
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-type polymer semiconductors incorporating heteroannulated benzothiadiazole†
Xiantao
Hu
 a,
Aniruddha
Basu
b,
Martina
Rimmele
a,
Adam V.
Marsh
b,
Filip
Aniés
a,
Qiao
He
*a,
Thomas D.
Anthopoulos
b and
Martin
Heeney
*ab
a,
Aniruddha
Basu
b,
Martina
Rimmele
a,
Adam V.
Marsh
b,
Filip
Aniés
a,
Qiao
He
*a,
Thomas D.
Anthopoulos
b and
Martin
Heeney
*ab
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, White City Campus, London W12 0BZ, UK. E-mail: q.he16@imperial.ac.uk; m.heeney@imperial.ac.uk
bDivision of Physical Sciences and Engineering and KAUST Solar Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 28th December 2022
Abstract
We report four novel n-type polymer semiconductors containing a 2,1,3-benzothiadiazole (BT) based acceptor annulated with a 2-(1,3-dithiol-2-ylidene)malonitrile (DTYM) group. Four copolymers with varying electron donor groups are investigated with regards to optoelectronic properties. Theoretical calculations and optical characterization indicate that the introduction of the DTYM group leads to backbone twisting and blue-shifted absorption in comparison to the unsubstituted BT analogues, but increases the dipole moment and electron affinity. All polymers exhibit n-type semiconducting character in organic field-effect transistors (OFETs) fabricated by blade coating, with the co-polymer containing a fluorinated bithiophene donor demonstrating the best performance. This work highlights the influence of the DTYM group on polymer semiconductors and further demonstrates its potential as a functional group to enhance electron affinity.
Introduction
Over the past few decades, organic semiconductors (OSCs) have attracted much attention for use in next-generation printed electronic devices because of their unique combination of mechanical flexibility, low potential fabrication costs, and synthetic tunability compared to traditional inorganic semiconductors.1–3 These materials can be used in a wide range of applications, from organic photovoltaics (OPVs),4–6 organic field-effect transistors (OFETs)7,8 to organic light-emitting diodes (OLEDs) and beyond.9 To realize the potential of these technologies, extensive efforts have been directed towards tuning the properties of OSCs, including their light-absorbing ability and charge carrier transport, primarily via the design and synthesis of novel semiconducting compounds.According to the type of charge carriers they transport, OSCs can be classified into p-type (majority hole transport) and n-type (majority electron transport) materials. Historically, the development of n-type OSCs lags significantly behind that of their p-type counterparts in terms of charge-carrier mobility and synthetic scalability,10,11 thus, significant effort has been put into developing n-type OSCs based on small-molecule and polymer materials. Polymeric n-type OSCs are particularly attractive due to their good solution rheology, enhanced film-forming properties, superior mechanical flexibility/stretchability, and improved device stability.12–15 State-of-the-art n-type polymers are often comprised of an alternating electron-rich unit (donor, D) and electron-deficient unit (acceptor, A). This donor–acceptor (D–A) design strategy is favourable to form a ‘push-pull’ electron system as a result of hybridization of frontier molecular orbitals, facilitating backbone planarization and delocalisation.2,16,17 It is well established that a rational choice of donor and acceptor monomers allows fine-tuning of the microstructure and electronic structure of the resulting polymers, as the energy levels of the polymers are influenced by the highest occupied molecular orbital (HOMO) energy level of the donor unit and the lowest unoccupied molecular orbital (LUMO) energy level of the acceptor unit.18–22
With the development of high-performance D–A polymer OSCs, a library of electron-rich donor units has been extensively studied. However, electron-deficient acceptor units are relatively underdeveloped and remain an active area of research.18,23 2,1,3-Benzothiadiazole (BT) has become one of the most widely investigated acceptor units used in constructing OSCs due to its planar and rigid geometry, heteroatom interactions, high absorption coefficient, electrochemical stability and electron-withdrawing ability.22,24–29 Nevertheless, polymers based on BT commonly function as p-type OSCs as they have relatively high-lying LUMO energy levels.30 Tremendous efforts have been taken to address this by structural modification of BT and significant progress has been made, for example: replacing the bridgehead sulphur atom with nitrogen,31 oxygen,32,33 and selenium34,35 to adjust the electronic structure, functionalization of the 5- and/or 6- positions of BT with electron-donating17,36,37 or electron-withdrawing groups10,21,38 to tune the frontier molecular orbital energy levels and heteroannulation of an additional ring on the 5,6-positions to extend the conjugation length.39–44
