
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branched phosphazenium salts as effective and versatile cocatalysts for epoxide/CO2 coupling†
Massimiliano
Brivio
a,
Lorenzo
Veronese
a,
Incoronata
Tritto
 a,
Paolo
Biagini
b,
Riccardo
Po’
b,
Laura
Boggioni
*a and
Simona
Losio
*a
a,
Paolo
Biagini
b,
Riccardo
Po’
b,
Laura
Boggioni
*a and
Simona
Losio
*a
aCNR-SCITEC, Istituto di Scienze e Tecnologie Chimiche “Giulio Natta”, via A. Corti 12, 20133, Milano, Italy. E-mail: simona.losio@scitec.cnr.it; laura.boggioni@scitec.cnr.it
bIstituto Eni Donegani, via Fauser 4, 28100, Novara, Italy
First published on 26th January 2023
Abstract
Branched phosphazenium salts of general formula [(Me2N)3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N]4P+X− (X− = Cl− or N3−) have been tested as cocatalysts for different catalyst systems, such as salen-type chromium and porphyrin cobalt complexes, in the alternating copolymerization of CO2 and epoxides (CHO and PO) under various experimental conditions. Higher monomer conversion than that obtained with the benchmark PPN+X− counterparts under the same conditions was achieved. The polymers obtained have comparable or greater molar masses. Thus, phosphazenium salts can be viable, if not superior, alternatives to onium salts in CO2/epoxide ROCOP.
N]4P+X− (X− = Cl− or N3−) have been tested as cocatalysts for different catalyst systems, such as salen-type chromium and porphyrin cobalt complexes, in the alternating copolymerization of CO2 and epoxides (CHO and PO) under various experimental conditions. Higher monomer conversion than that obtained with the benchmark PPN+X− counterparts under the same conditions was achieved. The polymers obtained have comparable or greater molar masses. Thus, phosphazenium salts can be viable, if not superior, alternatives to onium salts in CO2/epoxide ROCOP.
Introduction
Over the past decades, metal- and organocatalyzed polymerization reactions have attracted remarkable attention, leading to major advances in this field. The availability of huge combinations of both ligands and metal centers led to the rapid development of metal-based catalysts. Indeed, transition metal coordination complexes are at the heart of polymerization catalysis.1 In the past 30 years, relevant efforts have been made for the development of homogeneous and heterogeneous catalysts for the ring opening copolymerization (ROCOP) of CO2 and epoxides, such as cyclohexene oxide and propylene oxide (from now on CHO and PO, respectively).2 This reaction is an interesting method to achieve aliphatic polycarbonates, although different amounts of cyclic by-products can be obtained.3 Thanks to a thorough molecular design of ligands bound to metal centers, numerous catalysts show, at the same time, high activity and high selectivity towards the alternating insertion of CO2 and epoxides. In all these catalytic systems involving transition metals, chain growth occurs by prior coordination of epoxides to the metal complex followed by the ring opening and formation of an alkoxide species from which the growth of the chain for alternating insertion begins as shown in Scheme 1, where X is a nucleophile coming from the cocatalyst or already present on the catalyst.4–6 | ||
| Scheme 1 Mechanism for poly(propylenecarbonate) formation. | ||
The most investigated classes of catalytic systems consist of salen or porphyrin complexes with chromium, cobalt or aluminium as the metal together with an ionic or neutral nucleophilic co-catalyst, often 4-(N,N-dimethylamino)pyridine (DMAP) or soluble “onium” halide salts such as R4N+X− or Ph3PN+PPh3 (PPN+)X−.
Phosphazenes are well-known organic and uncharged superbases with almost no nucleophilicity with variable numbers of phosphorus atoms and topologies (linear, branched or cyclic) (Fig. S1 in the ESI†) and have been widely studied as organocatalysts for the ring-opening polymerization (ROP) of various kinds of cyclic monomers.7 The first known phosphazene base was synthesized in the early 1970s and had a phosphorus(V) atom with one imine group and three amine groups.8 Due to their highly basic and non-nucleophilic nature, phosphazenes can easily turn protic compounds into nucleophilic initiators by the deprotonation or activation of weak nucleophiles, which could further initiate fast and controlled ROP reactions of various types of monomers such as epoxides.9 Despite their relative popularity as ROP catalysts, there is a very limited number of examples of phosphazenes used in ring-opening copolymerization (ROCOP).10,11 In 2016, different phosphazene bases have been successfully used as organocatalysts for the alternating copolymerization of epoxides and other types of monomers such as ethylene oxide or styrene oxide and 3,4-dihydrocoumarin.12
With regard to the copolymerization of epoxides and CO2, the number of reported cases of phosphazene use is very small. Feng reported the effective all-organic copolymerization of cyclohexene (CHO) and propylene oxide (PO) and CO2 with Et3B as an initiator in the presence of phosphazenium alkoxides or phosphonium halides such as PPNCl.11 Recently, in a seminal work Liu and Li reported three phosphazenes as binary organocatalysts in combination with Et3B for the copolymerization of CO2 and different epoxides under mild conditions.13 Organocatalysis represents an alternative method for the synthesis of polymers under metal-free conditions, being the strategy used to manipulate the reactivity of the two monomers through their activation with a non-metal external activator such as TEB. Moreover, Coates achieved high-Mw enantiopure poly(propylene oxide) by combining tetrakis[(tri-1-pyrrolidinylphosphoranyliden)amino]phosphonium acetate with a bimetallic cobalt catalyst usually employed with CO2.14
