DOI:
10.1039/D2PY01545E
(Paper)
Polym. Chem., 2023,
14, 1743-1751
Bioactive polyethylene synthesized by ring opening metathesis polymerization for potential orthopaedic applications†
Received
9th December 2022
, Accepted 20th March 2023
First published on 21st March 2023
Abstract
Efficient synthesis and bioevaluation of novel brush-type polyethylene-peptide copolymers for potential applications in orthopaedic implants are described here. The brush-type copolymers containing pendant arms of polyethylene (PE) and PEGylated biomolecules including linear arginyl-glycyl-aspartic acid (RGD) and collagen fragments (Gly-Pro-Hyp)3 were synthesized by ring-opening metathesis polymerization (ROMP) using the well-defined 2nd generation Grubbs’ ruthenium catalyst. The random copolymerization of two separate norbornene-dicarboxylic anhydride macromonomers allowed for the incorporation of hydrophilic PEGylated biomolecules into hydrophobic polyethylene. Thermal stability of this polyethylene-peptide copolymer was also markedly improved for melt extrusion-based material processing. The copolymers were blended with ultra-high molecular weight polyethylene (UHMWPE), extruded into filaments and 3D-printed into sheets using fused filament fabrication methods. The ability of these polyethylene materials to enhance osteogenic activity whilst reducing inflammatory response compared to pure UHMWPE was evaluated by the in vitro alkaline phosphatase (ALP) assay and an in vivo murine model study, respectively. The results presented here serves as a promising guide for biofunctionalization of polyethylene materials for potential orthopaedic applications.
1 Introduction
Synthetic polymers such as polyethylene (PE), polycaprolactone (PCL) and poly(methyl methacrylate) (PMMA) are important materials used in the biomedical fields.1,2 Amongst these, polyethylene (PE) is the most common polymer used in medical implant fabrication due to its desired mechanical properties, inertness, and nontoxicity.3 Yet, foreign body responses on PE-based medical devices have been widely reported,4 resulting in extensive research into improving the biocompatibility of polyethylene.5 Some of the current methods used to improve the biocompatibility of PE include blending of biomolecules into the base polymer.6,7 However, this method has disadvantages of potential phase separation or premature thermal degradation of biomolecules during material processing that requires heat.8,9
The arginyl-glycyl-aspartic acid (RGD) sequence is the minimal binding domain for fibronectin to recognize cell surface integrins.10 Materials modified with RGD peptides have been reported to facilitate cell adhesion, migration and proliferation.11 Apart from RGD, collagen is found in the extracellular matrix, particularly in skin, cartilage and bone. The use of collagen in implants targeting tissue regeneration has been studied over the years. Collagen fragments bearing glycine, proline and hydroxyproline sequences (GPHyp)3 have also been shown to promote skin cell adhesion, migration, differentiation and proliferation.12–14 Despite the apparent usefulness of these biomolecules for improving biocompatibility and potentially inducing tissue regeneration in implants,15 these short peptide sequences tend to have low thermal stability, are hygroscopic and lack mechanical strength, making their use in medical devices challenging. Therefore, in order to improve the usability of these short peptides in medical device fabrication, methods to improve their stability would be necessary.
Ring-opening metathesis polymerization (ROMP) of norbornene (NB) derivatives is a versatile method for functional polymer synthesis. Grubbs and co-workers have demonstrated that a polynorbornene backbone is non-toxic16–18 and RGD-functionalized polynorbornenes constructed by ROMP can facilitate cell proliferation.11 Recently, we have reported on bioactive PCL-peptide and PLA-peptide brush copolymers for bone-tissue engineering.19 Inspired by these positive outcomes, we sought to utilize ROMP to construct peptide-containing bioactive polyethylene copolymers for use as bioadditives in orthopaedic materials. Herein, we report the preparation of a series of “bioactive polyethylene” brush copolymers via ROMP, consisting of PE side chains and PEGylated RGD or (GPHyp)3 (Fig. 1). The “bioactive polyethylene” was formulated with ultra-high molecular weight polyethylene (UHMWPE) and 3D-printed into coupons for biovalidation in vitro and in vivo. It functions as a carrier for thermally sensitive biomolecules in high-melting point polymers such as UHMWPE. Also, the biocompatibility of UHMWPE is improved using this “bioactive polyethylene” without premature degradation of the biomolecules RGD or (GPHyp)3, which are present in the final PE formulation.
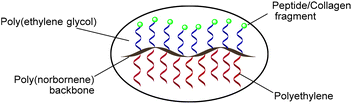 |
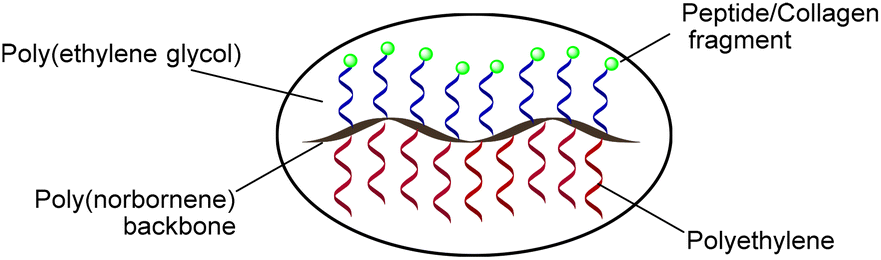
| | Fig. 1 Schematic of a bioactive polyethylene-peptide brush copolymer. | |
2 Experimental section
2.1 Polymer synthesis and characterization
Macromonomers (MMs) I and II, where I is a PEGylated RGD or (GPHyp)3 macromonomer, and II is a PE-based macromonomer, were created independently. I and II are then copolymerized together in a random fashion, using a 2nd generation Grubbs’ catalyst [G2], in a nitrogen-filled glovebox.20,21 Brief descriptions of the polymer syntheses are presented below. The conversion of I and incorporation of II in the final brush copolymers were determined by analyzing 1H NMR spectra according to the characteristic signals of the norbornene-terminated PE and PEG side chains, respectively. The PDIs and average molecular weights (Mn) of PE copolymers were calculated by high-temperature gel permeation chromatography (HT-GPC). For detailed chemical synthesis and characterization data, please refer to the ESI.†