Strong electron-withdrawing groups are typically incorporated into n-type OSCs as a low-lying LUMO energy level is required to achieve good charge injection from metal electrodes and to promote ambient stability.13,45 We were interested in the application of the electron-withdrawing group 2-(1,3-dithiol-2-ylidene)malonitrile (DTYM), which has been extensively explored to modify naphthalene diimide (NDI).46 Several reports have demonstrated that fusing the DTYM group to the NDI core can not only expand the π-conjugation and promote intermolecular π–π stacking and/or S⋯S interactions, but also lower the LUMO energy via its electron-withdrawing effect.47–49 The resulting small molecule NDI-DTYM and its derivatives display high electron mobilities and excellent air/operating stability in OFET devices.50–55 However, there are few examples of the modification of other acceptor units using DTYM. In our previous report, we reported a facile approach to heteroannulate the DTYM group to the 4,5-positions of a BT ring to produce an extended electron-withdrawing acceptor end group. This was coupled with an electron-rich indacenodithiophene (IDT) core to yield a medium gap, non-fullerene acceptor (NFA) material that achieved reasonable photovoltaic performance under low light conditions.38 However the influence of heteroannulation of the DTYM group to the 5,6-positions of BT has not been well explored, with only one report to the best of our knowledge, in which a co-polymer with benzodithiophene exhibited ambipolar transistor behaviour with mobility on the order of 10−4 cm2 V−1 s−1, whereas other copolymers were reported to show interesting solvatochromic behaviour.56
Inspired by the efficacy of the DTYM unit in the NFA, we hypothesised that its incorporation into p-type D–A polymer OSCs could potentially convert the charge transport from p-type to n-type in OFET devices.18 Herein, we report the synthesis and characterization of four n-type polymer OSCs (P1–4) using a DTYM-heteroannulated-BT accepting unit copolymerised with four donor units of varying electron-donating ability to fine-tune the energy levels. Among these polymers, polymer P3, containing a partially fluorinated bithiophene donor unit, exhibited the most red-shifted absorption and the lowest-lying LUMO energy level. We have also characterized the charge transport behaviour of these polymers in OFET devices, with P3 showing the best overall device performance.
Results and discussion
Synthesis and characterization
In an effort to ensure good solubility of the polymer incorporating the extended, annulated BT core, we flanked it with thiophene containing long, branched alkyl chains. The synthetic routes towards polymers P1–P4 are illustrated in Scheme 1. Bromination of the commercially available monomer 2-decyl-1-tetradecanol with N-bromosuccinimide (NBS), followed by organozinc formation, Negishi coupling and a final stannylation yielded compound 3 (yield: 78%). After Stille coupling with 4,7-dibromo-5,6-difluoro-2,1,3-benzothiadiazole, the resulting compound 4 (yield: 54%) was annulated with the 2-(1,3-dithiol-2-ylidene)malonitrile group to afford compound 5 (BT-DTYM) in high yield (90%). The facile nature of this reaction can be attributed to the good nucleophilicity of the 2,2-dicyanoethylene-1,1-dithiolate anion and the fact that nucleophilic aromatic substitution reactions are known to readily occur on fluorinated BT cores.57–60 Bromination of compound 5 afforded 6 (yield: 44%), which was polymerised by Stille coupling under microwave heating. Four donor monomers of differing electron-donating strength were investigated. The resulting four copolymers were purified by precipitation and subsequent washing with a range of non-solvents before extraction into chloroform. | ||
| Scheme 1 Synthetic routes to polymers P1, P2, P3, and P4. | ||
All polymers exhibited good solubility in common organic solvents including chloroform and chlorobenzene, even P3 despite the reduction in solubility often observed for fluorinated bithiophene. The molecular weights, as found by gel permeation chromatography (GPC) in chloroform against polystyrene standard, are summarised in Table S1.†P1–P3 were broadly comparable, with a similar number average molecular weight (Mn), weight average molecular weight (Mw) and dispersity (Đ = Mw/Mn). P4 was slightly lower, perhaps due to the low purity of the commercially available stannyl monomer. The structures of all materials were confirmed by a combination of nuclear magnetic resonance (NMR) spectroscopy and mass spectroscopy investigations (see ESI†).
