
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting retro oxa-Michael chemistry in polymers†
Karin
Ratzenböck
 ab,
Johanna M.
Uher
ab,
Susanne M.
Fischer
ab,
David
Edinger
ab,
Viktor
Schallert
ab,
Ema
Žagar
c,
David
Pahovnik
c and
Christian
Slugovc
*ab
ab,
Johanna M.
Uher
ab,
Susanne M.
Fischer
ab,
David
Edinger
ab,
Viktor
Schallert
ab,
Ema
Žagar
c,
David
Pahovnik
c and
Christian
Slugovc
*ab
aInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: slugovc@tugraz.at
bChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, 8010 Graz, Austria
cNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia
First published on 4th January 2023
Abstract
One way to obtain recyclable polymeric materials is to include reversible bonds in polymers. Herein, we study the reversibility of the oxa-Michael reaction and explore its scope and limitations in simple model systems and further in linear polymers as well as in polymer networks. The results show that the retro oxa-Michael reaction of sulfone, acrylate or acrylonitrile based adducts is considerably fast at elevated temperatures (>100 °C) if Brønsted bases (e.g. KOH) are used as catalysts. Under these conditions, alcohols can easily be exchanged in oxa-Michael adducts within minutes. Furthermore, poly(ether)s derived from oxa-Michael reactions can be depolymerized into small fragments in the presence of alcohols and show self-healing characteristics in networks.
Introduction
With a global recycling rate of plastics as low as 9% in 2019,1 multiple strategies from mechanical recycling to chemical recycling or upcycling need to be targeted to increase the recycling rate. Whereas in chemical recycling the polymer is degraded into its monomer,2 by upcycling new molecules, polymers or materials are obtained.3 Thus, implementing reversible reactions in polymer chemistry is key for forming recyclable materials. Upon depolymerization of polymers, building blocks suitable for subsequent polymerization can be obtained. Recently, a lot of research has been performed in the field of covalent adaptable networks (CANs) or vitrimers, which undergo reversible reactions under external stimuli such as pH, heat or UV light. Regarding Michael addition reactions, dynamic covalent exchange for thiol and amine-based Michael adducts has been reported. Reversibility is generally triggered by a base4 and/or elevated temperatures.5 The dynamic nature of thiol bonds has been demonstrated for small molecules,6e.g. by the transfer of a thiol from a ketone to an acrylate product at 90 °C7 (Scheme 1) or by the exchange of thiophenol with mercaptoethanol in a thiol–ketone or thiol–maleimide adduct at 90 °C.5,8 Moreover, a dynamic exchange of thia–Michael polymer networks is reported.7–9 Examples include thiol–maleimide linkers in RAFT-based materials, thiol–acrylate linkers in covalently crosslinked polyester networks or thiol–vinyl sulfone linkers in the polymeric network formed by radical polymerization.8–10 These materials exhibit reshaping and self-healing abilities at temperatures ranging from 90 to 100 °C. In addition, networks solely built up by the thiol–Michael reaction with a cinnamonitrile derivative show self-healing behaviour from 60 °C onward.11 Moreover, reversible thiol–yne crosslinking was demonstrated by the group of Du Prez.12 Regarding retro aza–Michael reactions, typically high temperatures (>150 °C) are required. Alternatively, for specific aza–Michael adducts, the retro-Michael reaction can also be promoted by the use of solvents at room temperature or catalysts (base) at 60 °C.13 Already in 1944, the reverse reaction of a diethanolamine–acrylonitrile adduct to diethanolamine and poly(acrylonitrile) was demonstrated at 150 °C (Scheme 1).14 Recent model studies show the amine transfer of an amine–alkylacrylate adduct to benzyl acrylate in the temperature range of 100–160 °C without an additional catalyst.15 This finding was further used to build CANs based on β-amino ester, which can then be reprocessed at 150 °C due to retro aza–Michael and transesterification reactions.15 Another example combines pH-sensitive amidation and heat-sensitive dynamic aza–Michael addition in CANs.16 Moreover, dynamic amino-yne click chemistry at 130 °C was reported in CANs.17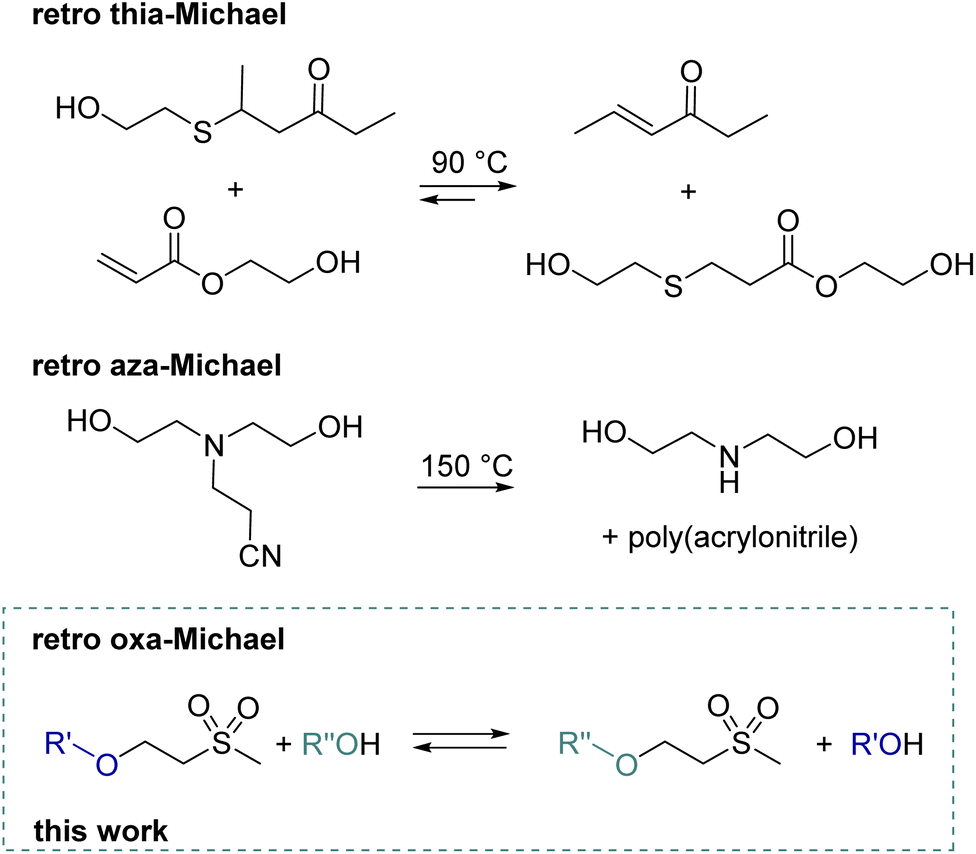 | ||