However, to the best of our knowledge, there is no available open literature to date on the use of phosphazenium salts as counterparts to the very effective PPN+X− (X = halides or azide) cocatalyst for the synthesis of polycarbonates mediated by common monometallic catalysts. In this paper, we report the high versatility and remarkable efficiency of two P5 branched phosphazenium salts of general formula [(Me2N)3PN]4P+X− (with X− = Cl− or N3−), namely tetrakis[tris(dimethylamino)phosphoranylideneamino]phosphonium chloride (from now on PPZCl) and azide (from now on PPZN3), under various synthetic conditions and in combination with different transition metal catalysts (Fig. 1). Part of this research work has been patented by some of the authors.15
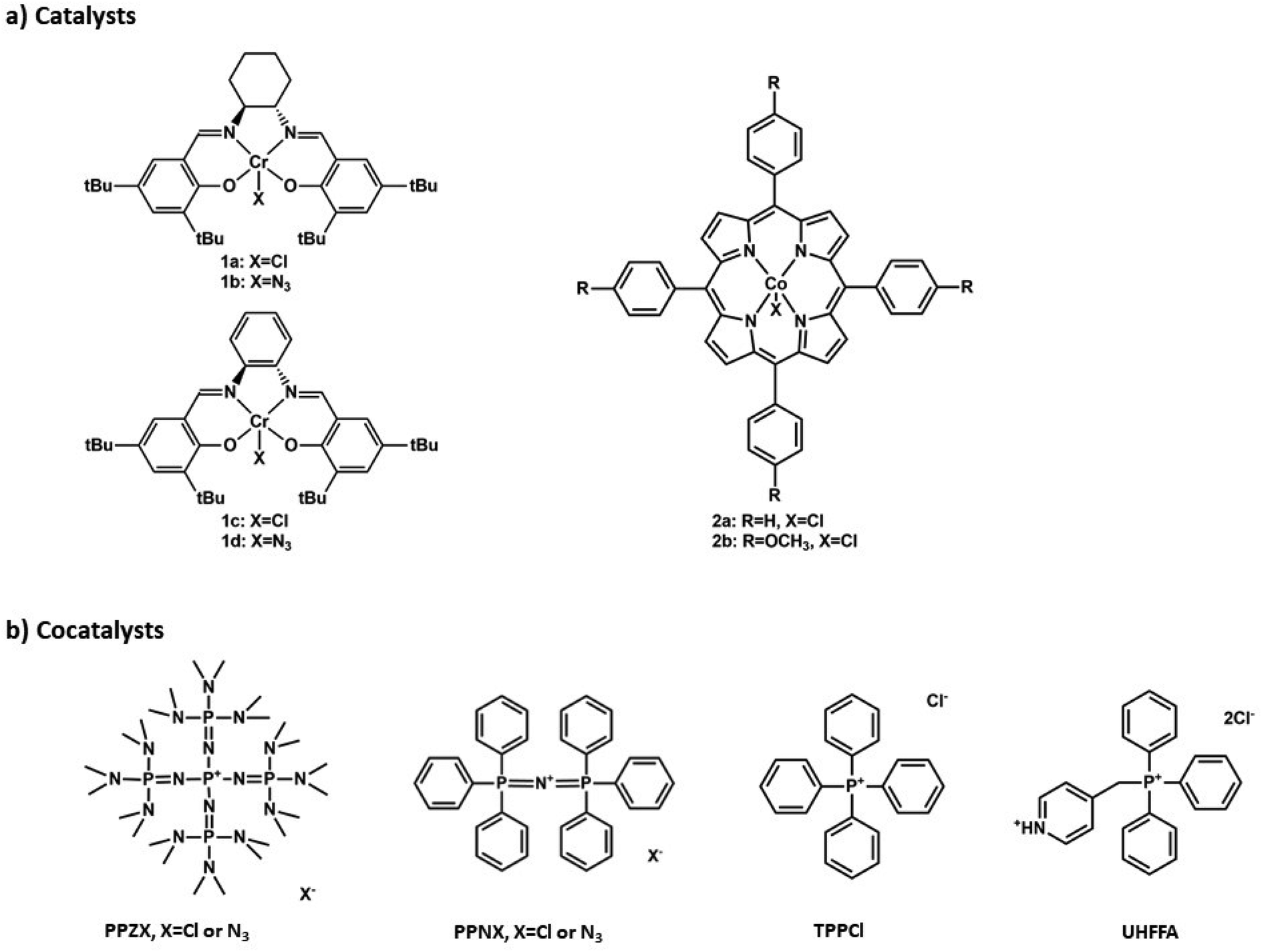 | ||
| Fig. 1 Salen (1a and 1b), salaphen (1c and 1d) and porphyrin (2a and 2b) derived metal catalysts and PPZX, PPNX, TPPCl and UHFFA cocatalysts used in this work. | ||
To make a thorough comparison with PPNX salts and to better assess the versatility of PPZX, the phosphazenium salts have been tested with salen-type chromium complexes, the most known (and employed) catalysts for the two considered epoxides (Fig. 1).
Some reactions have been conducted with porphyrin cobalt complexes as well, but entirely with PO, since, according to the literature, the successful copolymerization of CHO and CO2 to poly(cyclohexene carbonate) (from now on PCHC) is obtained with porphyrins only in the presence of neutral Lewis bases such as DMAP and not with onium salts.16,17
Experimental
Materials and methods
Synthesis of tetrakis[tris(dimethylamino)phosphoranylideneamino]phosphonium azide (PPZN3)
Equimolar amounts of PPZCl and NaN3 were dissolved in dry EtOH under nitrogen and stirred at room temperature for at least 5 h, and then filtered to remove NaCl. The solvent was evaporated under vacuum, and the crude product was dissolved again in a few drops of CH3CN under nitrogen and then precipitated in Et2O, affording a pure white powder in good yield (75%). ATR (cm−1): ν(N3) 1988, ν(PN) 1261. 1H NMR (CD2Cl2, 300 K, 600 MHz) δH (ppm) 2.59 (d, 72H). Anal. calcd for C24H72N19P5: C, 36.87; H, 9.28; N, 34.04. Found: C, 36.5; H, 9.1; N, 34.8.
Alternating copolymerization of cyclohexene oxide and CO2
The catalyst (1 eq.) and cocatalyst (1 eq.) were dissolved in 5 mL of CH2Cl2, stirred at room temperature for at least 1.5 hours, and then the solvent was removed under vacuum. 10 mL (2500 eq.) of neat cyclohexene epoxide were added to the solid residue and the solution was injected into a 25 mL steel autoclave at ambient temperature which was previously dried in vacuo at 80 °C overnight. The autoclave was charged with the desired pressure of CO2 and heated to the reaction temperature. After a period of 3.5 h, the autoclave was cooled to room temperature and vented in a fume hood. A portion of the crude polymer was collected for NMR analysis while the rest was extracted as a dichloromethane solution and precipitated in a MeOH/HCl (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution. The purification process was repeated three times and the purified polymer was dried in vacuo at 80 °C overnight.
:1) solution. The purification process was repeated three times and the purified polymer was dried in vacuo at 80 °C overnight.
Alternating copolymerization of propylene oxide and CO2
The catalyst (1 eq.) and cocatalyst (0.5 eq.) were dissolved in 5 mL of CH2Cl2 and stirred at room temperature for at least 1.5 hours, and then the solvent was removed under vacuum. 10 mL (2500 eq.) of neat propylene epoxide were added to the solid residue and the solution was added via the injection port into a 25 mL steel autoclave at ambient temperature which was previously dried in vacuo at 80 °C overnight. The autoclave was charged with the desired pressure of CO2 and heated to the reaction temperature. After the designed reaction time, the autoclave was cooled to room temperature and vented in a fume hood. A portion of the crude polymer was immediately collected and frozen for NMR analysis, while the rest was extracted as a dichloromethane solution and precipitated in a MeOH/HCl (9:1) solution. The so-obtained polymer was dissolved again in dichloromethane and precipitated in diethyl ether two times, and then the purified polymer was dried in vacuo at 40 °C overnight.
Polymer characterization
All the 1H and 13C NMR spectra were recorded on a Bruker DMX 600 MHz NMR spectrometer in CD2Cl2 at 300 K. Infrared spectra (FTIR-ATR) were recorded on a PerkinElmer Spectrum Two spectrometer. Size exclusion chromatography (SEC) analyses were performed on a Waters GPCV2000 system using THF as the mobile phase at 35 °C with a 0.6 mL min−1 flow. The sample concentration was set at 3 mg mL−1 and the injection volume at 150 μL. Polystyrene standards were used for the calibration of curves in the 162–380000 g mol−1 range. Calorimetric curves were obtained using a PerkinElmer DSC 8000 under a nitrogen atmosphere, with the samples being heated from −80 to 150 °C and cooled vice versa twice at 20 °C min−1. Analytical elemental analyses were performed at the Department of Chemistry, Materials and Chemical Engineering (CMIC) at Politecnico di Milano.