Theoretical calculations
To investigate the role of the DTYM group on the polymer energy levels and molecular geometries, we modelled our materials using density functional theory (DFT) with B3LYP level of theory and 6-31G (d, p) basis set. Relaxed potential energy scans were made of the intermonomer bond by systematically changing the dihedral bond angles between the benzothiadiazole and adjacent thiophene units, whilst allowing the rest of the molecule to relax to its minimum energy conformation. Long alkyl side chains were replaced with methyl groups to simplify the calculation process. Fig. 1 shows that BT is preferentially oriented in the trans conformation (where the thiophene sulfur atom points in the opposite direction to the benzothiadiazole sulfur atom), which is in agreement with literature calculations.61,62 In contrast, BT-DTYM exhibits an energy maximum in this conformation, with the cis conformation, where the BT-DTYM unit is approximately 45° out of the plane with respect to the flanking thiophenes, predicted to be the lowest energy conformer. It is also interesting to note that the energy difference between the trans and cis conformations is larger for BT-DTYM (∼4.1 kJ mol−1) than for BT (∼3.6 kJ mol−1). However, the energy barrier to rotation of BT-DTYM (∼6.6 kJ mol−1) is significantly lower compared to BT (∼20.2 kJ mol−1). DFT is known to have reliability issues with intermonomer torsional potentials because of basis set superposition errors,61,63 therefore, we corroborated these findings by performing similar calculations using second-order Møller–Plesset perturbation (MP2) theory and cc-pvdz basis set. To minimize the computational time, selected dihedral angles (0°, 45°, 90°, 135°, and 180°) were calculated. The MP2/cc-pvdz calculated barriers to rotation are consistent with the overall trend of the potential energy surface as calculated by DFT. Accordingly, due to the relatively lower rotation barriers, the polymer with fused BT-DTYM as comonomer is predicted to be more twisted than that with BT, which is reasonably ascribed to the large steric hindrance between the DTYM group and the adjacent thiophene rings.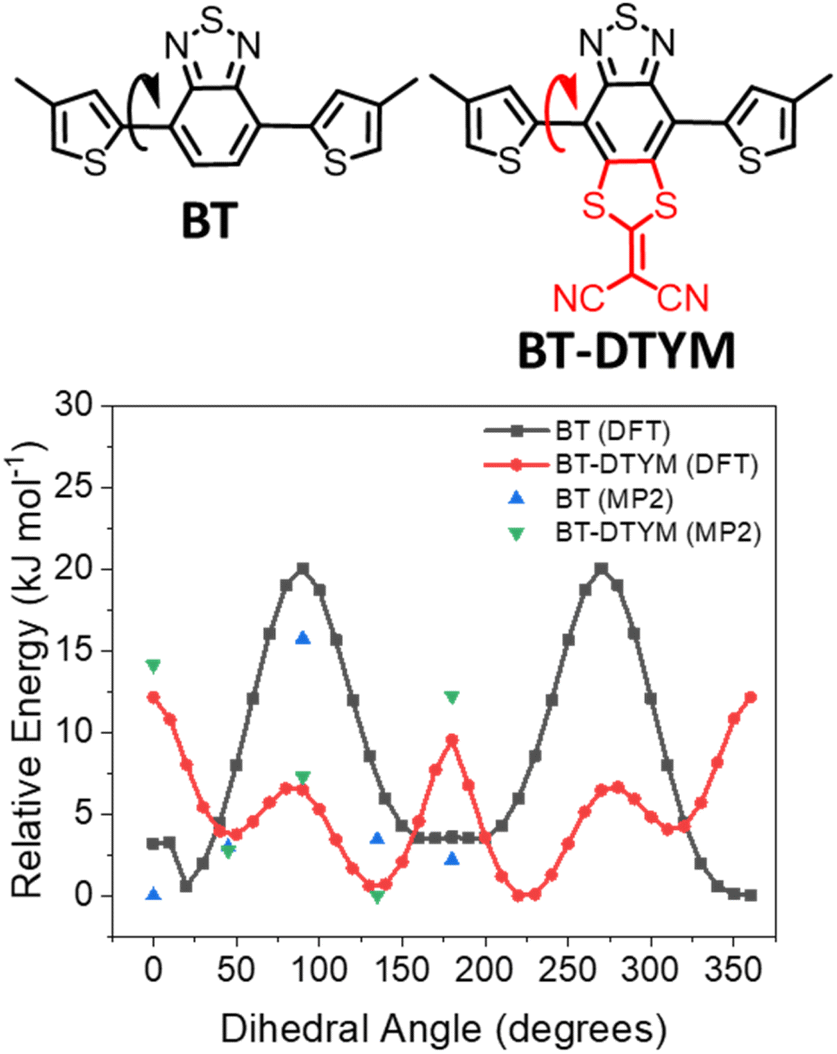 | ||
| Fig. 1 DFT and MP2-calculated relaxed potential energy scans of monomer BT and BT-DTYM. | ||
Fig. 2 shows the magnitude and direction of the dipole moment of the three conformations of BT and BT-DTYM at their minimum to maximum energy state. Although it is complex to relate the dipole properties to the device performance, increasing studies suggest that a large dipole moment is beneficial for self-assembly and molecular packing, in both solar and transistor devices.63–65 The dipole moments of BT-DTYM in the three conformations shown in Fig. 2 are consistently higher than those of non-substituted BT, which could be expected to enhance the intermolecular interactions.61,66 These calculations indicate that the introduction of DTYM groups in BT-DTYM induces a strong electron-withdrawing effect and generates a reverse dipole moment vector with respect to that found in BT.
 | ||
| Fig. 2 Comparison of the dipole moments of BT-DTYM (top) and BT (bottom) planar conformations. The blue arrows show the magnitude and direction of the dipole moments. The energies are converted into relative energies versus the energy minima. | ||
DFT calculations of the P1–P4 polymer repeat units (using methyl groups instead of long alkyl side chains to save computational time) were also investigated to assess co-planarity between co-monomer units, as well as estimate the HOMO and LUMO of the four polymer OSCs at their optimized geometry. As shown in Fig. 3, polymers P1 and P3 have a relatively smaller dihedral angle compared to the other two polymers, with polymer P3 showing the smallest dihedral angle (22.0°), which can be ascribed to the rational choice of 2,2′-bithiophene donor and non-covalent intramolecular interactions with the fluorine group.67 This may lead to a more compact packing behaviour in the solid state and extended π-conjugation for polymer P3.68,69 The delocalized HOMO is equally distributed over the donor and acceptor parts of the polymers, while the LUMO is mainly located on the acceptor parts, indicating efficient intramolecular charge transfer interactions. The four polymers share similar LUMO energy levels but polymer P4 has a slightly higher HOMO energy level compared to the other polymers, which suggests a stronger electron-donating ability from the benzodithiophene (BDT) unit.