| Scheme 1 Previous work demonstrating retro thia-7 and aza-Michael14 reactions in small molecules and the schematic representation of herein discussed retro oxa-Michael reaction. | ||
In contrast, reports on the utilization of retro carba-Michael18 and in particular retro oxa-Michael reactions in polymer chemistry are scarce. The reversibility of the oxa-Michael reaction is rather discussed as a major drawback in small molecule synthesis19 and decomposition products caused by the retro oxa-Michael reaction were observed.20 More explicitly, evidence for the reversibility of the oxa-Michael reaction is indicated by the exchange of methanol in β-methoxy ketone with d-methanol in the presence of PMe3.21 Similar studies show that the addition of deuterated methanol to a vinyl ketone is reversible as deuterium is incorporated in the α-position of the vinyl.22 Our group recently demonstrated that the attack and elimination of hydroxide to a vinyl sulfone is a fast, reversible process. By using deuterated water, we also observed partial deuteration of the vinyl.23 Examples for applying the retro oxa-Michael reaction include the evaporation of water from hydroxy ethyl sulfone derivatives at room temperature under basic conditions (pH = 8.5) to obtain reactive vinyl groups which can then further react with alcohols.24 However, this is described to be a very slow process, taking several days. In contrast, at 80 °C, 1,4-oxathiane 4,4-dioxide could be prepared selectively from bishydroxy ethyl sulfone under basic conditions within 24 h by the loss of water.23 Moreover, methylene malonate derivatives can be blocked with Michael donors such as alcohols or thiols. By the reverse Michael reaction, the alkene is liberated upon heat treatment (>200 °C). This allows either the deconstruction of polymers or the alkene can further take part in cross-linking and such formulations can be used in e.g. thermally activated coatings.25 However, no systematic study on the retro oxa-Michael reaction is available.
Therefore, we herein report a detailed study of the reversibility of the oxa-Michael reaction for simple model systems, identifying the ideal reaction conditions. The retro oxa-Michael reaction was demonstrated by showing the exchange of the alcohol in oxa-Michael adducts (Scheme 1). In other words, the retro oxa-Michael reaction is followed by the addition of another alcohol to the newly formed Michael acceptor giving a new oxa-Michael adduct. Furthermore, we demonstrate the scope and some limitations of the use of the retro oxa-Michael reaction in polymers and polymer networks.
Results and discussion
Retro oxa-Michael reaction in small molecules
To obtain a general understanding of the retro oxa-Michael reaction and its required conditions, simple oxa-Michael model compounds were synthesized in the first step. Vinyl sulfones were preferred over acrylates as Michael acceptors, because in the case of the latter, transesterification occurs as a competing side reaction.26 Model structures were synthesized from methyl vinyl sulfone and the respective alcohols under solvent-free conditions at room temperature with potassium hydroxide (KOH) as a catalyst (see the ESI†). In the initial study, a mixture of the benzyl alcohol adduct 1 and 3 equiv. of allyl alcohol was used to evaluate different catalysts for the reverse oxa-Michael reaction (Fig. 1a). As for the oxa-Michael addition of alcohols Lewis or Brønsted base catalysis is required,27,28 we expected that in the retro oxa-Michael reaction also a catalyst would be necessary. Various catalysts (10 mol% with respect to 1) ranging from pure Brønsted bases to pure Lewis bases were screened at a temperature of 