2.1.1 Synthesis of peptide-based macromonomer I.
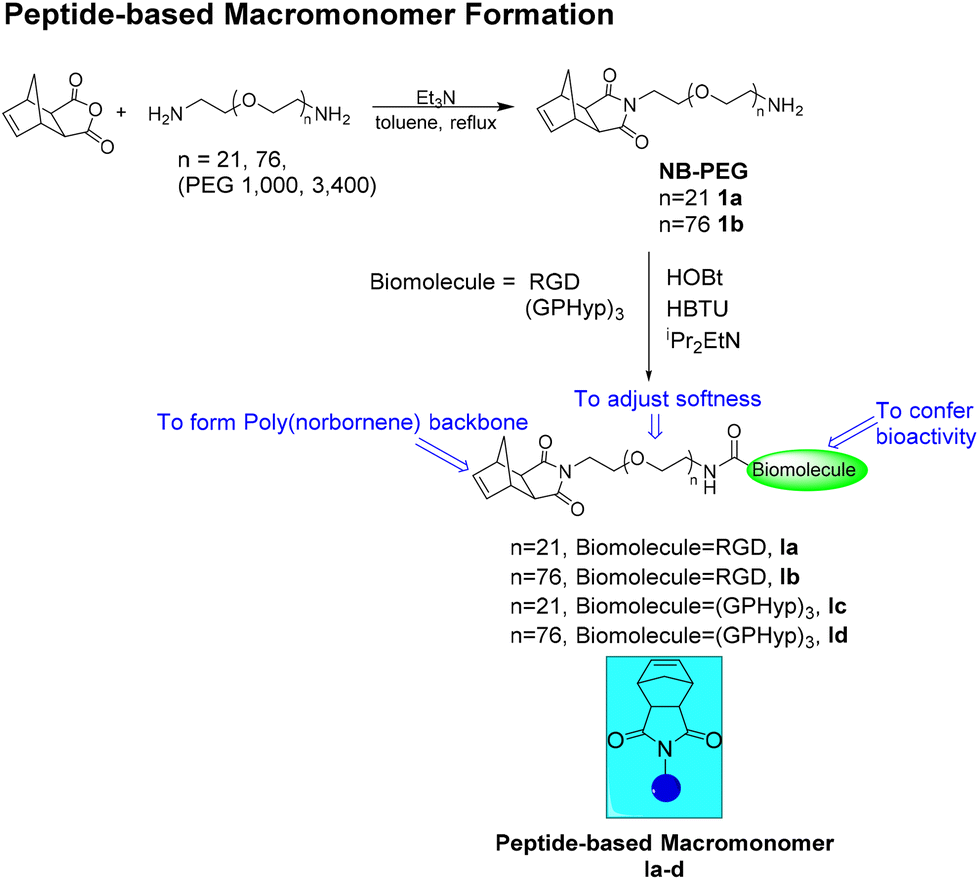
Polyethylene glycol (PEG) diamine of various chain lengths (MW 1000 and 3400) can be reacted with cis-norbornene-exo-2,3-dicarboxylic anhydride in refluxing toluene to create the main macromonomer body, NBPEG (1).22,23 Once NBPEG (1) is created, we add on the peptides by means of condensation reactions in the presence of hydroxybenzotriazole (HOBt), 3-[bis(dimethylamino)methyliumyl]-3H benzotriazol-1-oxide hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (iPr2EtN) to form an amide linkage between the amine terminal on NBPEG and the carboxylic acid terminal of the peptide. The molecular weights of the obtained peptide-based macromonomers (Ia–d) can be varied from 1500 to over 4000 by adjusting the PEG chain and the corresponding peptides (Scheme 1). Ia–d will serve as the bioactivity-conferring macromonomers in the subsequent brush copolymer formation by ROMP (Scheme 3).
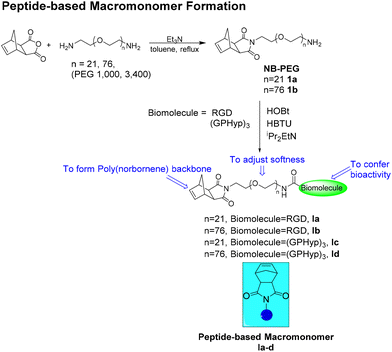 |
| | Scheme 1 Synthesis of peptide-based macromonomers Ia–d. | |
2.1.2 Synthesis of polyethylene-based macromonomer II.
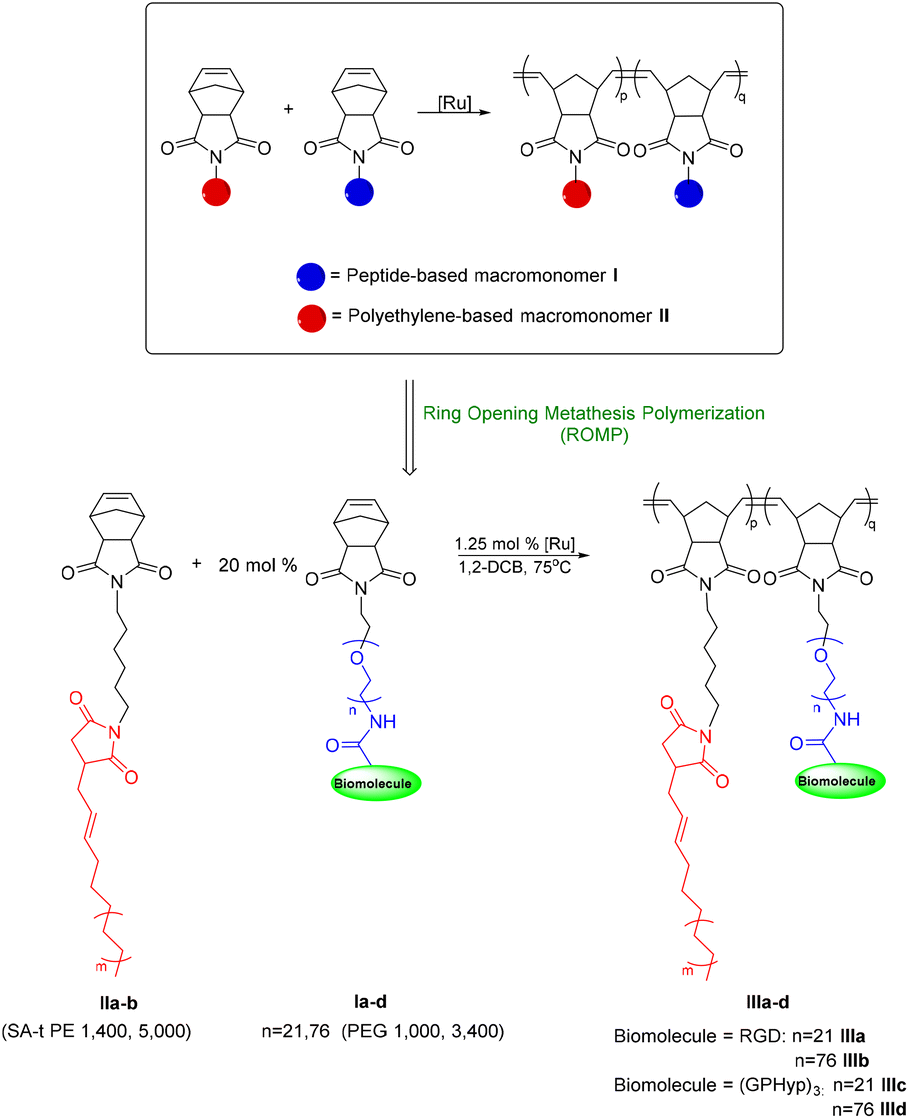
To obtain a primary amine terminus on polyethylene (PE) for macromonomer formation, a condensation reaction in refluxing toluene in the presence of hexamethylenediamine (HMDA) was carried out on succinic acid-terminated polyethylene (SA-t PE) (MW 1400 and 5000), which was prepared following literature procedures.24 Upon obtaining the amine-terminated PE (2), a second condensation reaction with cis-norbornene-exo-2,3-dicarboxylic anhydride afforded the PE-based macromonomers (NB-SA-t PE) (IIa and IIb) (Scheme 2) with a conversion of over 90% on SA-t PE1400.22IIa and IIb will function as synthetic macromonomers in the brush copolymer formation via ROMP, as highlighted in Scheme 3.