 | ||
| Fig. 3 Dihedral angle and frontier molecular orbitals of polymer units P1–P4 at their optimized geometries. | ||
Thermal properties
Thermal gravimetric analysis (TGA) was conducted to investigate the thermal stability of the polymers. All materials were stable up to 400 °C except for polymer P4 (265 °C) corresponding to a 5% weight loss at a constant heating rate of 10 °C min−1 (Fig. 4a), which is consistent with previously reported BDT-containing polymers.27,36 Nevertheless, the thermal stability of all polymers is adequate for device fabrication. The thermal behaviour of the polymers was investigated using differential scanning calorimetry (DSC), cycling between 25 °C and 300 °C. The endothermic transition in the heating cycle and the exothermic transition in the cooling cycle are ascribed to the polymer's melting temperature (Tm) and recrystallization temperature (Tc), respectively. As shown in Fig. 4b, polymers P1 and P3 show a crystallization peak at around 165 °C and 190 °C during the cooling process whereas polymer P2 does not exhibit a crystallisation peak after the endothermic peak around 209 °C, indicating that P1 and P3 are semi-crystalline but P2 forms a glass at this cooling rate.70 The lower Tm and Tc of P1 in comparison to P3 suggest that the less planar polymer backbone conformation in P1 disrupts the packing behaviour and reduces the intermolecular interaction, which is in good agreement with the theoretical calculations of the polymer unit dihedral angles (Fig. 3).71 The polymer P4 DSC trace was featureless with no thermal transitions observed in the temperature range of the experiment. | ||
| Fig. 4 (a) TGA traces of the polymers P1–P4 at a heating rate of 10 °C min−1 under nitrogen. (b) DSC traces of the polymers recorded at the first cycle with a heating and cooling rate of 10 °C min−1 under nitrogen. (c) UV-vis absorption spectra of the polymers in chloroform solution and as thin-films. (d) Cyclic voltammetry curves of the polymers. Measurements were performed in acetonitrile/[n-Bu4N]PF6 solutions (0.1 mol L−1). Scan rate: 0.1 V s−1. | ||
Optical and electrochemical properties
The optical properties of the four polymers were investigated both in chloroform solution and as thin films (Fig. 4c and Table 1). Both the solution and solid-state spectra exhibit a ‘dual band’ absorption, as observed for most D–A polymers, with a high energy band corresponding to the π–π* transition and a low energy band corresponding to intramolecular charge transfer (ICT) between the donor and acceptor comonomers.17,40,72 In chloroform, polymer P1 shows a broad absorption range from 300 nm to 700 nm, with the absorption maximum at 559 nm. For the P1 thin film, the maximum absorption peak red-shifts by 92 nm to 651 nm, which can be explained by solid-state planarization and packing. However, the absorption maximum is still blue-shifted compared to a reported polymer with an identical molecular structure except with fluorine groups substituted on the 5,6 positions on benzothiadiazole,73 which can be partly explained by the more twisted conformation observed in the computational investigation (Fig. 1). The large steric hindrance between the DTYM group and the adjacent thiophene ring leads to more twisting along the polymer backbone, resulting in a lowering of π-conjugation and a wider bandgap. Changing the donor unit to thieno[3,2-b]thiophene and 3,3′-difluoro-2,2′-bithiophene in corresponding polymers P2 and P3 results in a slight red-shift of the ICT band in chloroform (599 nm and 584 nm, respectively) but a similar position in the solid state (653 nm and 657 nm, respectively). Polymer P4, with strongly electron-donating BDT as comonomer, exhibits a slight blue-shift in the maximum absorption peak at 631 nm compared to P1, probably due to the weaker interchain interaction caused by the long side chains on both co-monomers.74 The optical bandgaps (Eoptg) of all polymers were calculated to be 1.56–1.68 eV by the onset of film absorption (λonset), using the following formula Eoptg = 1240/λonset (eV).75| UV-vis | DFT | CV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) Sol. | λ max (nm) Film | λ filmonset (nm) | E optg (eV) | E HOMO (eV) | E LUMO (eV) | E g (eV) | E oxonset (V) | E reonset (V) | E HOMO (eV) | E LUMO (eV) | E g (eV) | |
| P1 | 559 | 651 | 776 | 1.60 | −5.23 | −3.05 | 2.18 | 1.22 | −0.99 | −5.60 | −3.52 | 2.08 |
| P2 | 599 | 653 | 796 | 1.56 | −5.38 | −3.05 | 2.33 | 1.18 | −0.90 | −5.56 | −3.61 | 1.95 |
| P3 | 584 | 657 | 791 | 1.57 | −5.28 | −3.09 | 2.19 | 1.50 | −0.59 | −5.88 | −3.92 | 1.96 |
| P4 | 576 | 631 | 736 | 1.68 | −5.06 | −3.06 | 2.00 | 1.40 | −0.98 | −5.78 | −3.53 | 2.25 |
Cyclic voltammetry was used to study the electrochemical properties of the four polymers as thin films (Fig. 4d and Table 1). Samples were measured in acetonitrile/[n-Bu4N]PF6 solutions (0.1 mol L−1) with an Ag/Ag+ reference electrode. The HOMO energy levels of polymers P1 and P2 were calculated to be −5.60 eV and −5.56 eV from the oxidation onset at 1.22 V and 1.18 V respectively, based on the ferrocene/ferrocenium couple (EHomo = −[4.8 + Eox/reonset − EFco/+] eV). The LUMO energy levels were calculated from the reduction onset to be −3.52 and −3.61 eV for P1 and P2, respectively. Polymer P3 exhibits more negative energy levels (−5.88 and −3.92 eV for HOMO and LUMO, respectively) which can be explained by the strong electron-withdrawing ability of fluorine substituted on bithiophene. In contrast, polymer P4 shows a relatively high-lying LUMO energy level, arising from the electron-rich BDT unit.