Results and discussion
In spite of their widespread use as catalysts for epoxide homopolymerization, phosphazenium salts are almost unknown as cocatalysts for the ROCOP of epoxides and CO2 and only a very few examples of their use do exist in the literature.11,14 One of the most interesting examples of such a use of phosphazenium salts dates back to 2016, when Feng used Et3B and phosphazenium benzyloxides, generated in situ from t-Bu-Pn phosphazene bases and benzyl alcohol, to couple cyclohexene oxide or propylene oxide with CO2.11 In this work, a comparison between phosphazenium alkoxides and PPN halides is made, but no reaction using a phosphazenium halide is mentioned. Moreover, the resulting poly(propylene carbonate) (from now on PPC) and poly(cyclohexene carbonate) showed rather moderate TONs and molar masses compared to those obtained, within the same work, with other cocatalysts.The choice for this investigation then fell upon two salts of the large branched P5 [(Me2N)3PN]4P (PPZ) phosphazene, namely its chloride and its azide, the two most largely used anions of PPN salts. The phosphazenium salts have been tested for PO and CHO copolymerization with salen-type chromium complexes, and only for PO copolymerization with porphyrin cobalt complexes (Fig. 1).
Copolymerization of CO2 and cyclohexene oxide (CHO) catalyzed by chromium(III) salen complexes
In Table 1, the results of the copolymerization of CHO and CO2 with salen-type catalysts 1a and 1b and PPZX cocatalysts are summarized and compared with those obtained with the two onium PPN+ counterparts, used here as reference cocatalysts.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Cocatalyst | Conversionb (%) | Selectivityb (%) | Ether linkageb (%) | TONc | M n (g mol−1) | Theoretical Mne | Đ | T g (°C) |
|
a Catalyst:cocatalyst:CHO = 1:1:2500, T = 80 °C, PCO2 = 30 bar, and t = 3.5 h.
b Determined by 1H NMR spectroscopy of the crude product; conversion = moles of epoxide reacted, and selectivity = moles of polymer over cyclic carbonate.
c Moles of CHO consumed per mole of Cr.
d Determined by SEC using a polystyrene standard.
e Theoretical Mn = [conversion ratio (catalyst:CHO) × 98 g mol−1]/2.
f Determined from the DSC second heating cycle.
g From ref. 22a.
h Continuous flow of CO2.
|
||||||||||
| 1g | 1a | PPNCl | 65 | >99 | — | 1625 | 13300 |
79625 |
1.1 | 119 |
| 2h | PPZCl | 85 | >99 | — | 2125 | 14385 |
104125 |
1.1 | 117 | |
| 3 | PPNN3 | 85 | >99 | 2.8 | 2125 | 9400 | 104125 |
1.3 | 117 | |
| 4g | PPZN3 | 86 | >99 | 1.6 | 2150 | 14200 |
105350 |
1.2 | 119 | |
| 5 | 1b | PPNCl | 85 | >99 | 1.5 | 2125 | 9800 | 104125 |
1.1 | 116 |
| 6h | PPZCl | 83 | >99 | 1.8 | 2075 | 13100 |
101675 |
1.2 | 118 | |
| 7 | PPNN3 | 83 | >99 | 1.8 | 2075 | 8800 | 101675 |
1.2 | 111 | |
| 8 | PPZN3 | 84 | >99 | 1.0 | 2075 | 12600 |
102900 |
1.1 | 118 | |
All the reactions were carried out first in neat epoxide at 80 °C at 30 bar of CO2 for 3.5 hours, that is under the best conditions found in our study for the same copolymerization with the PPNCl cocatalyst.22a A pretreatment between the Cr(III) complex and the ionic phosphazene derivative was always carried out prior to copolymerization to ensure the maximum catalytic activity, by dissolving and stirring them in CH2Cl2 for a few hours, followed by removal of the solvent under vacuum. Reactions were generally performed by pressurizing CO2 to the desired pressure in the reactor and then leaving the polymerization medium stirring at a fixed temperature.22
When catalyst 1a (salenCrCl) is used, the employment of PPZCl with respect to PPNCl affords a remarkable improvement in the conversion, and subsequently in the TON, and only a slight increase in the molar mass is observed. The comparison of copolymers obtained with PPZN3vs.PPNN3 reveals a significant improvement in the molar mass given by the phosphazenium PPZ cation. The use of catalyst 1b (salenCrN3) allows instead a more stable trend in the conversion, which remains high, over 80%, with any cocatalyst. In this case, the advantage brought by the use of phosphazenium salts is visible in the increase of molar mass, affording copolymers with molar masses greater than a few thousand g mol−1. Along with remarkable conversions, selectivities also remain always higher than 99%, with the production of polycarbonate and a complete absence of cyclic carbonate and, with 1a and PPZCl, no or very few ether linkages, as clearly seen in the 1H NMR spectra of the crude products (Fig. S2 in the ESI†), completely flat in the 4.4–3.1 ppm region. Moreover, some polyether linkages, up to 1.8 mol%, are formed with all the cocatalysts and catalyst 1b (Fig. S3 in the ESI†). A narrow but bimodal dispersity of polycarbonates, determined by SEC measurements, is observed, indicating that the polymers have two distinct molar masses with each molar mass profile displaying a narrow dispersity (Fig. S4 in the ESI†). This dispersity may be due to a rapid and reversible chain transfer caused by trace amounts of water in the reaction mixture.23,24 The effectiveness of chain transfer is confirmed by the well-known discrepancy between the expected polymer Mn and that actually measured. The polydispersity indices are narrow and indicate that the chain transfer reactions occur more rapidly than propagation. The thermal analysis of the obtained samples shows Tgs values of around 111–119 °C, in agreement with those previously reported for pure PCHC (Fig. S5 in the ESI†).25
It is well known that ionic cocatalysts cannot affect the stereoselectivity of copolymerization. Indeed, only some salenCrX catalysts were found able to stereospecifically desymmetrize meso-epoxides; specifically, 1a was reported to be active only in the desymmetrization of 1-hexene,26 while CHO could only be partially (14.5 ee%) desymmetrized with S,S-salen-CrNO3 in the presence of N-heterocyclic Lewis bases.27 Thus, in our case only atactic polymers were in fact obtained, with the syndio and iso peaks each accounting for ca. 50% of the total signals in the 13C NMR spectrum as shown in Fig. 2.28
 | ||
| Fig. 2
13C NMR spectrum of PCHC (Table 1, entry 2), in which the signals due to the CO groups of the carbonate group appear. | ||
PPZCl has been studied at different temperatures (Table 2) with catalyst 1a to identify the effect of reaction conditions on conversion, molar mass, and selectivity. At 60 °C under our conditions, low conversion of the epoxide, around 17%, can be observed. It is clear that at such a temperature, the kinetics of the copolymerization is too slow. As the temperature is increased, the catalytic performance improves, and thus the best results in terms of CHO conversion and selectivity of polycarbonates are obtained at 80 °C both with PPZCl and PPNCl cocatalysts.22a In general, upon increasing the temperature, conversion and selectivity decrease. This trend is similar to that observed with UHFFA.22a Molar masses are low only at 60 °C (Fig. S6 in the ESI†). The lower Tg values observed at high temperatures may be related to lower selectivities (Fig. S7 in the ESI†).