 |
| | Scheme 2 Synthesis of polyethylene-based macromonomers IIa and IIb. | |
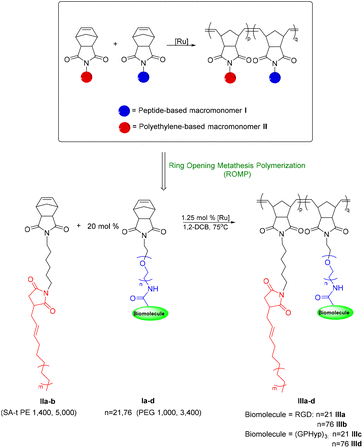 |
| | Scheme 3 General ROMP procedure for the preparation of PE-peptide brush copolymers (IIIa–d) where [Ru] refers to the 2nd generation Grubbs’ catalysts [G2]. | |
2.1.3 General ROMP procedure using I and II to prepare PE-peptide brush copolymers III.
With the macromonomers of PEGylated RGD or (GPHyp)3 (Ia–d) and norbornene-terminated polyethylene (IIa and IIb) ready, the final PE-peptide brush copolymers (IIIa–d) are prepared by ROMP using 1.25 mol% of 2nd generation Grubbs’ catalysts in 1,2-dichlorobenzene (1,2-DCB) at 75 °C with a molar ratio [I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :II] of 1:5 (Scheme 3).25 The reactions were terminated upon addition of ethyl vinyl ether. The copolymer was precipitated from methanol and repeatedly washed with acetone/H2O. PE-mPEG with no biomolecules, which is the brush copolymer with side chains of PE and methoxy-PEG, was prepared by the same ROMP strategy. It serves as a control for PE-peptide copolymers in the biological evaluation. The formation of the desired brush copolymer was characterized by 1H NMR spectroscopy, where the characteristic signals from the poly(ethylene glycol) (PEG) chain and polyethylene chains were observed, respectively.
:II] of 1:5 (Scheme 3).25 The reactions were terminated upon addition of ethyl vinyl ether. The copolymer was precipitated from methanol and repeatedly washed with acetone/H2O. PE-mPEG with no biomolecules, which is the brush copolymer with side chains of PE and methoxy-PEG, was prepared by the same ROMP strategy. It serves as a control for PE-peptide copolymers in the biological evaluation. The formation of the desired brush copolymer was characterized by 1H NMR spectroscopy, where the characteristic signals from the poly(ethylene glycol) (PEG) chain and polyethylene chains were observed, respectively.
2.2 Preparation of 3D-printed coupons
The obtained PE-peptide brush copolymers III were formulated as “bioadditive” (Fig. 2a) for UHMWPE supplied by Mitsui Chemicals Inc. as the base polymer to create the formulations [PE-peptide/UHMWPE]. Coupons CIII1400 and CIII5000 (Fig. 2c) were created for biological evaluation using these formulations in fused filament fabrication type 3D printing. A blending ratio of 10 wt% of III in UHMWPE was used, whilst filament extrusion was carried out using a Thermo Scientific 11 co-rotating twin-screw extruder to achieve an average filament diameter of 2.85 mm (Fig. 2b). Two types of PE coupons were 3D-printed using an Ultimaker S5 with printing parameters similar to those reported by Dontsov26 and Rocha27 but at a lower print speed to cater for in vitro and in vivo studies. The coupons for in vitro assays were formed with a dimension of 10 × 10 × 0.2 mm3, while the coupons for in vivo assays were formed with a dimension of 5 × 5 × 0.2 mm3.