Transistor device performance
The charge transport properties of these polymer materials were investigated using organic field-effect transistors (OFETs) in a top-gate/bottom-contact (TGBC) architecture on glass substrates (Fig. 5a). The polymer thin films were deposited using a blade coating technique (details provided in the ESI†). The transfer and output characteristics of the transistors are displayed in Fig. 5b, c and Fig. S22,† and a comparison of the transfer curves is shown in Fig. S23a.† These figures clearly indicate that the devices utilizing polymer P3 in the channel layer possess higher on-current compared to the devices with other polymers, whereas polymer P4 has the lowest on-current among them. To compare the saturation field-effect electron mobility (μe,sat) values of the devices, gate-dependent mobility curves (Fig. S23b†) are plotted for the best-performing devices of each polymer along with a comparison of the average μe,sat of all the devices (Fig. 5d). From these data it is evident that the polymer P3 showed significantly better performance in terms of μe,sat, which can be explained by the relatively lower LUMO energy level resulting from the strong electron-withdrawing abilities of the DTYM in combination with fluorine groups, and the more coplanar backbone conformation.1,76 However the average threshold voltage (VTh) value is slightly higher (Fig. S23c†) and the average on–off ratio (Ion–off) is marginally lower (Fig. S23d†) compared to the other polymers. The overall device performance of the four polymers is summarized in Table 2. | ||
| Fig. 5 OFET performance of the polymers. (a) OFET device structure. (b and c) Transfer and output characteristics of the polymer P3. (d) Comparison of average μe,sat of polymers P1–P4. | ||
| Polymer | μ e,sat (cm2 V−1 s−1) | V Th (V) | I on/Ioff |
|---|---|---|---|
| P1 | 1.31 (±0.07) × 10−3 | 68.15 (±9.1) | 4.2 (±2.6) × 104 |
| P2 | 5.23 (±3.3) × 10−4 | 70.87 (±17.9) | 1.5 (±0.9) × 104 |
| P3 | 5.2 (±0.46) × 10−3 | 77.71 (±2.5) | 0.5 (±0.15) × 104 |
| P4 | 7.54 (±1.28) × 10−5 | 67.71 (±3.9) | 0.01 (±0.006) × 104 |
Morphology and crystallinity investigation
To gain insight into the difference in charge carrier mobilities among the polymers, the morphology and the crystallinity of the spin-coated films was investigated using atomic force microscopy (AFM) and X-ray diffraction (XRD) analysis. As displayed in Fig. 6, the films of polymers P1–P3 look similar in terms of polymer grain size, whereas polymer P4 shows a smaller grain size accompanied by more grain boundaries. Analysis of the height distribution of the thin films (Fig. S24†) reveals that all materials had a flat surface and a very low surface roughness with a route mean square (RMS) roughness of under 2 nm for P1–P3 and 2.88 nm for P4. The superior morphology of P1–P3 may significantly contribute to the relatively higher charge carrier mobility compared to P4, which is in good agreement with the OFET device performance (Table 2). Thin film XRD was measured using a lab-based diffractometer and the resulting patterns are shown in Fig. S25.† No peaks were observed for P2 and P4. However, P3 exhibited several peaks at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ ≈6.99°, 10.36° and 13.74° and P1 exhibited weak peaks at 7.08 and 10.56° corresponding to the presence of crystalline regions. These observations correlate with the observed charge carrier mobility trends observed for the TFT devices.