140 °C and the conversion towards 2 was monitored by 1H-NMR spectroscopy after 1 h and 24 h. In the presence of strong Brønsted bases such as KOH, potassium tert-butoxide (KOtBu) and the phosphazene base 1-tert-butyl-2,2,4,4,4-pentakis-(dimethylamino)-2λ5,4λ5-catenadi-(phosphazen) (P2-tBu), about 60% of the newly formed adduct 2 could be detected after 1 h (Fig. 1b). The remaining 40% were unreacted 1. Additionally, free benzyl alcohol was observed in the reaction mixture, whereas methyl vinyl sulfone could not be detected. This indicates that methyl vinyl sulfone is only intermediately formed and is quickly trapped by the excessive alcohol. The use of the distinctly weaker Brønsted base sodium carbonate (Na2CO3) as a catalyst resulted in a much slower reaction, as only 6% conversion towards 2 was noted after 1 h, which further increased to 46% after 24 h. Using catalysts that are Lewis bases and considerably strong Brønsted bases at the same time led to differing results. While 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) gave similar conversions to the discussed pure strong Brønsted bases, the use of 1,1,3,3-tetramethylguanidine (TMG) resulted in only 23% conversion towards 2 in 24 h (14% in 1 h). The similar basicity of DBU (pKa = 13.5 ± 1.5) and TMG (pKa = 13.0 ± 1.0) cannot explain their diverging performance.29 Instead it is conceivable that DBU is less prone to degradation than TMG under the given reaction conditions.30 Less basic catalysts triethylamine (Et3N), 1,4-diazabicyclo[2.2.2]octane (DABCO) and triphenyl phosphine (PPh3) are almost or completely inactive in catalysing the retro oxa-Michael reaction under the studied conditions. The same applies to the uncatalysed reaction as no evidence for a reverse reaction could be observed after heating for 24 h at 140 °C. The study confirms the necessity of a Brønsted-basic catalyst for performing the retro oxa-Michael reaction with reasonable speed at the temperature under investigation. The catalyst is believed to deprotonate the Cα-atom next to the electron withdrawing group, in this case the sulfone. The corresponding carbanionic species in the following eliminates the corresponding alkoxide and forms the respective alkene. Nucleophilic substitution is a conceivable mechanistic alternative for the formation of 2 from 1 under the used reaction conditions. However, allyl benzyl ether or 2-(methylsulfonyl)ethanol, the other potential products of a nucleophilic substitution reaction, have never been observed in any of the herein presented reactions. Thus, a nucleophilic substitution mechanism is unlikely to take place. After 24 h, the ratio of 1 to 2 changed slightly in favour of 1. A gradual loss of the relatively volatile allyl alcohol (bp = 97 °C vs. benzyl alcohol: bp = 205 °C) over time might cause a readjustment of the equilibrium between 1 and 2.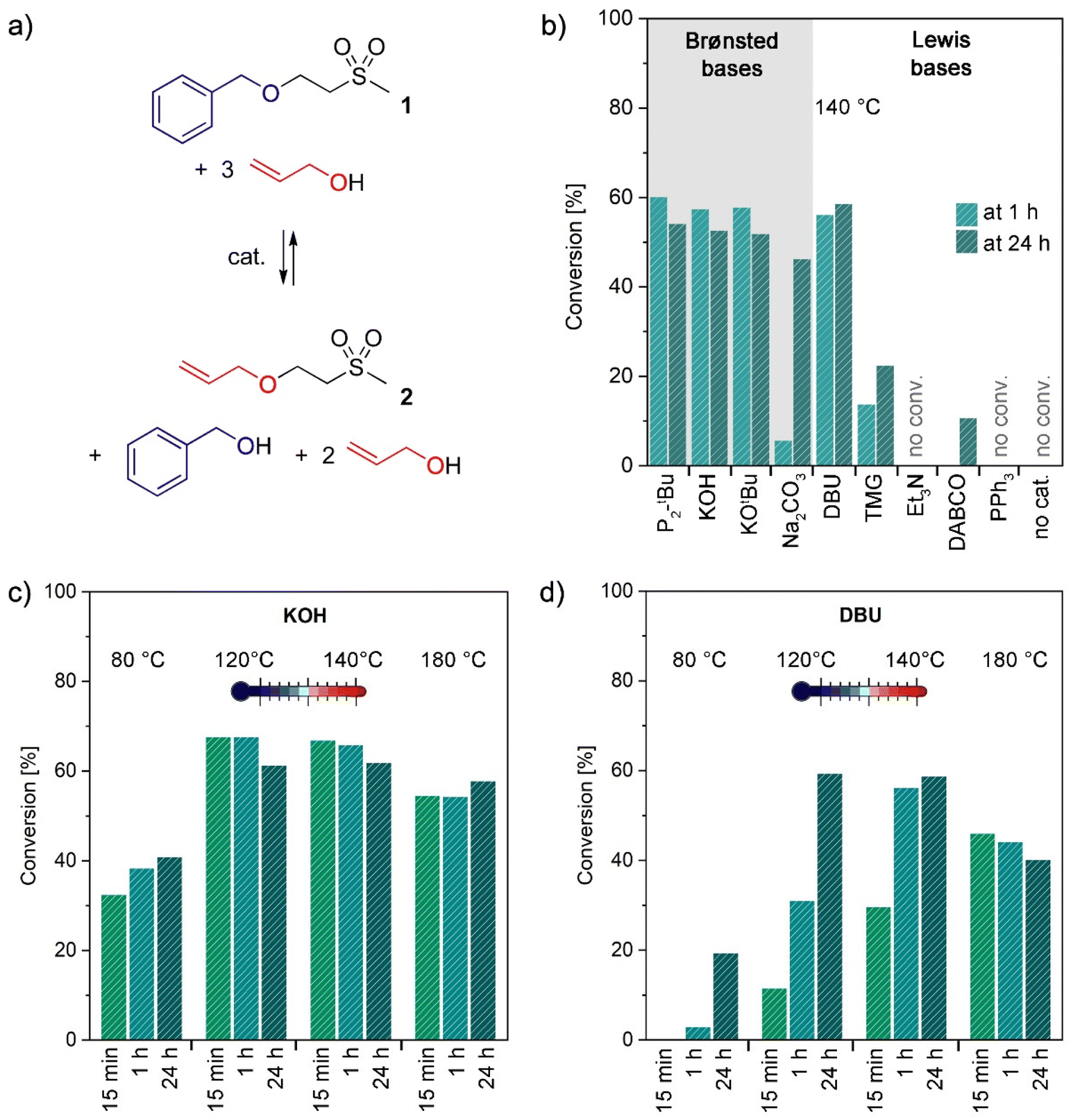 | ||
| Fig. 1 (a) Chemical equation of the test reaction; (b) catalyst screening for the conversion of 1 towards 2; reaction conditions: 0.10 equiv. catalyst, 3 equiv. allyl alcohol, 140 °C for either 1 h or 24 h; (c) temperature screening for the conversion of 1 towards 2; reaction conditions: 0.05 equiv. KOH, 3 equiv. allyl alcohol, conversion after 15 min, 1 h or 24 h; (d) temperature screening for the conversion of 1 towards 2; reaction conditions: 0.10 equiv. DBU, 3 equiv. allyl alcohol, conversion after 15 min, 1 h or 24 h. | ||