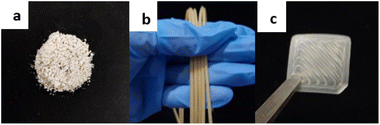 |
| | Fig. 2 (a) Bioactive polyethylene; (b) PE5000-RGD/UHMWPE filaments (FIII5000); (c) a 3D-printed PE5000-RGD/UHMWPE coupon (CIII5000). | |
2.3
In vitro alkaline phosphatase (ALP) assay
Alkaline phosphatase (ALP) is a widely recognized biochemical marker for osteoblast activity. The osteo-inductivity of exogenously applied BMP-2 (bone morphogenetic protein-2) can be measured in vitro using a murine myoblast cell line (C2C12). 3D-printed PE coupons CIII were placed in 100% ethanol for 5 minutes. Excess ethanol was removed, and the coupons were allowed to air-dry for 1 hour under a TC hood. The coupons were incubated with 100 ng BMP-2 in 200 μL of PBS for 20 minutes at room temperature with gentle agitation. The BMP-2 solution was removed, and the coupons were washed twice with 500 μL of PBS. Cells (C2C12) were harvested and seeded on the coupons at a density of 20000 cells per cm2 in DMEM with 5% FCS. The total protein was isolated from cells at day 4 using an ice-cold RIPA buffer. The cells were incubated for 20 minutes with the RIPA buffer, and then the cells and coupons were homogenized using a cell scraper. The insoluble material was removed via centrifugation (12000g, 4 °C, 10 minutes), and the total protein content of the supernatant was quantified by the BCA assay. A total of 7.5 μg of protein from each sample was analysed in triplicate after the addition of pNPP solution (Thermo Fischer) and incubation at 37 °C for 1 hour or longer. Absorbance at 405 nm was measured using a Hidex Sense spectrophotometer. Absorbance values were normalized to no-treatment control and represent mean ± S.D. from duplicate conditions in a single experiment. The procedure was performed as previously reported.28
2.4
In vivo biocompatibility study
The in vivo biocompatibility study of the 3D-printed PE coupons CIII5000 was performed using murine models. Eight-week-old female C57BL/6 wild-type mice were purchased from InVivos Pte Ltd and randomly assigned to one of the following four groups: (1) sham, (2) PE-blank, (3) PE-RGD and (4) PE-GPHP. All groups had at least 4 mice. Briefly, each mouse was anaesthetized with ketamine/xylazine, and a small incision was made on the upper dorsal back. Coupons CIII5000 were then inserted into the subcutaneous space under the skin. Four weeks post-implantation, all mice were sacrificed. Mouse skin surrounding the implant was harvested, fixed in formalin and embedded in paraffin for histological studies. Hematoxylin and eosin (H&E) staining and immunohistochemistry staining for CD3 (clone SP162) were performed on these skin sections. Appropriate controls were included, and images were captured using a Zeiss Axio Scan.Z1 slide scanner. All animal handling procedures were approved by the Institutional Animal Care and Use Committee and conformed to the National Advisory Committee for Laboratory Animal Research Guidelines (IACUC #201550).
3 Results and discussion
3.1 Polymer synthesis and characterization
The random brush copolymers, where two separate macromonomers were copolymerized via ROMP were designed to chemically incorporate hydrophilic biomolecules into hydrophobic PE. Notably, PEG was introduced here as a linker and to adjust softness and hydrophilicity of the final copolymers. The PEG chain also ensures that the bioactive peptide motif remains sterically accessible in the final brush structure and is not buried in the matrix of the synthetic PE side chains. We term such PE copolymers as “bioactive polyethylene” (Fig. 1). This bioactive polyethylene will find much use in biomedical applications where it possesses both the bioactivity of the appended biomolecules and the thermomechanical properties of the synthetic PE side chains. In fact, incorporation of hydrophilic macromonomers into brush polymers with PE side chains is technically difficult due to the challenge of balancing the poor solubility of PE with macromonomers of widely differing properties and the high demands of techniques for its characterization. Brush-type PE copolymers are rare in the literature. Although Hadjichristidis and co-workers29 reported one example of PE-based homobrushes, the brush polymer was homopolymerized with only norbornyl PE-based macromonomers with no biomolecules incorporated.
Due to the differing properties of peptide-based macromonomers I and PE macromonomers II, optimization of the ROMP reaction conditions for copolymerization of I and II was challenging, on its own. We began screening the copolymerization using NB-SA-t PE1400 (IIa) and NB-PEG1000-RGD (Ia) as representative macromonomers (Table 1). Due to the limited solubility of IIa in common organic solvents such as dichloromethane, THF and methanol, copolymerization had to be carried out in solvents compatible with IIa such as benzene and toluene at elevated temperature. Benzene at 75 °C worked as the best solvent for this ROMP, providing 13% incorporation of NB-PEG1000-RGD (Ia) into copolymer IIIa with quantitative conversion relative to IIa (entry 1). A ROMP using highly active 3rd generation bis(pyridine) Grubbs’ catalyst [G3] at 1.25 mol% with other factors unchanged was attempted but resulted in low conversion of I and incorporation of Ia. This is likely due to the low thermal stability of [G3]. Higher [G2] concentrations did not yield significant improvements in conversion or Ia incorporation (entries 2 and 3). Interestingly, Ia incorporation halved upon prolonged stirring from 24 h to 40 h (entry 4). This may be due to secondary metathesis reactions occurring during the prolonged reaction in the presence of [G2]. MMs with a lower molar ratio of [I:II] of [1:10] was attempted for reaction sustainability (entry 5). However, a decrease in Ia incorporation was observed. Hence, the MMs molar ratio was maintained at [1:5]. In view of the differing reactivities of these two MMs, we also tested the addition of Ia at two time points (t = 0 and 2 h) (entry 6). Only 6% incorporation of Ia was obtained. Thus, it is noteworthy that the reactivity of Ia in non-polar solvents is challenging, even with the gradual addition of the bio-macromonomer. We also explored the possibility of carrying out this reaction under atmospheric conditions. However, the incorporation of Ia is lowered to 4% (entry 7). Homopolymerization of these two types of macromonomers Ia and IIa under the same conditions was also carried out separately, to examine their reactivity (entries 8 and 9). Adaptability of Ia and IIa in this ROMP system varied. The PE macromonomer IIa showed comparatively efficient conversion (92%) compared to PEGylated macromonomer Ia (66%). This is likely due to better solubility of hydrophobic IIa in benzene compared to that of hydrophilic Ia. Hence, it is within expectations that ROMP of these two MMs together would be even more challenging.
Table 1 ROMP condition optimizationa
| Entry |
Catalyst (mol%) |
[Ia]0:[IIa]0 |
Conversionb (%) |
Ia Incorporationc (mol%) |
|
Reaction was conducted in 75 °C benzene in the presence of [G2] under an N2 atmosphere with [IIa]0 = 0.05 M for 24 h.
Conversion was calculated by 1H NMR analysis according to norbornyl protons.
Incorporation of Ia was determined by 1H NMR analysis according to the PEG signal of Ia.
Reaction was conducted for 40 h.
10 mol% × 2, added respectively at t = 0 and 2 h.
Reaction was conducted under an air atmosphere.
Homopolymerization of NB-PEG1000-RGD.
Homopolymerization of NB-SA-t PE1400.
|
| 1 |
1.25 |
[1:5] |
>99 |
13 |
| 2 |
1.75 |
[1:5] |
89 |
6 |
| 3 |
2.00 |
[1:5] |
88 |
6 |
| 4d |
1.25 |
[1:5] |
73 |
3 |
| 5 |
1.25 |
[1:10] |
88 |
5 |
| 6 |
1.25 |
[1:5]e |
83 |
6 |
| 7f |
1.25 |
[1:5] |
86 |
4 |
| 8 |
1.25 |
[100:0]g |
66 |
— |
| 9 |
1.25 |
[0:100]h |
92 |
— |
Overall, by balancing the challenges of poor solubility of polyethylene and differing properties between Ia and IIa, we were able to achieve the optimized ROMP conditions using 1.25 mol% 2nd generation Grubbs’ catalyst [G2] in 75 °C benzene with a molar ratio of [I:II] at [1:5], to achieve the optimum conversion of IIa and incorporation of Ia into the copolymer. However, in consideration of the carcinogenicity of benzene, we sought to employ a more benign solvent such as 1,2-dichlorobenzene (1,2-DCB). Fortunately, we were able to replicate the reaction outcomes under the same reaction conditions.