θ ≈6.99°, 10.36° and 13.74° and P1 exhibited weak peaks at 7.08 and 10.56° corresponding to the presence of crystalline regions. These observations correlate with the observed charge carrier mobility trends observed for the TFT devices.
 | ||
| Fig. 6 Topography AFM images of (a) P1 (b) P2 (c) P3 and (d) P4. All thin films were thermally annealed at 80 °C. Scale bar in 500 nm in all images. | ||
Conclusions
In summary, we developed the acceptor unit, BT-DTYM, and copolymerised it with different donor units of varying electron-donating strength to yield four new n-type semiconducting polymers (P1–P4). We investigated their differences in backbone geometry, optical and electrochemical properties, morphology and OFET device performance to understand the impact of DTYM incorporation. Theoretical calculations demonstrated that the introduction of the DTYM group onto the BT acceptor unit increased the molecular dipole moment but reduced backbone planarity in comparison to the unsubstituted BT. Of the range of co-polymers studied, P3, containing a partially fluorinated bithiophene donor group, exhibited a more red-shifted absorption combined with lower-lying HOMO and LUMO levels, which can be explained by the strong electron-withdrawing ability of fluorine in combination with DTYM. All polymers demonstrated n-type semiconductor behaviour in organic field-effect transistors, with reasonable saturation mobility up to 5.2 × 10−3 cm2 V−1 s−1 for polymer P3. Our work demonstrates a facile approach to DTYM-heteroannulation for increasing the electron affinity of BT-based monomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physics Science Research Council (EPSRC) (EP/V048686/1 and EP/T028513/1) and the Royal Society and Wolfson Foundation for financial support. This work was supported by King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under award OSR-2019-CRG8-4095.References
- H. Jiang, S. Zhu, Z. Cui, Z. Li, Y. Liang, J. Zhu, P. Hu, H.-L. Zhang and W. Hu, Chem. Soc. Rev., 2022, 51, 3071–3122 RSC.
- Z.-G. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422–4433 CrossRef CAS PubMed.
- C. J. Kousseff, R. Halaksa, Z. S. Parr and C. B. Nielsen, Chem. Rev., 2022, 122, 4397–4419 CrossRef CAS PubMed.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- M. Moser, A. Wadsworth, N. Gasparini and I. McCulloch, Adv. Energy Mater., 2021, 11, 2100056 CrossRef CAS.
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- X. Guo, L. Han and X. Hou, Mater. Chem. Front., 2021, 5, 6760–6778 RSC.
- F. Tenopala-Carmona, O. S. Lee, E. Crovini, A. M. Neferu, C. Murawski, Y. Olivier, E. Zysman-Colman and M. C. Gather, Adv. Mater., 2021, 33, 2100677 CrossRef CAS PubMed.
- P. Kafourou, B. Park, J. Luke, L. Tan, J. Panidi, F. Glöcklhofer, J. Kim, T. D. Anthopoulos, J.-S. Kim, K. Lee, S. Kwon and M. Heeney, Angew. Chem., Int. Ed., 2021, 60, 5970–5977 CrossRef CAS PubMed.
- T. Hodsden, K. J. Thorley, A. Basu, A. J. P. White, C. Wang, W. Mitchell, F. Glöcklhofer, T. D. Anthopoulos and M. Heeney, Mater. Adv., 2021, 2, 1706–1714 RSC.
- H. Sun, H. Yu, Y. Shi, J. Yu, Z. Peng, X. Zhang, B. Liu, J. Wang, R. Singh, J. Lee, Y. Li, Z. Wei, Q. Liao, Z. Kan, L. Ye, H. Yan, F. Gao and X. Guo, Adv. Mater., 2020, 32, 2004183 CrossRef CAS PubMed.
- H. Sun, X. Guo and A. Facchetti, Chem, 2020, 6, 1310–1326 CAS.
- Q. Qi, X. Guo, B. Zhu, P. Deng, H. Zhan and J. Yang, Org. Electron., 2020, 78, 105616 CrossRef CAS.
- K. Feng, J. Huang, X. Zhang, Z. Wu, S. Shi, L. Thomsen, Y. Tian, H. Y. Woo, C. R. McNeill and X. Guo, Adv. Mater., 2020, 32, 2001476 CrossRef CAS PubMed.
- W. Zhong, S. Sun, L. Ying, F. Liu, L. Lan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 7315–7321 CrossRef CAS PubMed.
- A. Creamer, A. Casey, A. V. Marsh, M. Shahid, M. Gao and M. Heeney, Macromolecules, 2017, 50, 2736–2746 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- K. Iguchi, T. Mikie, M. Saito, K. Komeyama, T. Seo, Y. Ie and I. Osaka, Chem. Mater., 2021, 33, 2218–2228 CrossRef CAS.
- C. Zhang, W. L. Tan, Z. Liu, Q. He, Y. Li, J. Ma, A. S. R. Chesman, Y. Han, C. R. McNeill, M. Heeney and Z. Fei, Macromolecules, 2022, 55, 4429–4440 CrossRef CAS.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- A. Balan, D. Baran and L. Toppare, Polym. Chem., 2011, 2, 1029–1043 RSC.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- W. Meng, J. Lv, T. Duan, Z. Kan, S. Lu, X. Dai and Z. Li, Mater. Chem. Phys., 2020, 247, 122874 CrossRef CAS.
