with good control over the targeted molecular weights. Significantly, a series of control experiments validated that the polymerization proceeds solely through a Cu(0)-RDRP mechanism while the possibility of degenerative transfer was excluded. The polymerizations showed linear pseudo-first order kinetics with low final dispersity values and our approach could be expanded to a range of solvents and monomers. Our work expands the scope of copper-mediated polymerization and highlights the possibility of alkyl iodides to be used as efficient initiators generating iodide-containing polymers under mild conditions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. A. gratefully acknowledges ETH Zurich for financial support. N. P. T. acknowledges the award of a DECRA Fellowship and DP from the ARC (DE180100076 and DP200100231). KP thanks Onasis Foundation as this scientific paper was partially supported by the Onassis Foundation – Scholarship ID: FZQ051-1/2020-2021. We thank Dr Richard Whitfield for MALDI-Tof-MS measurement.
References
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
-
D. H. Solomon, E. Rizzardo and P. Cacioli, Polymerization process and polymers produced thereby, U.S. Pat., 4581429, 1986 Search PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsujii and H. Kaji, Macromolecules, 2011, 44, 8709–8715 CrossRef CAS.
- C.-G. Wang, A. M. L. Chong, H. M. Pan, J. Sarkar, X. T. Tay and A. Goto, Polym. Chem., 2020, 11, 5559–5571 RSC.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- A. J. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- V. Bonometti, E. Labbé, O. Buriez, P. Mussini and C. Amatore, J. Electroanal. Chem., 2009, 633, 99–105 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- J. Mosnáček and M. T. Ilčíková, Macromolecules, 2012, 45, 5859–5865 CrossRef.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox and V. Percec, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- K. Parkatzidis, N. P. Truong, R. Whitfield, C. E. Campi, B. Grimm-Lebsanft, S. R. Buchenau, M. A. Rübhausen, S. Harrisson, D. Konkolewicz and S. Schindler, J. Am. Chem. Soc., 2023, 145(3), 1906–1915 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572–7573 CrossRef CAS.
- G. Szczepaniak, M. Łagodzińska, S. Dadashi-Silab, A. Gorczyński and K. Matyjaszewski, Chem. Sci., 2020, 11, 8809–8816 RSC.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- R. Whitfield, K. Parkatzidis, K. G. Bradford, N. P. Truong, D. Konkolewicz and A. Anastasaki, Macromolecules, 2021, 54, 3075–3083 CrossRef CAS.
- K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Polym. Chem., 2021, 12, 5583–5588 RSC.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef CAS PubMed.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4417 RSC.
- D. Konkolewicz, P. Krys, J. R. Gois, P. V. Mendonca, M. Zhong, Y. Wang, A. Gennaro, A. A. Isse, M. Fantin and K. Matyjaszewski, Macromolecules, 2014, 47, 560–570 CrossRef CAS.
- P. Krys, Y. Wang, K. Matyjaszewski and S. Harrisson, Macromolecules, 2016, 49, 2977–2984 CrossRef CAS.
- S. Harrisson and J. Nicolas, ACS Macro Lett., 2014, 3, 643–647 CrossRef CAS PubMed.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 5517–5525 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 6421–6432 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef.
- H. S. Wang, K. Parkatzidis, S. Harrisson, N. P. Truong and A. Anastasaki, Chem. Sci., 2021, 12, 14376–14382 RSC.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- S. Lanzalaco, M. Fantin, O. Scialdone, A. Galia, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2017, 50, 192–202 CrossRef CAS.
- C. Y. Lin, M. L. Coote, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12762–12774 CrossRef CAS PubMed.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 10468–10482 CrossRef CAS PubMed.
- K. A. Davis and K. Matyjaszewski, J. Macromol. Sci., Part A: Pure Appl.Chem., 2004, 41, 449–465 CrossRef.
- L. Lei, M. Tanishima, A. Goto, H. Kaji, Y. Yamaguchi, H. Komatsu, T. Jitsukawa and M. Miyamoto, Macromolecules, 2014, 47, 6610–6618 CrossRef CAS.
- K. Matyjaszewski, S. Gaynor and J.-S. Wang, Macromolecules, 1995, 28, 2093–2095 CrossRef CAS.
- S. G. Gaynor, J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 8051–8056 CrossRef CAS.
- A. Goto, K. Ohno and T. Fukuda, Macromolecules, 1998, 31, 2809–2814 CrossRef CAS.
- Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 2420–2424 CrossRef CAS.
- Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 3543–3549 CrossRef CAS.
- Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 6746–6751 CrossRef CAS.
- H. Uegaki, Y. Kotani, M. Kamigaito and M. Sawamoto, ACS Symp. Ser., 2000, 760, 196–206 CrossRef CAS.
- I. Onishi, K. Y. Baek, Y. Kotani, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2033–2043 CrossRef CAS.
- M. Kamigaito, I. Onishi, S. Kimura, Y. Kotani and M. Sawamoto, Chem. Commun., 2002, 2694–2695 RSC.
- A. Goto, Y. Tsujii and T. Fukuda, Polymer, 2008, 49, 5177–5185 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS PubMed.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Leonardo
de Haro Amez
,
Leonardo
de Haro Amez