Upon optimizing the ROMP conditions of NB-SA-t PE1400 (IIa) and NB-PEG1000-RGD (Ia), we extended the reaction conditions to longer chain NB-SA-t PE5000 macromonomer (IIb), longer PEG chain (MW 3400) and other PEGylated peptide macromonomers (Ib–d) such as collagen fragment (GPHyp)3 (Table 2, entries 2–4). Good conversion of II (>70%) and incorporation of I (>3%) were obtained. Overall, bioactive PE-peptide brush copolymers III could be afforded by ROMP with a conversion of over 90% on NB-SA-t PE1400 and 70% on NB-SA-t PE5000, with NB-PEG-peptide incorporation of up to 13%, using the presented strategies.
Table 2 ROMPa conversion and incorporation of PE-peptide brush copolymers III
| Entry |
PE MMs II |
PEG MMs I |
Brush copolymer III |
Conversionb (%) |
I incorporationc (mol%) |
|
Reaction was conducted in 75 °C 1,2-dichlorobenzene (1,2-DCB) in the presence of 1.25 mol% [G2] with [II]0 = 0.05 M under an N2 atmosphere for 24 h.
Conversion of II was calculated by 1H NMR analysis according to the norbornyl protons of II.
Incorporation of I was determined by 1H NMR analysis according to the PEG signal of I.
|
| 1 |
NB-SA-t PE1400 |
NB-PEG1000-RGD |
IIIa
|
>99 |
13 |
| 2 |
NB-SA-t PE5000 |
NB-PEG3400-RGD |
IIIb
|
72 |
3 |
| 3 |
NB-SA-t PE1400 |
NB-PEG1000-(GPHyp)3 |
IIIc
|
>99 |
6 |
| 4 |
NB-SA-t PE5000 |
NB-PEG3400-(GPHyp)3 |
IIId
|
77 |
4 |
| 5 |
NB-SA-t PE1400 |
NB-methoxy-PEG1000 |
IVa
|
>99 |
12 |
| 6 |
NB-SA-t PE5000 |
NB-methoxy-PEG5000 |
IVb
|
99 |
4 |
Alongside the above, PE (MW 1400 and 5000)–methoxy-PEG (MW 1000 and 5000) copolymers without biomolecules (IV) were also prepared under the same conditions (Table 2, entries 5 and 6). Copolymers IV serve as controls in the subsequent biovalidation. The higher conversion and incorporation of I into IV is due to the higher solubility of NB-methoxy-PEG in 1,2-dichlorobenzene (DCB). The syntheses of brush copolymers III and IV were subsequently scaled up to 2 g with high consistency in both conversion and biomacromonomer incorporation for further material processing.
We have also explored an alternative synthetic route to obtain IIIa as shown in Scheme 4. First, condensation of cis-norbornene-exo-2,3-dicarboxylic anhydride with hexamethylenediamine (HMDA) was performed to obtain a monomer (NB-HMDA) for ROMP with Ia, followed by grafting of SA-t PE onto the brush polymer in the final step. However, the ROMP step only gave a low yield of less than 25%. We noticed a significant amount of unreacted Ia and NB-HMDA after the ROMP reaction. Although the subsequent PE chain “grafting-on” can still be carried out with SA-t PE (MW 1400), it is noted that this route to obtain IIIa is impractical.
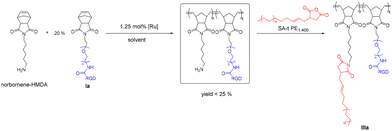 |
| | Scheme 4 Alternative synthetic route to form PE1400-RGD copolymer IIIa, where [Ru] refers to the Grubbs’ catalysts. | |
The formation of the PE-peptide copolymer was evident from 1H NMR spectroscopy. The NMR characterization of PE polymers is challenging due to its limited solubility in common deuterated organic solvents at room temperature. Finally, we managed to determine the copolymer structure from spectra recorded using d4-dichlorobenzene (1,2-C4D6Cl2) at 120 °C on a Bruker ASCEND cryo-400 NMR spectrometer. Using 1H NMR spectra of the PE1400-RGD copolymer IIIa (Fig. 3) as a reference, chemical shifts of vinyl protons (5.48 and 5.31 ppm), PE chains (1.29 ppm), PEG chains (3.55 ppm), the norbornyl skeleton (5.80 ppm) and the terminal –CH3 of PE chains (0.84 ppm) could be identified. Repeated purification of the copolymer using acetone/H2O ensured that there is no unreacted NB-PEG1000-RGD (Ia) left. Thus, the chemical shift at 3.55 ppm due to PEG is indicative of PEGylated biomolecule incorporation into the final brush copolymer. However, separation of PE-based macromolecules is known to be extremely challenging and impractical on a large scale. Due to the very limited solubility of PE in common organic solvents at moderate temperatures, we are still investigating methods to separate unreacted II from III. It is heartening to note from our bioassays that the unreacted NB-SA-t PE1400 (IIa) is non-cytotoxic. Furthermore, from the 1H NMR, only a trace amount of the norbornyl protons (6.06 ppm) from NB-SA-t PE1400 (IIa) was observed. 1H NMR spectra for all other polymers and calculation of PE MM I conversion and PEGylated MM II incorporation can be found in the ESI.†
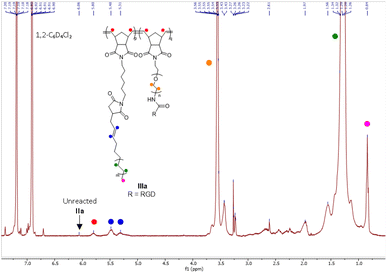 |
| | Fig. 3
1H NMR spectrum of the PE1400-RGD copolymer IIIa. | |
Gel permeation chromatography (GPC) analysis of polymers containing both very hydrophobic PE and highly polar PEG-peptide is challenging in terms of finding a suitable GPC eluent and temperature. Thus, it is understandable that establishment of a realistic MW distribution and measurement of the true MW of the polymers are expected to be difficult. The values of various absolute molecular weights shown in Table 3 were calculated on the assumption that the relation of the first-order equation holds between the measured logarithmic value of the absolute molecular weight and the holding time. A certain amount of granular insoluble components was observed in a sample solution of IIIb during the GPC operation. This is consistent with the relatively lower Mn of IIIb observed since the insoluble components were filtered away prior to the GPC operation. These insoluble components are likely to be the PE5000-RGD copolymer of greater molecular size. Alongside these observations, it is also noteworthy that due to the limited solubility in solvents, there is no way to separate the unreacted NB-SA-t PE macromonomer from the final brush copolymer. However, since the NB-SA-t PE macromonomer is present in a very small amount, the effect on the GPC analyses is marginal for PE1400 copolymers IIIa and IIIc. However, a slight degree of inaccuracy in the MW determination for PE5000 copolymers IIIb and IIId is expected due to lower conversion in this ROMP reaction. Repeated washing of mixtures with acetone/H2O is the method we currently use for further purification. Investigation on the feasible method for PE derivative separation is underway. GPC traces including peptide-based macromonomers Ia–d are shown in the ESI.†
Table 3 GPC data of NB-SA-t PE MMs II and copolymers III
| Polymers |
M
n
(KDa) |
PDI |
|
Determined by HT-GPC in 1,2,4-trichlorobenzene at 160 °C.