- S. Wang, Y. Hu, J. Yang and W. Huang, Synth. Met., 2019, 250, 94–98 CrossRef CAS.
- R. Ratha, A. Singh, M. A. Afroz, R. K. Gupta, M. Baumgarten, K. Müllen and P. K. Iyer, Synth. Met., 2019, 252, 113–121 CrossRef CAS.
- C. R. Belton, A. L. Kanibolotsky, J. Kirkpatrick, C. Orofino, S. E. T. Elmasly, P. N. Stavrinou, P. J. Skabara and D. D. C. Bradley, Adv. Funct. Mater., 2013, 23, 2792–2804 CrossRef CAS.
- C. L. Chochos, N. Leclerc, N. Gasparini, N. Zimmerman, E. Tatsi, A. Katsouras, D. Moschovas, E. Serpetzoglou, I. Konidakis, S. Fall, P. Lévêque, T. Heiser, M. Spanos, V. G. Gregoriou, E. Stratakis, T. Ameri, C. J. Brabec and A. Avgeropoulos, J. Mater. Chem. A, 2017, 5, 25064–25076 RSC.
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M.-G. Kim, A. Facchetti and X. Guo, Adv. Mater., 2019, 31, 1905161 CrossRef CAS PubMed.
- C. Zhu, J. Yuan, F. Cai, L. Meng, H. Zhang, H. Chen, J. Li, B. Qiu, H. Peng, S. Chen, Y. Hu, C. Yang, F. Gao, Y. Zou and Y. Li, Energy Environ. Sci., 2020, 13, 2459–2466 RSC.
- J. K. Gallaher, S. N. Pugliese, M. A. Uddin, T. H. Lee, J. Y. Kim, H. Y. Woo and J. M. Hodgkiss, J. Phys. Chem. C, 2021, 125, 26590–26600 CrossRef CAS.
- P. Cai, X. Xu, J. Sun, J. Chen and Y. Cao, RSC Adv., 2017, 7, 20440–20450 RSC.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- S. Das, P. B. Pati and S. S. Zade, Macromolecules, 2012, 45, 5410–5417 CrossRef CAS.
- R. Ratha, M. A. Afroz, R. K. Gupta and P. K. Iyer, New J. Chem., 2019, 43, 4242–4252 RSC.
- X. Gong, G. Li, C. Li, J. Zhang and Z. Bo, J. Mater. Chem. A, 2015, 3, 20195–20200 RSC.
- X. Hu, R. Datt, Q. He, P. Kafourou, H. K. H. Lee, A. J. P. White, W. C. Tsoi and M. Heeney, J. Mater. Chem. C, 2022, 10, 9249–9256 RSC.
- B. Hu, M. Li, W. Chen, X. Wan, Y. Chen and Q. Zhang, RSC Adv., 2015, 5, 50137–50145 RSC.
- L. Lan, B. Zhao, J. Zhang, Y. Li, Y. Liu, Y. Mai, B. Liao and C. Gao, New J. Chem., 2019, 43, 3565–3571 RSC.
- K. Kranthiraja, K. Gunasekar, S. H. Park, I.-N. Kang, J. Y. Lee, M. Song and S.-H. Jin, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2668–2679 CrossRef CAS.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22–36 RSC.
- J. H. Vella, L. Huang, N. Eedugurala, K. S. Mayer, T. N. Ng and J. D. Azoulay, Sci. Adv., 2021, 7, eabg2418 CrossRef CAS PubMed.
- L. Huang, N. Eedugurala, A. Benasco, S. Zhang, K. S. Mayer, D. J. Adams, B. Fowler, M. M. Lockart, M. Saghayezhian, H. Tahir, E. R. King, S. Morgan, M. K. Bowman, X. Gu and J. D. Azoulay, Adv. Funct. Mater., 2020, 30, 1909805 CrossRef CAS.
- T. Hodsden, K. J. Thorley, J. Panidi, A. Basu, A. V. Marsh, H. Dai, A. J. P. White, C. Wang, W. Mitchell, F. Glöcklhofer, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2020, 30, 2000325 CrossRef CAS.
- Y. Yamashita, T. Suzuki, G. Saito and T. Mukai, J. Chem. Soc., Chem. Commun., 1986, 1489–1491 RSC.
- X. Gao, C.-A. Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697–3699 CrossRef CAS PubMed.
- Y. Hu, X. Gao, C.-A. Di, X. Yang, F. Zhang, Y. Liu, H. Li and D. Zhu, Chem. Mater., 2011, 23, 1204–1215 CrossRef CAS.
- X. Chen, J. Wang, G. Zhang, Z. Liu, W. Xu and D. Zhang, New J. Chem., 2013, 37, 1720–1727 RSC.
- Y. Hu, Z. Wang, X. Zhang, X. Yang, H. Li and X. Gao, Chin. J. Chem., 2013, 31, 1428–1438 CrossRef CAS.
- Y. Hu, Y. Qin, X. Gao, F. Zhang, C.-A. Di, Z. Zhao, H. Li and D. Zhu, Org. Lett., 2012, 14, 292–295 CrossRef CAS PubMed.