| Entry | T (°C) | Conversionb (%) | Selectivityb (%) | TONc | M n (g mol−1) | Đ | Theoretical Mne | T g (°C) |
|---|---|---|---|---|---|---|---|---|
|
a Catalyst = 1a, cocatalyst = PPZCl, catalyst:cocatalyst:CHO = 1:1:2500, PCO2 = 30 bar, and t = 3.5 h.
b Determined by 1H NMR spectroscopy of the crude product; conversion = moles of epoxide reacted, and selectivity = moles of polymer over cyclic carbonate.
c Moles of CHO consumed per mole of Cr.
d Determined by SEC using a polystyrene standard.
e Theoretical Mn = [conversion ratio (catalyst:CHO) × 98 g mol−1]/2.
f Determined from the DSC second heating cycle.
|
||||||||
| 9 | 60 | 17 | >99 | 425 | 4500 | 1.2 | 20825 |
110 |
| 2 | 80 | 85 | >99 | 2125 | 14385 |
1.1 | 104125 |
117 |
| 10 | 100 | 76 | 93 | 1900 | 15900 |
1.2 | 93100 |
114 |
| 11 | 120 | 75 | 88 | 1875 | 15700 |
1.3 | 91875 |
113 |
Salaphen catalysts, 1c and 1d, were also tested, although as already reported by Darensbourg et al.,18 in combination with other cocatalysts, their electron-poor diimine backbone is less effective with CHO, leading to much lower conversions and to copolymers with much lower molar masses. This remains true in any case, and even the usage of high performing PPZX phosphazenium salts cannot overcome the drawbacks caused by the catalyst skeleton, leading to halved conversions (ca. 45% and 40%) and molar masses (ca. 7000 g mol−1 and 5600 g mol−1) with PPZCl and PPZN3, respectively, also producing a tiny but finite amount, about 2–3 mol%, of cyclic carbonate. The presence of a Cl group rather than a N3 nucleophilic group on the catalyst or the cocatalyst leads to minimum differences that can be easily neglected.
Copolymerization of CO2 and propylene oxide (PO) catalyzed by chromium(III) salaphen complexes
It is well recognized that, depending on the monomer, different salen-type catalysts should be used, due to the electronic effect of the skeleton. In particular, electron-rich diamine backbones are highly efficient with CHO, while with PO no reaction occurs or only a cyclic carbonate is formed.29,30 Metal complex catalysts with electron poor aromatic rings are very effective with PO, and thus catalysts 1c and 1d have been used in the copolymerization of propylene oxide and CO2 to study the phosphazenium salts as cocatalysts.All of the copolymerization reactions were carried out in neat epoxide at 60 °C, at 30 bar of CO2, for 24 h. A catalyst to cocatalyst ratio of 1:0.5 has been used to afford the best polymerization rate, according to the results reported in our previous paper.22a The copolymers were characterized by 1H NMR spectroscopy and SEC analysis and the main results are presented in Table 3.
| Entry | Catalyst | Cocatalyst | Conversionb (%) | Selectivityb (%) | Ether linkageb (%) | TONc | M n (g mol−1) | Theoretical Mne | Đ | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Catalyst:cocatalyst:PO = 1:0.5:2500, T = 60 °C; P = 30 bar, and t = 24 h.
b Determined by 1H NMR spectroscopy of the crude product; conversion = moles of epoxide reacted, and selectivity = moles of polymer over cyclic carbonate.
c Moles of PO consumed per mole of Cr.
d Determined by SEC using a polystyrene standard.
e Theoretical Mn = [conversion ratio (catalyst:PO) × 58 g mol−1]/2.
f Determined from the DSC second heating cycle (Fig. S8 in the ESI†).
g From ref. 22a.
|
||||||||||
| 12 | 1c | PPNCl | 78 | 76 | — | 1950 | 38500 |
56550 |
1.2 | 38 |
| 13 | PPZCl | 82 | 82 | — | 2050 | 41000 |
59450 |
1.2 | 42 | |
| 14 | PPNN3 | 78 | 80 | — | 1950 | 30700 |
56550 |
1.2 | 40 | |
| 15 | PPZN3 | 84 | 82 | — | 2100 | 38200 |
60900 |
1.2 | 42 | |
| 16g | 1d | PPNCl | 73 | 64 | 10 | 1800 | 26000 |
52925 |
1.3 | 34 |
| 17 | PPZCl | 79 | 85 | 5 | 1975 | 32300 |
57275 |
1.3 | 38 | |
| 18 | PPNN3 | 58 | 89 | — | 1450 | 20771 |
42050 |
1.2 | 41 | |
| 19 | PPZN3 | 88 | 99 | 6 | 2200 | 29200 |
63800 |
1.2 | 35 | |
In general, good results in terms of conversion, selectivity and molar mass are obtained with all cocatalysts. Conversion values vary considerably and increase when PPZ salts are used; moreover, a certain percentage of polyether is always obtained in all samples from 1d, that is with the salaphen catalyst having N3 as a ligand, as shown in Fig. 3 by the 1H NMR peak at 3.5 ppm (Fig. S9 and S10 in the ESI†). PPZN3 showed the greatest selectivity toward polycarbonate formation (99%) (Table 3, entry 19). Molar masses greater than those obtained with the benchmark PPNX are obtained with PPZCl, and Mn values of 41000 and 32300 g mol−1 are obtained with catalyst 1c and 1d, respectively (Table 3, runs 13 and 17, and Fig. S11 in the ESI†).
 | ||
| Fig. 3 1H NMR spectrum of the obtained crude PPC (Table 3, entry 17). Signals assigned to cyclic propylene carbonate (PC) and unreacted PO signals are visible. | ||
Copolymerization of CO2 and propylene oxide (PO) catalyzed by cobalt(III) porphyrin complexes
Cobalt(III) tetraphenylporphyrin chloride (TPPCoCl) has been proved to be an active catalyst for the alternating copolymerization of CO2 with both CHO and PO, as first discovered by Wang31 and Sugimoto.16 Later, modifications of the porphyrin ligand with the introduction of a series of electron-withdrawing groups in the para-position of the meso-ring showed a slight increase in the catalytic activity in the copolymerization of CO2 with PO.20Thus, we decided to perform some PO/CO2 copolymerization reactions in the presence of PPZCl with 2a, the prototypical TPPCoCl, and 2b, [5,10,15,20-tetra(para-methoxy)phenylporphyrin]cobalt(III) chloride, catalysts and to compare the polymerization results with those obtained with the benchmark PPNCl (Table 4). In addition, some reactions have been conducted with two quaternary phosphonium salts, namely TPPCl and UHFFA (Fig. 1), recently reported as effective cocatalysts in the alternating copolymerization of carbon dioxide with epoxides.20 Both these cocatalysts in combination with N,N′-bis(3,5-di-tert-butylsalicylidene)-phenylenediimine chromium(III) azide exhibit catalytic activity with PO (in neat epoxide at 60 °C, P = 30 bar, t = 24 h), with tetraphenylphosphonium chloride affording the best results in terms of selectivity, exceeding the values obtained with the reference PPNCl cocatalyst.