For further screenings, KOH and DBU were chosen as the preferred catalysts. P2-tBu was not further investigated as it shows no advantage over KOH and is much more expensive and toxic. DBU was considered as an interesting alternative as it is a liquid and thus has advantages over the solid KOH under certain circumstances. Lower catalyst loadings (2 and 5 mol% with respect to 1) were studied for KOH and DBU (Table S2†). In the case of KOH, the best results were obtained with 5 mol% with 65% conversion to 2 after 1 h. Lower catalyst loadings diminish the retro oxa-Michael reaction. In the case of DBU, a loading of 10 mol% (56% of 2 after 1 h) outperforms 5 mol% (44% of 2 after 1 h). In the following, either 5 mol% KOH or 10 mol% DBU was used to assess the impact of the reaction temperature. The aforementioned reaction was additionally performed at 80, 120, 140 and 180 °C with 5 mol% KOH or 10 mol% DBU. The results are depicted in Fig. 1c and d and show the conversion to product 2 after 15 min, 1 h and 24 h. For KOH, equally good conversions are obtained at 120 °C and 140 °C. A slightly worse conversion was observed at 180 °C. Even at 80 °C, the retro oxa-Michael reaction takes place; however, at a slower pace. Moreover, the maximum conversion is already reached at 120 °C after 15 min. As the retro oxa-Michael reaction occurred fast with KOH, the reaction progress was monitored in shorter intervals at 140 °C (Fig. S15†). It was already found that after a reaction time of 1 min, 65% conversion to 2 is obtained. Thus, the retro oxa-Michael reaction is a considerably fast reaction under KOH catalysis at elevated temperatures. In contrast, with the amidine base DBU, longer reaction times and higher temperatures are needed to reach similar conversions. At 80 °C, hardly any reverse reaction occurs and also at 120 °C and 140 °C the reverse reaction is distinctly slower than with KOH. The results at 180 °C show trends similar to those with KOH.
To elucidate the impact of different alcohols in the retro oxa-Michael reaction, Michael adducts with primary alcohols of allylic (1 and 2) or aliphatic (3 and 4) nature and secondary alcohols (5) were screened in the exchange reaction with allyl alcohol, benzyl alcohol, 1-butanol, 2-methylpropan-1-ol or cyclopentanol. The alcohol was added in excess (3 equiv.) and the reaction was carried out with 5 mol% KOH at 140 °C. The results after reaction times of 15 min and 24 h are depicted in the heatmap plot shown in Fig. 2a. As a first result, it can be seen that in most experiments, the equilibrium is already reached within the first 15 min as the conversion towards the exchanged products does not significantly change after a reaction time of 24 h. Secondly, the alcohol exchange is the lowest with compound 5 as the starting material and gradually increases from row to row in Fig. 2a reaching the highest exchange with adduct 2. In the next step, the corresponding equilibrium constants (K) were calculated based on the values obtained after a reaction time of 24 h. The equilibrium constants for the forward reaction and the corresponding reverse reaction are shown in Fig. 2b. In the case of the reaction of allyl alcohol, benzyl alcohol or 1-butanol with the corresponding adducts 1–3, the equilibrium can be reached from either side of the exchange reaction. In contrast, 2-methylpropan-1-ol and cyclopentanol show a different behaviour. Depending on the starting point, different equilibrium constants are noticed, i.e. an ideal behaviour is not observed. For example, the reaction of adduct 5 and allyl alcohol results in a K5→2 of 0.004 (reverse reaction: K2→5 = 250) while the reaction of adduct 2 with cyclopentanol gives a K2→5 of 2.74 (reverse reaction: K5→2 = 0.365). In the following, DFT calculations of Gibbs free energies were performed. The thereby determined equilibrium constants (for details on the calculations, see ESI, Fig. S16†) revealed values between 0.5 and 2 for all combinations. Accordingly, it can be assumed that the conversion of 4 and 5 towards their exchange products is more hindered compared to the formation of 4 and 5 from adducts 1–3 with 2-methylpropan-1-ol or cyclopentanol. In the latter cases, the expected equilibrium concentrations are reached. The reason for the observed misfit is yet unknown but might be due to a deactivation of the base by an unidentified side reaction before the equilibrium was reached. The screening demonstrates that not each alcohol component is equally suited for providing the desired reversible exchange reaction.