- B. Leng, D. Lu, X. Jia, X. Yang and X. Gao, Org. Chem. Front., 2015, 2, 372–377 RSC.
- Y. Hu, D. X. Cao, A. T. Lill, L. Jiang, C.-A. Di, X. Gao, H. Sirringhaus and T.-Q. Nguyen, Adv. Electron. Mater., 2018, 4, 1800175 CrossRef.
- J. Panidi, J. Kainth, A. F. Paterson, S. Wang, L. Tsetseris, A.-H. Emwas, M. A. McLachlan, M. Heeney and T. D. Anthopoulos, Adv. Funct. Mater., 2019, 29, 1902784 CrossRef.
- H. Li, Y. Shi, G. Han, J. Liu, J. Zhang, C. Li, J. Liu, Y. Yi, T. Li, X. Gao, C. Di, J. Huang, Y. Che, D. Wang, W. Hu, Y. Liu and L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 4380–4384 CrossRef CAS PubMed.
- M. A. Kolaczkowski, A. Garzón-Ruiz, A. Patel, Z. Zhao, Y. Guo, A. Navarro and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 53328–53341 CrossRef CAS PubMed.
- A. Creamer, C. S. Wood, P. D. Howes, A. Casey, S. Cong, A. V. Marsh, R. Godin, J. Panidi, T. D. Anthopoulos, C. H. Burgess, T. Wu, Z. Fei, I. Hamilton, M. A. McLachlan, M. M. Stevens and M. Heeney, Nat. Commun., 2018, 9, 3237 CrossRef PubMed.
- S. Cong, A. Creamer, Z. Fei, S. A. J. Hillman, C. Rapley, J. Nelson and M. Heeney, Macromol. Biosci., 2020, 20, 2000087 CrossRef CAS PubMed.
- M. Comí, D. Patra, R. Yang, Z. Chen, A. Harbuzaru, Y. Wubulikasimu, S. Banerjee, R. P. Ortiz, Y. Liu and M. Al-Hashimi, J. Mater. Chem. C, 2021, 9, 5113–5123 RSC.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- A. Casey, Y. Han, Z. Fei, A. J. P. White, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. C, 2015, 3, 265–275 RSC.
- E. Collado-Fregoso, P. Boufflet, Z. Fei, E. Gann, S. Ashraf, Z. Li, C. R. McNeill, J. R. Durrant and M. Heeney, Chem. Mater., 2015, 27, 7934–7944 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- Q. Tao, M. Xiao, M. Zhu, L. Shao, Z. Sui, P. Wang, G. Huang, Y. Pei, W. Zhu and F. Huang, Dyes Pigm., 2017, 144, 142–150 CrossRef CAS.
- H. S. Kim, C. E. Song, J.-W. Ha, S. Lee, S. Rasool, H. K. Lee, W. S. Shin and D.-H. Hwang, ACS Appl. Mater. Interfaces, 2019, 11, 47121–47130 CrossRef CAS PubMed.
- M. Saito, T. Fukuhara, S. Kamimura, H. Ichikawa, H. Yoshida, T. Koganezawa, Y. Ie, Y. Tamai, H. D. Kim, H. Ohkita and I. Osaka, Adv. Energy Mater., 2020, 10, 1903278 CrossRef CAS.
- H. Yu, Y. Wang, H. K. Kim, X. Wu, Y. Li, Z. Yao, M. Pan, X. Zou, J. Zhang, S. Chen, D. Zhao, F. Huang, X. Lu, Z. Zhu and H. Yan, Adv. Mater., 2022, 34, 2200361 CrossRef CAS PubMed.
- B. Yin, Z. Chen, S. Pang, X. Yuan, Z. Liu, C. Duan, F. Huang and Y. Cao, Adv. Energy Mater., 2022, 12, 2104050 CrossRef CAS.
- Z. Pang, W. Zhang, J. Wu, Y. Luo, J. Liu, S. Zhao, Z. Xu, Z. Lu and Y. Huang, Dyes Pigm., 2020, 181, 108562 CrossRef CAS.
- Y. Li, W. K. Tatum, J. W. Onorato, S. D. Barajas, Y. Y. Yang and C. K. Luscombe, Polym. Chem., 2017, 8, 5185–5193 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- H.-S. Chung, W.-H. Lee, C. E. Song, Y. Shin, J. Kim, S. K. Lee, W. S. Shin, S.-J. Moon and I.-N. Kang, Macromolecules, 2014, 47, 97–105 CrossRef CAS.
- S. P. Prakoso, S. Kumar, S.-L. Wu, G.-T. Ciou, Y.-J. Ke, S. Venkateswarlu, Y.-T. Tao and C.-L. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 1169–1178 CrossRef CAS PubMed.
- K. Zhou, H. Dong, H.-l. Zhang and W. Hu, Phys. Chem. Chem. Phys., 2014, 16, 22448–22457 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01430k |
| This journal is © The Royal Society of Chemistry 2023 |