| Entry | Catalyst | Cocatalyst | Conversionb (%) | Selectivityb (%) | Ether linkageb (%) | TONc | M n (g mol−1) | Theoretical Mne | Đ | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Catalyst:cocatalyst:PO = 1:0.5:2500, T = 25 °C; P = 20 bar, and t = 24 h.
b Determined by 1H NMR spectroscopy of the crude product; conversion = moles of epoxide reacted, and selectivity = moles of polymer over cyclic carbonate.
c Moles of PO consumed per mole of Cr.
d Determined by SEC using a polystyrene standard (Fig. S12 in the ESI†).
e Theoretical Mn = [conversion ratio (catalyst:PO) × 58 g mol−1]/2.
f Determined from the DSC second heating cycle (Fig. S13 in the ESI†).
|
||||||||||
| 20 | 2a | PPNCl | 60 | 93 | 7 | 1500 | 34300 |
43500 |
1.1 | — |
| 21 | PPZCl | 67 | 34 | 7 | 1675 | 56400 |
48575 |
1.4 | 43 | |
| 22 | PPZN3 | 65 | 41 | 14 | 1425 | 32570 |
47125 |
1.3 | 41 | |
| 23 | TPPCl | 28 | >99 | — | 700 | 10900 |
20300 |
1.1 | 36 | |
| 24 | UHFFA | 22 | >99 | — | 550 | 17100 |
15950 |
1.1 | 40 | |
| 25 | 2b | PPNCl | 71 | 66 | 7 | 1775 | 40700 |
51475 |
1.3 | 41 |
| 26 | PPZCl | 90 | 66 | 11 | 2250 | 54500 |
65250 |
1.3 | 42 | |
| 27 | PPZN3 | 75 | 41 | 8 | 1875 | 49500 |
54375 |
1.4 | 40 | |
| 28 | TPPCl | 75 | 64 | 25 | 1875 | 34200 |
54375 |
1.3 | 40 | |
| 29 | UHFFA | 42 | >99 | — | 1050 | 15200 |
30450 |
1.1 | 35 | |
The copolymerization reactions have been conducted at 20 bar of CO2 pressure, at 25 °C, and for 24 hours (Table 4). These conditions have been chosen since by increasing the reaction temperature and CO2 pressure, a gradual loss of the catalytic activity can be observed; moreover, at higher temperatures, the formation of cyclic carbonate due to the chain-back biting reaction increases.16,31
Of all the analyzed cocatalysts, TPPCl and UHFFA show a detrimental effect on the conversion and molar mass, but show high selectivity in PPC copolymerization with 2a as a catalyst, compared with PPNCl. In contrast, the presence of electron-donating fragments in the 2b catalyst has a beneficial effect on the catalytic activity of this complex, with the classical PPNCl, the phosphazenium salt and the UHFFA cocatalyst, since the introduction of the methoxy group modulates the electronic environment at the cobalt center, increasing the copolymer yield compared with that from the unsubstituted 2a catalyst. These results are in agreement with those reported by Rieger with a series of 5,10,15,20-tetra(p-alkoxy)porphyrin cobalt(III) chlorides and by Inoue for Al-porphyrins substituted with methoxy groups.20,32 A similar increase in the catalyst activity has been reported by Darensbourg with the introduction of methoxy groups into the salen-phenolate rings.18
In addition, moving from phosphonium to phosphazenium salt, both catalysts exhibit a sensible increase in the molar mass, above 54000 g mol−1, when PPZCl is used. With catalyst 2a and both cocatalysts slightly higher conversions are achieved even with significantly lower selectivity, while with catalyst 2b, higher conversion with no changes in the selectivity can be observed with PPZCl (Fig. S14–S17 in the ESI†).
The quaternary phosphonium salts show for both catalysts a clear decrease in the conversion along with a sensible increase in the selectivity up to 99 mol%; however, molar masses up to 17000 g mol−1 are obtained. It is worth noting that in light of the low conversion, the obtained molar masses, even though lower than those observed with salen catalysts, show a value of about 50% of the theoretical Mn value.
Comparison of the effects of branched phosphazenium salts as cocatalysts on the copolymerization of CO2 and CHO and PO
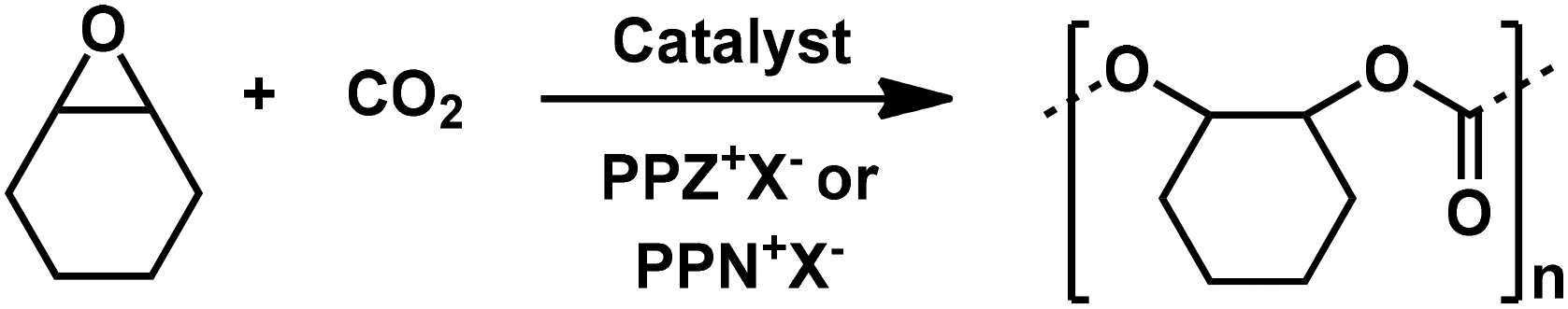
In order to find a rationale for the effects of branched phosphazenium salts as cocatalysts on the copolymerization of CO2 and CHO and PO, Mnvs. conversion profiles for copolymerization of CHO with salen-type catalysts 1a and 1b and for PO copolymerization with salaphen-type catalysts 1c and 1d and different cocatalysts are compared in Fig. 4. For this comparison, our data obtained under the same conditions but with cocatalysts different from those shown in Tables 1–3 have also been included.22a | ||
| Fig. 4 M n vs. conversion of CHO copolymerization with salen-type catalysts 1a and 1b (top) and of PO copolymerization with salaphen-type catalysts 1c and 1d (bottom) and different cocatalysts. | ||
It is evident that higher molar masses and monomer conversion with PPZ+X− than those with the benchmark PPN+X− counterparts under the same conditions are achieved. These differences are more visible in PO copolymerization, which shows higher molar mass than CHO copolymerization. We hypothesized that greater molar masses of PO copolymers are due to the lower propensity of PO towards chain transfer.22a It is also evident that PPZCl gives better results than PPZN3, in terms of molar mass, and PPZN3, in terms of conversion. Moreover, in general with both PPZX salts and chlorine catalysts salenCrCl (1a) and salaphenCrCl (1c), better copolymer results than with salenCrN3 (1b) and salaphenCrN3 (1d) are obtained.