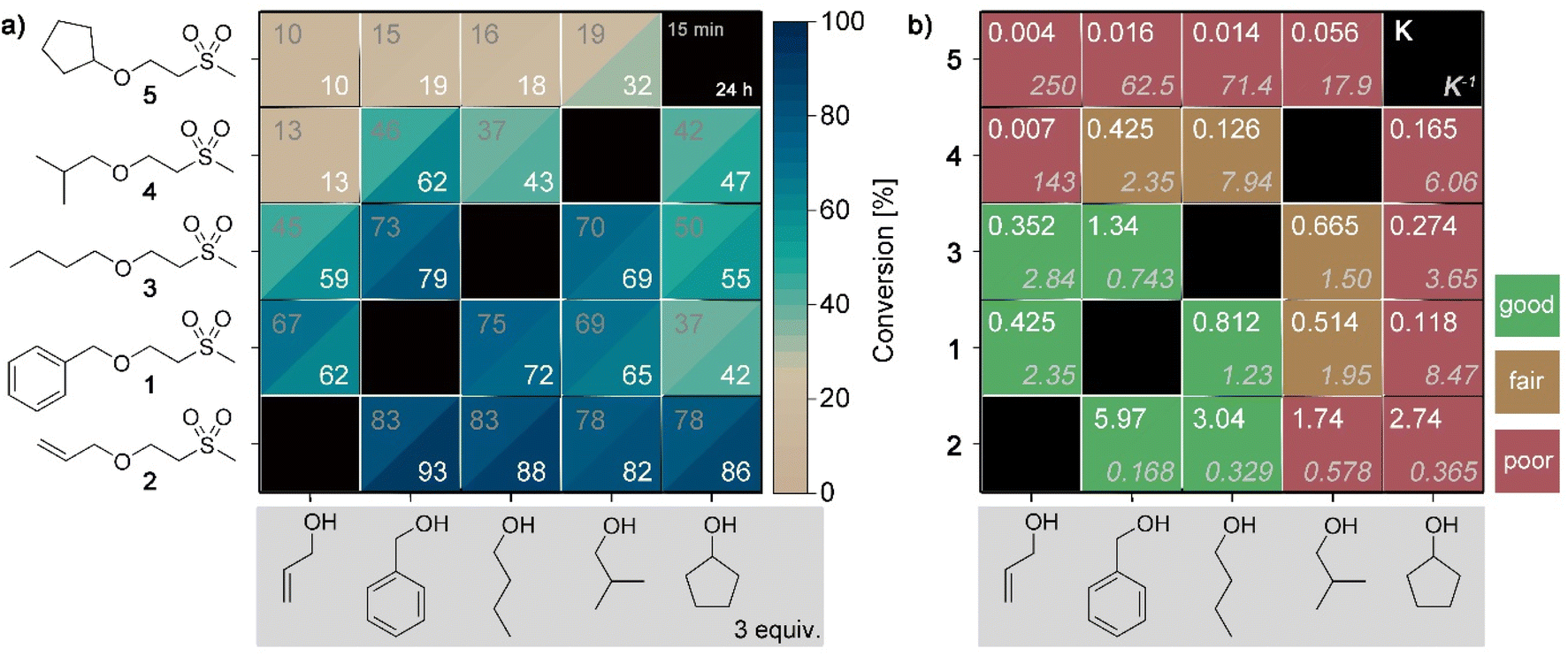 | ||
| Fig. 2 (a) Heatmap plot visualizing the conversion towards the respective product by the exchange of the alcohol by retro oxa-Michael reaction; reaction conditions: alcohol (3 equiv.), 140 °C, 5 mol% KOH, monitored after 15 min (upper, left corner) and after a reaction time of 24 h (lower, right corner); (b) matrix visualizing the corresponding equilibrium constants given for the forward (K) and the reverse reactions (K−1); colour code: green = good correlation (deviation of K and K−1 less than factor 3); brown = fair correlation (deviation of K and K−1 more than factor 3 but less than factor 15) and red = poor correlation (deviation of K and K−1 more than factor 15). | ||
In the following, we wanted to demonstrate the retro oxa-Michael reaction of acrylate-based models as especially in oxa-Michael polymerizations, acrylates are widely used.26,31 Benzyl 3-(benzyloxy)propanoate (6) was reacted with 3 equiv. of allyl alcohol at 140 °C with KOH or DBU as the catalyst (Fig. 3 and Fig. S20†). The three products resulting from the retro oxa-Michael reaction and transesterification (6a–6c) could be observed by 1H-NMR spectroscopy (Fig. S19†). For acrylates, DBU seems to be a more efficient catalyst in the retro Michael reaction. However, still only 20% of the newly formed allyl alcohol Michael adducts (6a and 6b) could be observed after a reaction time of 1 h, whereas two to three times as much transesterification occurred (6b and 6c, Fig. 3). Over 24 h, further retro Michael reaction took place leading to 35% of the newly formed oxa-Michael product. Instead, with KOH only about 10% of Michael adducts 6a and 6b were observed after 1 h, and the conversion did not increase significantly over time. The amount of transesterification products was similar to the case of DBU (Fig. S20†). The results show that the retro oxa-Michael reaction can take place with a variety of Michael acceptors. However, in the case of acrylates, transesterification is an omnipresent side reaction.