|
| 1 |
NB-SA-t PE1400IIa |
3.0 |
1.38 |
| 2 |
NB-SA-t PE5000IIb |
4.2 |
2.41 |
| 3 |
PE1400-RGD copolymer IIIa |
5.7 |
12.78 |
| 4 |
PE5000-RGD copolymer IIIb |
6.4 |
3.49 |
| 5 |
PE1400-(GPHyp)3 copolymer IIIc |
20.8 |
4.18 |
| 6 |
PE5000-(GPHyp)3 copolymer IIId |
26.7 |
3.79 |
3.2 Thermal stability
Upon synthesis and chemical characterization, thermal gravimetric analyses (TGA) of the copolymers were conducted to ascertain their thermal stability before we proceeded with filament extrusion and 3D printing of coupons for biovalidation (Fig. 4). From the representative TGA graph of IIIa [3], we can see improved thermal stability of the bioactive PE1400-RGD copolymer IIIa [3], over that of pure RGD peptide [1] and the PEGylated peptide macromonomer (NB-PEG1000-RGD) Ia [2]. It was observed that the pure RGD [1] starts degrading slightly above 100 °C, whereas Ia [2] shows a 2-phase mass loss between 200 and 350 °C, likely due to the loss of RGD followed by the degradation of PEG. On the other hand, IIIa [3] had only lost about 5% of its weight at around 400 °C, likely from RGD loss. The significant improvement in the thermal stability of IIIa over that of either pure RGD or Ia allows most material processing methods such as melt extrusion-based 3D printing to be used on IIIa. Apart from TGA analyses, inductively coupled plasma mass spectrometry (ICP-MS) analysis of the isolated brush copolymers was also carried out to check and ensure that the residual Ru content in the copolymers falls below 1 ppm (Table S3†) for their acceptable use in tissue-contact medical device fabrication.30
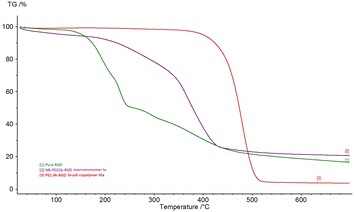 |
| | Fig. 4 TGA of pure RGD, Ia and IIIa. | |
3.3
In vitro osteogenic activity
Osteogenic and BMP-2 binding properties are important in orthopaedic implant materials. BMP binding is important for both osteogenesis and chondrogenesis. Alkaline phosphatase (ALP) assays using C2C12 myoblast cells and BMP-2 were performed on the 3D-printed coupon CIIIa of PE1400-RGD/UHMWPE (Fig. 5). The ALP activity of C2C12s cultured on the PE1400-RGD/UHMWPE coupon CIIIa (“PE1kRGD”) was over 20% higher than that of neat UHMWPE (“PE blank”), suggesting that the incorporation of bioactive PE1400-RGD copolymer into UHMWPE improves the osteogenic differentiation of C2C12 murine myoblasts in vitro.
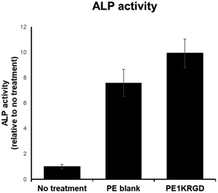 |
| | Fig. 5 ALP activity of C2C12 murine myoblast cells cultured on the 3D-printed PE1400-RGD/UHMWPE coupon CIIIa. | |
ALP activity of the C2C12 cells with BMP-2 was also analyzed on the 3D-printed PE5000-peptide/UHMWPE coupons CIIIb (“PE5k-RGD”) and CIIId (“PE5k-GPHP”) (Fig. 6). From the data, it is clear that all the 3D-printed coupons are able to support BMP-2-mediated ALP activity. However, little difference in ALP activity is observed between PE5000-RGD/UHMWPE CIIIb (“PE5k-RGD”) and PE5000-mPEG/UHMWPE CIVb (“PE5k-mPEG”) after the addition of BMP-2. The lower ALP activity improvement over neat UHMWPE (“PE-blank”) in CIIIb may be due to lower RGD content in PE5000-RGD/UHMWPE CIIIb since the blending with UHMWPE is by weight. Between PE5000-RGD/UHMWPE CIIIb (“PE5k-RGD”) and PE5000-(GPHyp)3/UHMWPE CIIId (“PE5k-GPHP”), it is clear that CIIIb (“PE5k-RGD”) is able to enhance osteogenic activity more, indicating that the incorporation of RGD peptide into brush copolymer III is able to enhance BMP-2-mediated signals to a greater extent compared to that of the collagen fragment (GPHyp)3. Based on the GPC characteristics of IIId, there is unseparated UHMWPE contained in the final CIIId, resulting in the poor ALP assay performance. The outcome here showed that PE5000-(GPHyp)3IIId is less promising to be the additive for orthopaedic applications.