MALDI-ToF-MASS spectra may be helpful to demonstrate which is the end group or real initiator and to better understand the polymerization mechanism. An investigation of the effect of different catalytic systems on the chain structures and end groups of CO2-based polycarbonates by MALDI-TOF mass spectrometry has been carried out. The spectra are complex and the discussion of results is the object of a manuscript in preparation.35 However, in Fig. 5, the MALDI-TOF spectrum collected for entry 8, prepared with catalyst 1b and PPZN3 as a cocatalyst, is shown.
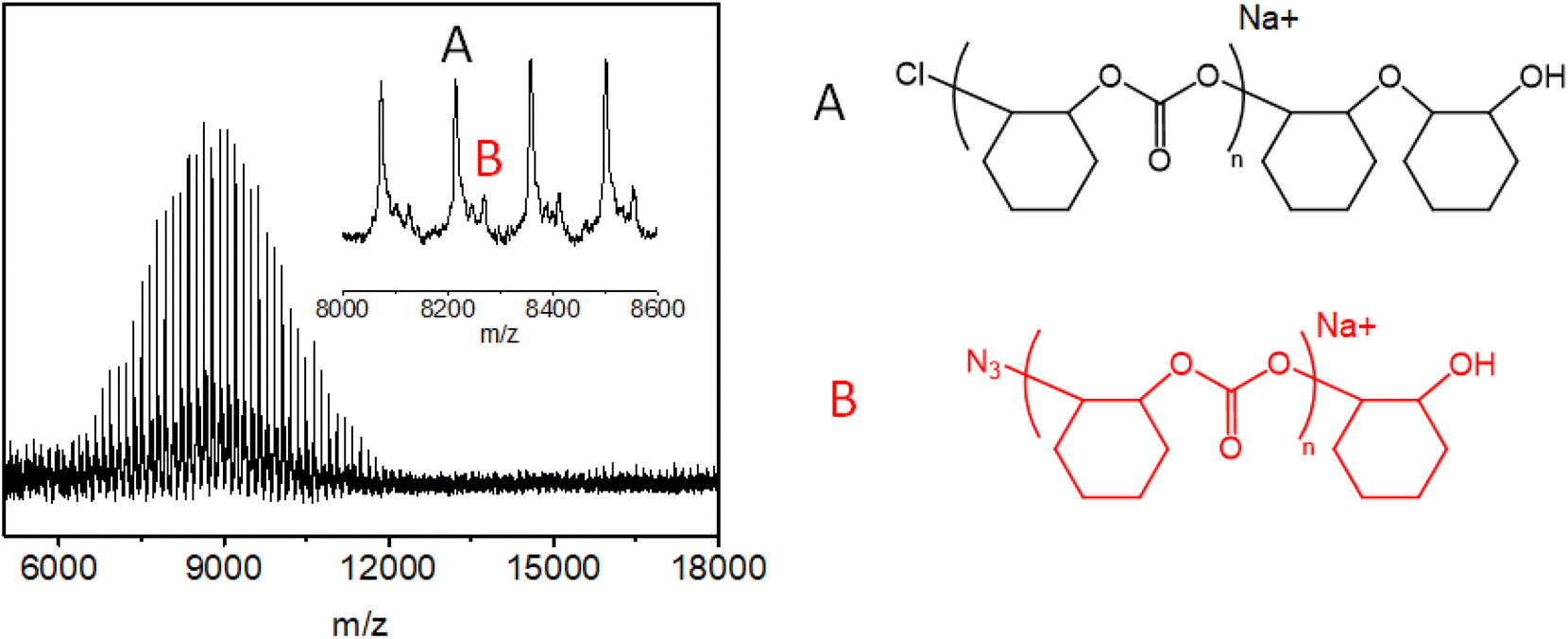 | ||
| Fig. 5 MALDI-TOF spectrum collected for entry 8 (zoomed in from m/z = 8600 to m/z = 9000), prepared with catalyst 1b and PPZN3 as a cocatalyst. | ||
Two signals named A and B, one end-capped with a chloride group and a hydroxyl group and the other one with a hydroxyl group and an azide, can be observed. Although N3 is present on both the catalyst 1b and cocatalyst (entry 8 in Table 1) signal B is the smallest. The presence of Cl at one end probably originates from the pretreatment of the catalyst and cocatalyst in dichloromethane.23 The presence of the high A signal is a clear indication that there is an equilibrium between the N3 anion and Cl and that the equilibrium is shifted towards chlorine.
Regarding selectivities in CHO copolymerization, all cocatalysts used in this work show selectivities over 99%, while selectivities vary considerably in PO copolymerization (Fig. 6). In these copolymerization reactions, there is an almost direct relationship between monomer conversion and selectivity. Interestingly PPZN3 which exhibits the highest conversion gives the highest selectivity with salaphenCrN3 (1d).
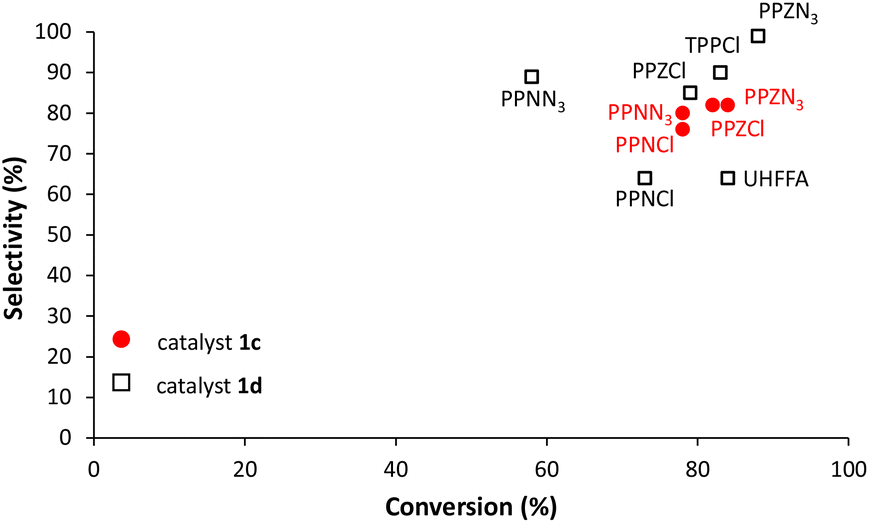 | ||
| Fig. 6 Selectivity vs. conversion of PO copolymerization with salaphen-type catalysts 1c and d and different cocatalysts. | ||
This confirms our previous observation that cocatalysts less efficient in PO copolymerization have lower selectivities and/or higher ether linkages.
This behavior was explained by considering that with less effective cocatalysts polymeric species spend more time in the resting state, which makes more probable the formation of cyclic carbonate and ether linkages (Scheme S1 in the ESI†).35
Comparison of the results of PO copolymerization with porphyrins in Fig. 7 is more difficult to rationalize at first sight.