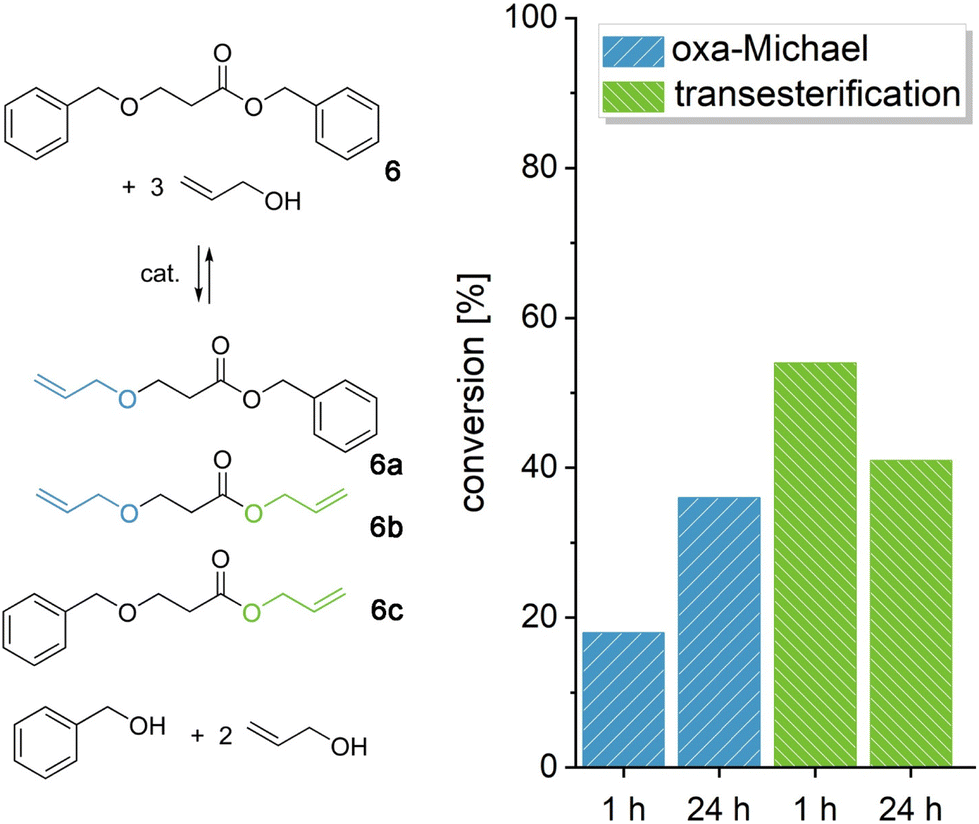 | ||
| Fig. 3 Reaction of acrylate-based model 6 with 3 equiv. allyl alcohol at 140 °C with 0.10 equiv. DBU leading to oxa-Michael and transesterification products 6a–6c. The conversion towards oxa-Michael and transesterification products after 1 h and 24 h is depicted on the right. | ||
Besides exchanging the alcohol in oxa-Michael adducts, the transfer of alcohol from one to another Michael acceptor was monitored by investigating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methyl vinyl sulfone derived adduct 1 and 3-(allyloxy)propanenitrile (7). Scrambling of the adducts occurred already within a reaction time of 15 min at 140 °C in the presence of 0.05 equiv. of KOH. The corresponding allyl alcohol adduct of methyl vinyl sulfone 2 as well as the corresponding benzyl alcohol adduct of acrylonitrile were identified (Fig. S21 and Scheme S1†). Moreover, the free Michael acceptors, methyl vinyl sulfone and acrylonitrile, were observed together with the free alcohols. Scrambling as described above could also be observed in a 1:1 mixture of methyl vinyl sulfone derived adduct 1 and ((2-(allyloxy)ethyl)sulfonyl)benzene (8) (Scheme S2†). The corresponding products as well as free vinyl sulfones and alcohols were noted by 1H-NMR spectroscopy (Fig. S22†). These findings further support the claim that the exchange reaction proceeds via the retro oxa-Michael reaction, which is a dissociative mechanism.
:1 mixture of methyl vinyl sulfone derived adduct 1 and 3-(allyloxy)propanenitrile (7). Scrambling of the adducts occurred already within a reaction time of 15 min at 140 °C in the presence of 0.05 equiv. of KOH. The corresponding allyl alcohol adduct of methyl vinyl sulfone 2 as well as the corresponding benzyl alcohol adduct of acrylonitrile were identified (Fig. S21 and Scheme S1†). Moreover, the free Michael acceptors, methyl vinyl sulfone and acrylonitrile, were observed together with the free alcohols. Scrambling as described above could also be observed in a 1:1 mixture of methyl vinyl sulfone derived adduct 1 and ((2-(allyloxy)ethyl)sulfonyl)benzene (8) (Scheme S2†). The corresponding products as well as free vinyl sulfones and alcohols were noted by 1H-NMR spectroscopy (Fig. S22†). These findings further support the claim that the exchange reaction proceeds via the retro oxa-Michael reaction, which is a dissociative mechanism.
Retro oxa-Michael reaction in polymerisation
To further study the reversibility in polymers, poly(ether-sulfone)s and poly(ether-ester)s were synthesized by oxa-Michael reactions. Poly1 and poly2 were prepared by reacting divinyl sulfone with ethylene glycol or (Z)-2-butene-1,4-diol, respectively, at room temperature over 24 h with 5 mol% KOH as a catalyst (see the ESI†).32 Moreover, 2-hydroxyethyl acrylate was polymerized according to a modified literature procedure (see the ESI†) with 5 mol% DBU at room temperature (denoted as poly3).27 In the case of poly1, the catalyst was removed by acidic work-up. The samples poly2 and poly3 were used without further purification. Depolymerisation of poly1 prepared from divinyl sulfone and ethylene glycol32 was studied by adding an excess of benzyl alcohol (3 equiv.) and 5 mol% KOH to the polymer and heating the mixture to 140 °C under stirring. After a reaction time of 1 h, depolymerisation to bis(2-benzyloxyethanyl)sulfone and free ethylene glycol was noted by NMR spectroscopy (Fig. S25 and S26†). In contrast, if no catalyst was added and poly1 was solely stirred with benzyl alcohol at 140 °C, no depolymerisation took place and poly1 was unaffected (Fig. S27 and S28†). However, etherification of benzyl alcohol to dibenzyl ether was observed under these conditions. According to the literature, at 120 °C and under acidic conditions, etherification of benzyl alcohol can take place.33 As in poly1 the catalyst was removed by an acidic work-up (ESI†); traces of acid might have catalysed the etherification of the alcohol. Thus, oxa-Michael polymers can easily be depolymerized into small molecules albeit not into their monomer divinyl sulfone.In the next step, the thermal stability of oxa-Michael polymers in the presence and absence of a base was investigated. Poly1 was synthesized with various catalysts (5 mol%) including Brønsted bases (KOH and KOtBu) and Lewis bases (DMAP and PPh3) (see the ESI for details†). Thermal degradation of the obtained polymers (still including the initial catalyst) was studied by thermogravimetric analysis (TGA) using a heating rate of 10 °C min−1. In the case of KOH and KOtBu, a mass loss of 5% was observed at 194 ± 1 °C (Fig. 4a). As evident from the first derivative of the curve shown in Fig. 4b, decomposition starts at about 160 °C and continues until about 300 °C. At 340 °C and 260 °C, two further inflection points were observed, pointing to at least three different degradation mechanisms (Fig. 4b). Poly1 obtained from nucleophiles was slightly more stable with a mass loss of 5% at 215 °C and 232 °C with PPh3 and DMAP, respectively (Fig. 4a and Table S3†). In these analyses also three different degradation processes are indicated, albeit most of the degradation occurred at temperatures above 300 °C. For comparison, poly1 was purified to remove the basic catalyst (KOH). Thereafter, poly1 exhibited a much higher thermal stability with a degradation onset around 320 °C. The inflection point of the by far most important degradation was found at 360 °C. A minor mass loss of purified poly1 around 100 °C is due to the solvent traces in the polymer resulting from purification (Fig. S23†). The solvent could not be removed completely under high vacuum. However, harsher drying conditions were not applied because heat treatment prior to the TGA measurement might lead to ambiguous results.