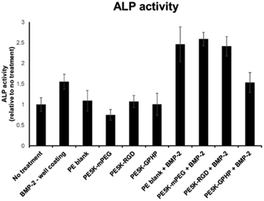 |
| | Fig. 6 ALP activity of C2C12 murine myoblast cells with BMP-2 cultured on the 3D-printed PE5000-peptide/UHMWPE coupons CIIIb and CIIId. | |
3.4
In vivo biocompatibility
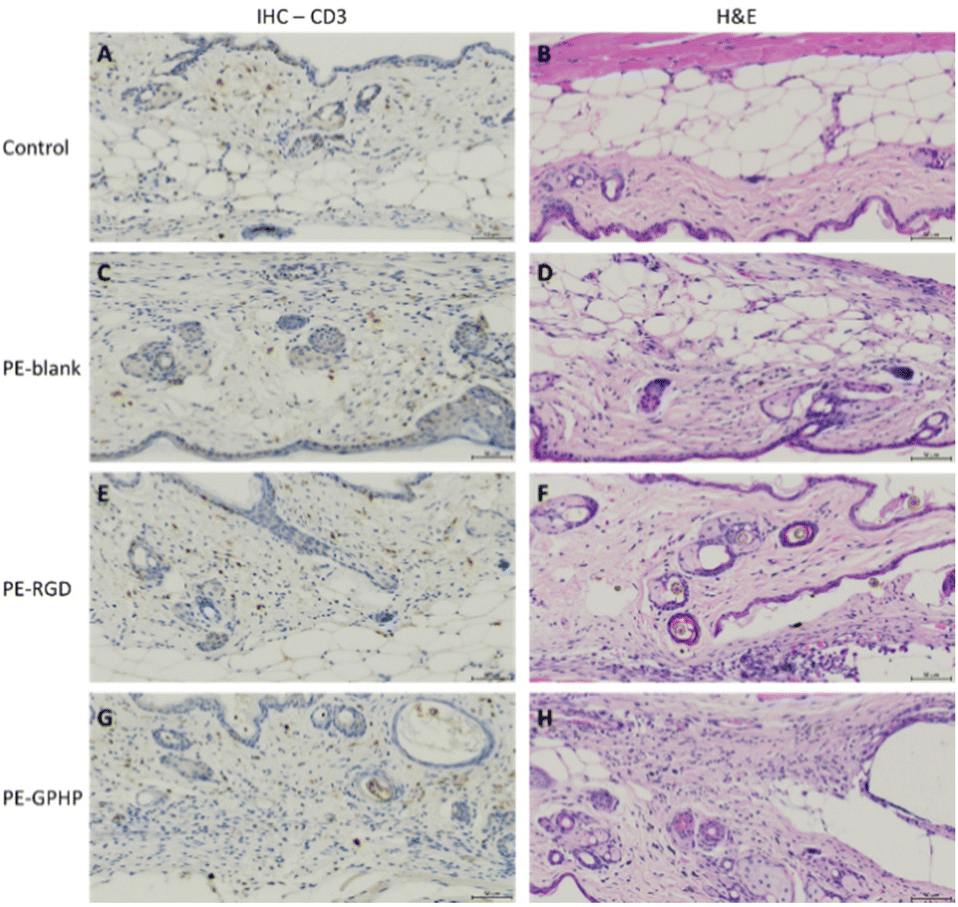
In vivo biocompatibility evaluation of the 3D-printed PE5000-peptide/UHMWPE coupons CIIIb and CIIId was performed using murine models. 3D-printed coupons CIIIb (“PE-RGD”) and CIIId (“PE-GPHP”) were inserted into the subcutaneous space under the skin via a small incision made on the upper dorsal back of the mouse. Histological analyses of the tissues at the implantation site after animal sacrifice at the end of week 4 showed increased inflammation of tissues due to the neat UHMWPE coupon (“PE-blank”) (Fig. 7C and D) and CIIId coupon (“PE-GPHP”) (Fig. 7G and H). Notably, a reduction in inflammation was observed for the CIIIb coupon (“PE-RGD”) (Fig. 7E and F). This inflammation reduction of CIIIb (“PE-RGD”) is important for its use in orthopaedic implants since the UHMWPE is a popular material for articulating surfaces of metal joint implants, but it is also known to cause significant inflammatory responses in the human body.31,32 The ability of IIIb to reduce the inflammatory response in vivo and its ability to bind BMP-2 makes it a promising polymer additive for use in polyethylene-based orthopaedic implant materials.
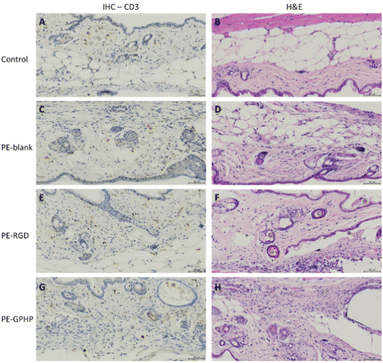 |
| | Fig. 7 Comparison of the bio-implanted murine skin tissues. (A, C, E and G) Immunohistochemical (IHC) staining for CD3 + cells, with cell nuclei (blue) and CD3 (brown) differentially labelled. (B, D, F and H) Haematoxylin and eosin (H&E) staining, with nuclear component (haematoxylin) and cytoplasmic components (eosin) differentially stained. Pathological assessment reports 2+ for (A and B) negative control, 3+ for (C and D) PE-blank, 2+ (E and F) PE-RGD and 3+ (G and H) PE-GPHP. All images shown are representative of at least 4 C57BL/6 mice per group. Scale bar = 50 μm. | |
4 Conclusions
In summary, we report a modular and efficient strategy for the synthesis of bioactive PE-peptide copolymers via ROMP. Synthesis and characterization of brush polymers containing PE side chains and PEGylated peptides have been presented. This method can potentially be extended to various peptides to provide access to versatile biofunctionalized brush copolymers. By incorporating integrin-binding peptides such as RGD and collagen mimics such as (GPHyp)3 into the PE-based brush copolymers, osteogenic activity and inflammatory response reduction could be introduced. The improvement of the thermal stability of the peptides in the brush copolymers allow these polymers to function as biomolecule carriers in high melting point UHMWPE. The reduction in inflammatory response and BMP-2 binding capability of our bioactive PE/UHMWPE formulation make the material an attractive candidate for use in PE-based orthopaedic devices such as articulating surfaces of joint implants. Optimization of formulations and further studies on the use of these bioactive PE materials using in vivo large animal models for tissue repair are ongoing.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We acknowledge the generous support for this research from the Institute of Materials Research (IMRE) and Engineering and the Institute of Molecular and Cell Biology (IMCB), A*STAR, Singapore. This work was funded by Wound Care Innovation in the Tropics (grant H1701a00N9) and A*STAR GAP projects (ACCL190291, 200322). All succinic-acid terminated polyethylene and UHMWPE were obtained from Mitsui Chemicals Inc. (MCI) via materials transfer agreements between A*STAR and MCI. We thank Dr Wang Hongbo from the Mitsui Chemicals Singapore R&D Center, Dr Song Xiaolu from Jiangsu University, China, and Mr Heng Teck Huat from IMRE, for their contributions in the early phase of this work.