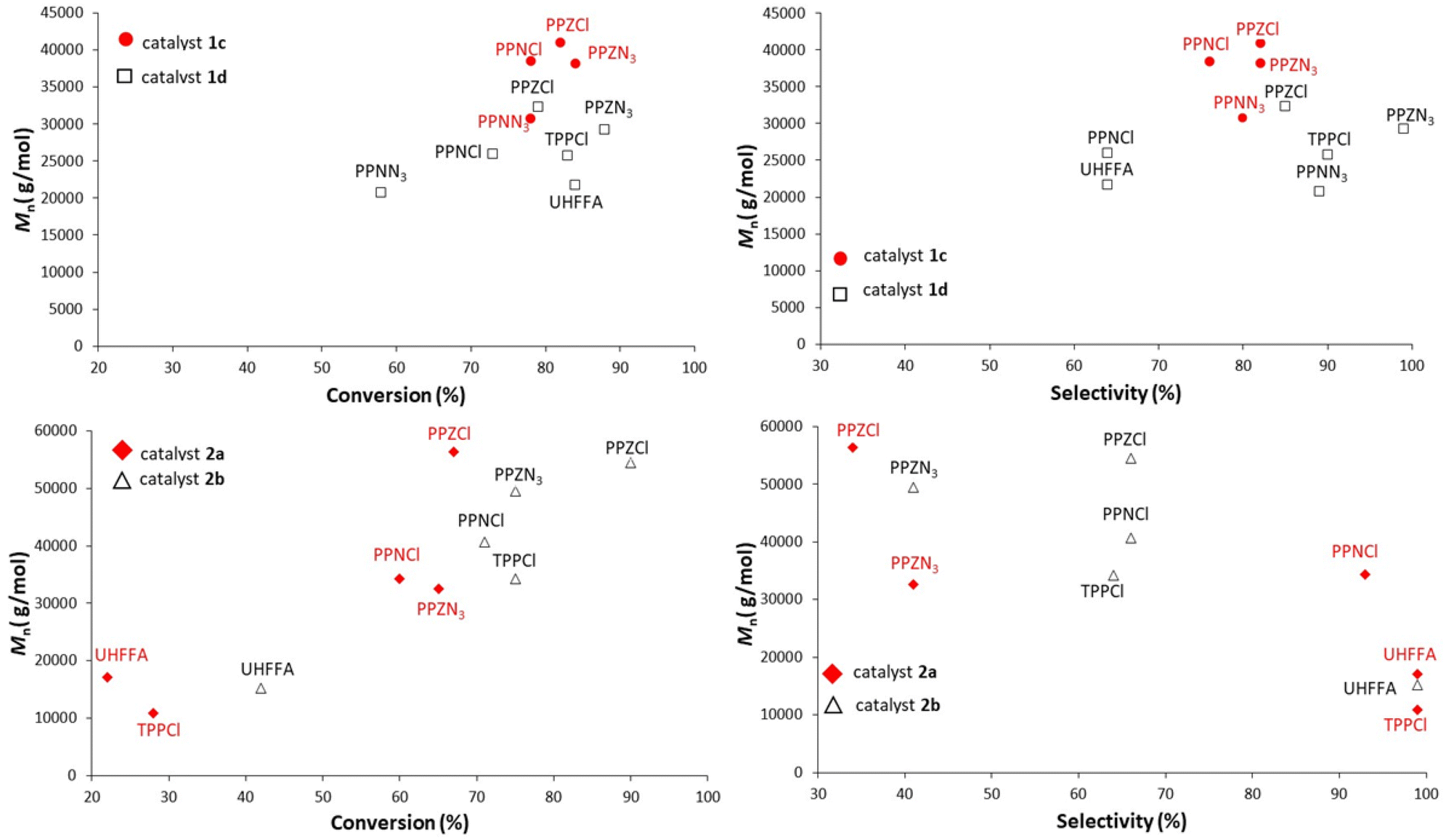 | ||
| Fig. 7 M n vs. conversion (left) and vs. selectivity (right) of PO copolymerization with salaphen-type catalysts 1c and 1d (top) and with porphyrin-type 2a and 2b catalysts and different cocatalysts (bottom). | ||
However, porphyrin catalysts also give better results with PPZX cocatalysts than with PPNX cocatalysts. Moreover, if we consider only the results obtained with branched PPZX cocatalysts, PPZCl and PPZN3 give a very high molar mass with catalyst 2b and PPZCl gives the same also with catalyst 2a. Moreover, for the same copolymerization there is again a correspondence between selectivities and conversion. It is not clear if results with PPZN3 and catalyst 2a are outliers.
UHFFA, TPPCl, and PPNCl give the highest selectivities with porphyrin catalysts, but give lower conversions and molar masses. Probably steric hindrances of catalysts and cocatalysts play a role in these copolymerization reactions.
Copolymerization of epoxides and CO2 involves the coordination of the epoxide to the metal activated by X, a nucleophile coming from the cocatalyst, which favours the epoxide insertion by facilitating the removal of the catalyst ligand Nu (Scheme S1 in the ESI†). If we consider ionic cocatalyst salts such as PPN+X− and PPZ+X− where X− is Cl or N3, the following equilibria have to be taken into account:
| PPZ+X− ⇌ PPZ+ + X− |
| PPN+X− ⇌ PPN+ + X− |
The size of the cation influences the strength of the anion–cation interaction. PPZ+ has greater steric hindrance (see Table S1 in the ESI†) and greater ability to stabilize the positive ion pair than PPN+ which shifts the above equilibria more to the right, allowing X− to activate the metal of the catalyst. This difference seems to explain the greater efficiency of branched phosphazenium salts as cocatalysts. The influence of the size of the cation on the propagation rate and thus on the activity and selectivity is quite common for anionic ROP.36
Conclusions
Large branched phosphazenium salts of general formula [(Me2N)3PN]4P+X−, where X is chloride or azide, the two most largely used anions of PPN salts, have been tested as cocatalysts for different catalyst systems in the alternating copolymerization of CO2 and CHO with salen-type chromium complexes, salenCrCl (1a) and salenCrN3 (1b), under various experimental conditions. A careful examination of the catalytic activity of the various complexes and of the molar mass indicated that the use of PPZX salts as cocatalysts has a beneficial effect on the copolymer production, with conversions at least comparable or even superior to those obtained from onium salts. Higher monomer conversion and greater molar masses than those obtained with the benchmark PPN+X− counterparts under the same conditions were achieved. PPZCl gives better results than PPZN3 in terms of molar mass, and PPZN3 in terms of conversion, and in general with both PPZX salts, salenCrCl (1a) gives better copolymerization results than salenCrN3 (1b).
The phosphazenium salts have also been tested for PO and CO2 copolymerization with salaphenCrCl (1c) and salaphenCrN3 (1d), as well as with porphyrin cobalt complexes tetra phenylporphyrinCoCl (2a) and the tetramethoxysubstituted one (2b). With both cocatalysts higher molar masses and comparable or even superior conversions and selectivities are mostly obtained with respect to the onium cocatalysts. In particular, when porphyrin catalysts are used, molar masses above 50000 g mol−1 are achieved. Thus, phosphazenium salts are viable, or even superior, alternatives to onium salts in CO2/epoxide ROCOP.
If we consider the reaction mechanisms proposed by Darensbourg33 and Coates,34 cations which are able to easily delocalize positive charge should be the most active for the copolymerization, because of better stabilization of the separated ion pair. Moreover, the phosphazenium positive effect is also probably due to their higher steric hindrance that favours the epoxide's interaction with the catalyst and subsequently its ring opening. It is well known that the epoxide ring opening is the rate-determining step. In conclusion, the greater possibility of phosphazenium cations to delocalize the positive charge due to their conjugated double bond structure along with the great steric hindrance of the bulky branched structure allow them to overcome onium cations.