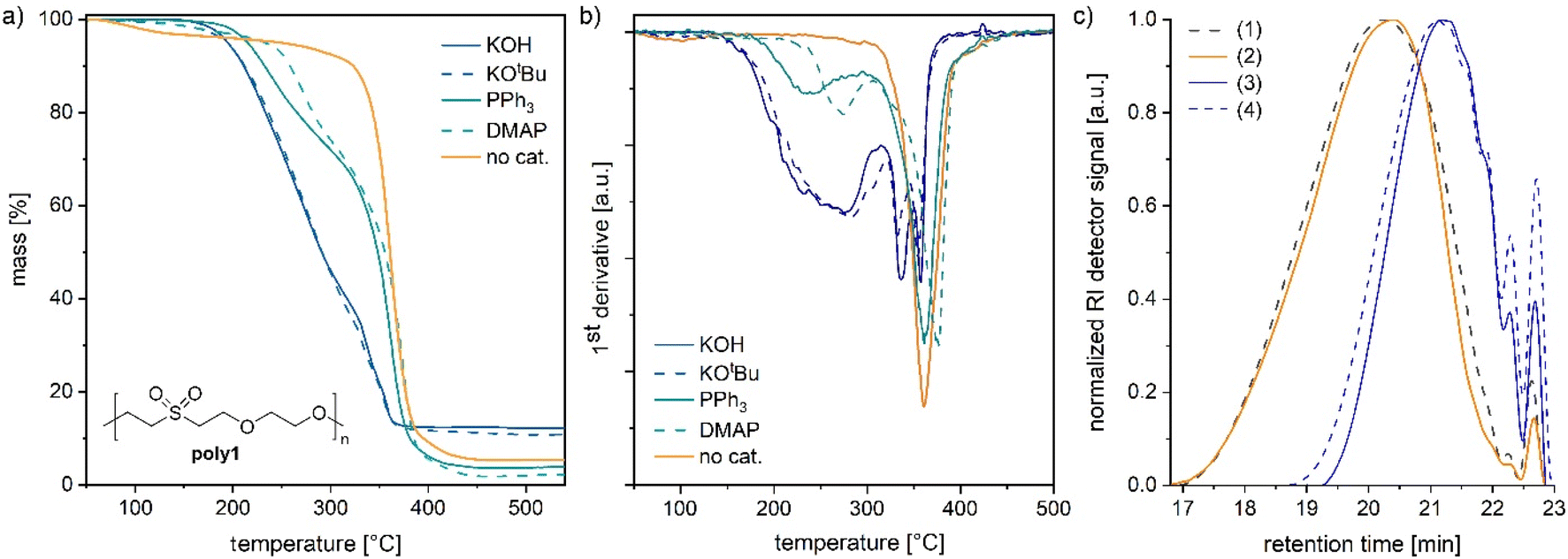 | ||
| Fig. 4 (a) Thermogravimetric investigation of poly1 in the presence of KOH or KOtBu (blue), nucleophiles PPh3 or DMAP (turquoise) or in the absence of a catalyst (orange); (b) 1st derivative of the mass–temperature plots shown in a; (c) size exclusion chromatograms of poly1 after preparation and purification (1), after heating for 1 h at 140 °C (2), after heating for 1 h at 140 °C in the presence of KOH (3), and after heating for 1 h at 140 °C in the presence of KOH and stirring for 24 h at 25 °C (4). | ||
A similar degradation behaviour, i.e. accelerated decomposition in the presence of Lewis or Brønsted bases used in the preparation of the polymer, has been reported for the oxa-Michael polymer of vinylsulfonylethanol.34 It can be concluded that Brønsted bases cause the decomposition of the polymers at lower temperatures and polymers prepared via Lewis base initiation have sufficient Brønsted-basic character due to the presence of zwitterionic species that show a similar effect. The results suggest that the retro oxa-Michael reaction occurs in the presence of Brønsted bases. However, the Brønsted-base catalysed degradation of alkyl sulfones via carbon–sulphur bond cleavage is also known and might be responsible for the other observed degradation steps.35 Furthermore, it has to be considered that using different catalysts might result in different polymer architectures, which in turn might cause different TGA results. Accordingly, the features of the TGAs of poly1 prepared with PPh3 or DMAP are not necessarily exclusive due to the above-mentioned Brønsted-basic character of the materials.