References
- A. J. T. Teo, A. Mishra, I. Park, Y. J. Kim, W. T. Park and Y. J. Yoon, ACS Biomater. Sci. Eng., 2016, 2, 454–472 CrossRef CAS PubMed.
- A. Bharadwaz and A. C. Jayasuriya, Mater. Sci. Eng., C, 2020, 110, 110698 CrossRef CAS PubMed.
-
V. R. Sastri, in 3 - Materials Used in Medical Devices, ed. V. R. Sastri, William Andrew Publishing, Oxford, 2014, pp. 19–31 Search PubMed.
- E. Zolotarevova, G. Entlicher, E. Pavlova, M. Slouf, D. Pokorny, F. Vesely, J. Gallo and A. Sosna, Acta Biomater., 2010, 6, 3595–3600 CrossRef CAS PubMed.
- A. Gigante, C. Bottegoni, V. Ragone and L. Banci, J. Funct. Biomater., 2015, 6, 889–900 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- R. Langer, Biomaterials in Drug Delevery and Tissue Engeneering: One Laboratory's Experience, Acc. Chem. Res., 2000, 33, 94–101 CrossRef CAS PubMed.
- I. M. Weiss, C. Muth, R. Drumm and H. O. K. Kirchner, BMC Biophys., 2018, 11, 2 CrossRef PubMed.
- S. L. Perry, Curr. Opin. Colloid Interface Sci., 2019, 39, 86–97 CrossRef CAS.
- P. M. Stephen and A. H. Jeffrey, An RGD spacing of 440 nm is sufficient for integrin alpha V beta 3- mediated fibroblast spreading and 140 nm for focal contact and stress fiber formation, J. Cell Biol., 1991, 114(5), 1089–1100 CrossRef PubMed.
- P. R. Patel, R. C. Kiser, Y. Y. Lu, E. Fong, W. C. Ho, D. A. Tirrell and R. H. Grubbs, Biomacromolecules, 2012, 13, 2546–2553 CrossRef CAS PubMed.
- S. Y. Yoo, M. Kobayashi, P. P. Lee and S. W. Lee, Biomacromolecules, 2011, 12, 987–996 CrossRef CAS PubMed.
- H. Ceylan, S. Kocabey, H. Unal Gulsuner, O. S. Balcik, M. O. Guler and A. B. Tekinay, Biomacromolecules, 2014, 15, 2407–2418 CrossRef CAS PubMed.
- K. M. Hennessy, B. E. Pollot, W. C. Clem, M. C. Phipps, A. A. Sawyer, B. K. Culpepper and S. L. Bellis, Biomaterials, 2009, 30, 1898–1909 CrossRef CAS PubMed.
- G. Bohn, B. Liden, G. Schultz, Q. Yang and D. J. Gibson, Adv. Text. Wound Care, 2016, 5, 1–10 CrossRef PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chemistry-Based Functional Proteomics Reveals Novel Members of the Deubiquitinating Enzyme Family, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- E. M. Kolonko, J. K. Pontrello, S. L. Mangold and L. L. Kiessling, General Synthetic Route to Cell-Permeable Block Copolymers via ROMP, J. Am. Chem. Soc., 2009, 131(21), 7327–7333 CrossRef CAS PubMed.
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nusslein and G. N. Tew, Antimicrobial polymers prepared by ROMP with unprecedented selectivity: a molecular construction kit approach, J. Am. Chem. Soc., 2008, 130(30), 9836–9843 CrossRef CAS PubMed.
- Y. C. Teo, E. J. Park, J. Guo, A. Asyraf, A. Smith, D. Goh, J. P. S. Yeong, S. Cool and P. Teo, ACS Appl. Bio Mater., 2022, 5(10), 4770–4778 CAS.
-
V. Héroguez, A. Chemtob and D. Quemener, in ROMP in Dispersed Media, 2015, pp. 25–44 Search PubMed.
-
C. Slugovc, in Synthesis of Homopolymers and Copolymers, 2015, pp. 1–23 Search PubMed.
- J. B. Matson and R. H. Grubbs, Synthesis of Fluorine-18 Functionalized Nanoparticles for Use as in Vivo Molecular Imaging Agents, J. Am. Chem. Soc., 2008, 130, 6731–6733 CrossRef CAS PubMed.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, Synthesis of Norbornenyl Polymers with Bioactive Oligopeptides by Ring Opening Metathesis Polymerization, Macromolecules, 2000, 33(17), 6239–6248 CrossRef CAS.
- A. V. Sesha Sainath, M. Isokawa, M. Suzuki, S. Ishii, S. Matsuura, N. Nagai and T. Fujita, Macromolecules, 2009, 42, 4356–4358 CrossRef.
- M. S. Sanford, J. A. Love and R. H. Grubbs, Mechanism And Activity Of Ruthenium Olefin Metathesis Catalysts, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- Y. V. Dontsov, S. V. Panin, D. G. Buslovich and F. Berto, Materials, 2020, 13 Search PubMed.
- C. R. Rocha, A. R. Torrado Perez, D. A. Roberson, C. M. Shemelya, E. MacDonald and R. B. Wicker, J. Mater. Res., 2014, 29, 1859–1866 CrossRef CAS.
- R. A. A. Smith, S. Murali, B. Rai, X. Lu, Z. X. H. Lim, J. J. L. Lee, V. Nurcombe and S. M. Cool, Biomaterials, 2018, 184, 41–55 CrossRef CAS PubMed.
- H. Zhang and N. Hadjichristidis, Macromolecules, 2016, 49, 1590–1596 CrossRef CAS.
- ICH guideline Q3D (R1) on elemental impurities 2019, March 22.
- I. Fernandez-Bueno, S. Di Lauro, I. Alvarez, J. C. Lopez, M. T. Garcia-Gutierrez, I. Fernandez, E. Larra and J. C. Pastor, J. Ophthalmol., 2015, 904096 Search PubMed.
- E. Gibon, L. A. Cordova, L. Lu, T. H. Lin, Z. Yao, M. Hamadouche and S. B. Goodman, J. Biomed. Mater. Res., Part B, 2017, 105, 1685–1691 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Eun Ju
Park
a,
Yew Chin
Teo
a,
Eun Ju
Park
a,
Yew Chin
Teo