Author contributions
M. Brivio and L. Veronese, investigation; P. Biagini and R. Po’, conceptualization and validation; I. Tritto, writing – review and editing; and S. Losio and L. Boggioni, supervision, conceptualization, writing – review and editing.Conflicts of interest
The patent WO2020229965A1 covers part of the work presented in this paper.Acknowledgements
The authors thank Eni S.p.A., Rome, Italy, for the financial support (contract no. 3500042562). We are grateful to Ms F. Greco, Mr A. Giacometti Schieroni, Dr F. Bertini, and Dr R. Chiarcos for the acquisition of the NMR spectra, SEC and DSC measurements, and MALDI-ToF MASS analysis, respectively.Notes and references
- (a) M. Bochmann, Organometallics, 2010, 29, 4711 CrossRef CAS; (b) N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363 RSC; (c) Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1 CrossRef CAS.
- S. Inoue, H. Koinuma and T. J. Tsuruta, Polym. Sci., Part B: Polym. Lett., 1969, 7, 287 CrossRef CAS.
- (a) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721 CrossRef CAS PubMed; (b) G. Trott, P. Saini and C. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed; (c) S. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990 RSC; (d) X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839 CrossRef CAS PubMed; (e) C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565 CrossRef CAS; (f) A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed; (g) C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037 CrossRef CAS.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579 RSC.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212 RSC.
- T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728 CrossRef CAS PubMed.
- (a) J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170 CrossRef CAS PubMed; (b) Y. Xia, J. Shen, H. Alamri, N. Hadjichristidis, J. Zhao, Y. Wang and G. Zhang, Biomacromolecules, 2017, 18, 3233 CrossRef CAS PubMed; (c) X. Wang, Y. Liu, Z. Li, H. Wang, H. Gebru, S. Chen, H. Zhu, F. Wei and K. Guo, ACS Macro Lett., 2017, 6, 1331 CrossRef CAS PubMed; (d) S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Chem. Phys., 2018, 39, 1800485 Search PubMed; (e) J. Shi, Z. Liu, N. Zhao, S. Liu and Z. Li, Macromolecules, 2022, 55, 2844 CrossRef CAS.
- (a) K. Issleib and M. Lischewski, Synth. Inorg. Met.-Org. Chem., 1973, 3, 255 CrossRef CAS; (b) P. Haasemann and J. Goubeau, Z. Anorg. Allg. Chem., 1974, 408, 293 CrossRef CAS; (c) H. Goldwhite, P. Gysegem, S. Schow and C. Swyke, J. Chem. Soc., Dalton Trans., 1975, 12 RSC; (d) R. Appel and M. Halstenberg, Angew. Chem., Int. Ed. Engl., 1977, 16, 263 CrossRef.
- (a) A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS PubMed; (b) C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2012, 33, 1938 CrossRef CAS PubMed; (c) M. J. Ji, M. Q. Wu, J. Y. Han, F. J. Zhang, H. W. Peng and L. H. Guo, Curr. Org. Chem., 2021, 25, 272 CrossRef CAS.
- (a) H. Zhang, S. Hu, J. Zhao and G. Zhang, Macromolecules, 2017, 50, 4198 CrossRef CAS; (b) H. Li, J. Zhao and G. Zhang, ACS Macro Lett., 2017, 6, 1094 CrossRef CAS PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117 CrossRef CAS PubMed.
- (a) S. Y. Hu, G. X. Dai, J. P. Zhao and G. Z. Zhang, Macromolecules, 2016, 42, 4462 CrossRef; (b) H. X. Zhang, S. Y. Hu, J. P. Zhao and G. Z. Zhang, Macromolecules, 2017, 50, 4198 CrossRef CAS; (c) H. X. Zhang, S. Y. Hu, J. P. Zhao and G. Z. Zhang, Eur. Polym. J., 2017, 95, 693 CrossRef CAS.
- J. Zhang, L. Wang, S. Liu and Z. Li, Angew. Chem., Int. Ed., 2021, 61, e212111197 Search PubMed.
- P. C. B. Widger, S. M. Ahmed and G. W. Coates, Macromolecules, 2011, 44, 5666 CrossRef CAS.
- P. Biagini, R. Po, S. Losio and M. Brivio, WO2020229965A1, 2020.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312 CrossRef CAS.
- W. Xia, PhD thesis, Technische Universität München, 2015, https://d-nb.info/1071370049/34 Search PubMed.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024 CrossRef CAS PubMed.
- J. L. Leighton and E. N. Jacobsen, J. Org. Chem., 1996, 61, 389 CrossRef CAS.
- C. E. Anderson, S. I. Vagin, W. Xia, H. Jin and B. Rieger, Macromolecules, 2012, 45, 6840 CrossRef CAS.
- K. D. Demadis, T. J. Meyer and P. S. White, Inorg. Chem., 1998, 37, 3610 CrossRef CAS PubMed.
- (a) L. Veronese, M. Brivio, P. Biagini, R. Po, I. Tritto, S. Losio and L. Boggioni, Organometallics, 2020, 39, 2653 CrossRef CAS; (b) P. Biagini, C. Perego, R. Po, L. Boggioni, M. Cozzolino, S. Losio, A. Flamigni, A. Colombo, C. Dragonetti, F. Fagnani, P. Matozzo and D. Roberto, Inorg. Chim. Acta, 2022, 532, 120753 CrossRef CAS.
- H. Sugimoto, H. Otsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172 CrossRef CAS.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972 CrossRef CAS.
- G. Wu, S. Jiang, X. Lu, W. Ren and S. Yan, Chin. J. Polym. Sci., 2012, 30, 487 CrossRef CAS.
- E. N. Jacobsen, M. Tokunaga and J. F. Larrow, PCT Int. Appl, WO 00/09463, 2000 Search PubMed.
- B. Li, R. Zhang and X.-B. Lu, Macromolecules, 2007, 40, 2303 CrossRef CAS.
- (a) B. O. Li, G. P. Wu, W. M. Ren, Y. M. Wang, D. Y. Rao and B. X. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102 CrossRef CAS; (b) K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008 CrossRef CAS; (c) M. Cheng, N. A. Darling, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2000, 2007 RSC; (d) C. T. Cohen, C. M. Thomas, K. L. Peretti, E. B. Lobkovsky and G. W. Coates, Dalton Trans., 2006, 237 RSC.
- (a) D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622 CrossRef CAS PubMed; (b) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS PubMed.
- D. J. Darensbourg;, R. M. Mackiewicz;, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831 CrossRef PubMed.
- Y. Qin, X. Wang, S. Zhang, X. Zhao and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5959 CrossRef CAS.
- H. Sugimoto, T. Aida and S. Inoue, Macromolecules, 1990, 23, 2869 CrossRef CAS.
- D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026 CrossRef CAS PubMed.
- C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182 CrossRef CAS.
- As suggested by reviewers, one case, at least, of the MALDI-ToF-MASS spectrum should be present and discussed in this work.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Słomkowski, Prog. Polym. Sci., 2007, 32, 247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01471h |
| This journal is © The Royal Society of Chemistry 2023 |