To gain further information on the decomposition processes, differently treated polymer samples were investigated by size exclusion chromatography (SEC) and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS). A base-free sample of poly1 characterized by a number average molar mass (Mn) of 1750 g mol−1 and a dispersity (Đ) of 1.7 was heated for 1 h at 140 °C whereupon no significant change in Mn or Đ was noted (Fig. 4c). Thereafter, 10 mol% KOH (with respect to the repeat unit) was added to poly1 and the mixture was heated for 1 h at 140 °C. By this procedure, the polymer's Mn value was significantly reduced to 930 g mol−1 (Đ = 1.4). Upon stirring the same sample for 24 h at room temperature, no further change in the molar mass or dispersity was observed (Table S4†). Another polymer, namely poly2 prepared from divinyl sulfone and (Z)-2-butene-1,4-diol,28,32 showed a similar behaviour. Upon the first thermal treatment in the presence of KOH, the Mn-value decreased from 1530 g mol−1 (Đ = 3.2) to 1100 g mol−1 (Đ = 2.6, Fig. S29 and Table S5†). Two subsequent heat treatments (1 h at 140 °C, followed by 1 h at 25 °C) resulted in no further significant changes of Mn or Đ. Accordingly, it can be assumed that the retro oxa-Michael reaction is reversible under these conditions. The decrease in mass observed after the first heat cycle might be due to a few yet unknown repeat units, which lead to irreversible chain breakage under basic conditions. To further elucidate the origin of the molar mass reduction upon heating, poly2 samples before and after thermal treatment in the presence of KOH were investigated by MALDI-TOF MS (Fig. S30 and S31†). Before MALDI-TOF MS analysis, each sample was quenched upon the addition of acetic acid. Poly2 (before any heat treatment) is terminated by vinyl sulfone and alcohol end groups as expected. Oligomer chains terminated twice by (Z)-butenediol were found to be the most abundant species. Peculiarly, the second most abundant series showed acetylation of one of the hydroxyl end groups, which results from quenching with acetic acid. The presence of the acetate ester group was also confirmed by 1H-NMR spectroscopy (Fig. S32†). New series terminated by sulfinic acid and vinyl ether groups were identified in poly2 after the heating cycle in the presence of KOH (Fig. S31†). Such end groups can be traced back to the base catalysed thermal decomposition of aliphatic sulfones.35,36 Accordingly, reduction in the molar mass (detected by SEC) in the case of poly1 and poly2 is most likely not a result of a retro oxa-Michael reaction, but rather of an irreversible thermal carbon–sulphur bond cleavage in a yet unidentified repeat group.
Similar experiments were performed with an acrylate-based polymer, namely the homopolymer of 2-hydroxyethyl acrylate (HEA, denoted poly3) prepared with DBU as the catalyst. As shown before, DBU is the preferred catalyst for the retro-Michael reaction of acrylates. Thus, another 5 mol% DBU was added to poly3 and the reaction mixture was stirred at 140 °C for 1 h. The SEC analysis shows a decrease in Mn from the initial 1810 g mol−1 (Đ = 1.8) to 570 g mol−1 (Đ = 1.3) after heating (Fig. S33†). The 1H-NMR spectrum of the degraded sample revealed the presence of free ethylene glycol, unreacted acrylate groups and Rauhut–Currier repeat units when compared to the corresponding 1H-NMR spectrum of poly3 before the heating step (Fig. S35†).27 Afterwards, at room temperature no increase in Mn was noted over a period of 2 weeks. A possible explanation for this observation could be the formation of macrocycles. A further possible degradation reaction might be the saponification of the ester groups with residual water. However, by 1H-NMR and IR spectroscopy no carboxylic acid formation could be identified. Moreover, Rauhut–Currier units formed by two acrylates are no longer accessible for oxa-Michael reactions. Therefore, the stoichiometry is shifted in favour of the alcohol eventually leading to shorter polymer strands. Finally, the observation of the Rauhut–Currier units prompts the question why such equivalent groups were not detected in divinyl sulfone-based polymers poly1 and poly2. It can be speculated that such a divinyl sulfone derived repeat group is a better Michael acceptor than the acrylate-derived Rauhut–Currier unit and might still react with alcohols. Thus, divinyl sulfone-based oxa-Michael polymers might undergo branching and those branching points might be particularly labile under basic conditions and elevated temperatures.
Self-healing/reprocessing of an oxa-Michael network
Experiments with linear polymers indicate a fast, reversible oxa-Michael reaction at high temperatures under basic conditions. In polymer networks such a behaviour is interesting as it enables self-healing or reprocessability. Therefore, the oxa-Michael polymer network poly4 was synthesized with divinyl sulfone, triethanolamine and DBU as the catalyst at 80 °C and was post-cured at 140 °C. The slightly yellow, flexible polymer was characterized by IR spectroscopy, TGA and differential scanning calorimetry (DSC). The IR spectra and thermal properties of poly4 cured at 80 °C and post-cured at 140 °C are identical (Fig. S41†). Its degradation onset temperature with a mass loss of 5% (Td 95%) is 215 ± 8 °C and the glass transition temperature (Tg) is between 5 and 8 °C (Fig. S42 and S43†). The network is insoluble in dichloromethane, dioxane and toluene and the highest swelling is observed in dichloromethane (106%) compared to 16–17% swelling in the other solvents. Accordingly, the sol–gel analysis was carried out with dichloromethane revealing a soluble fraction of poly4 of approx. 3–4 wt%. Oligomers terminated with vinyl groups and unreacted divinyl sulfone were identified in the extract (Fig. S40†).To demonstrate the self-healing properties of the oxa-Michael polymers, poly4 was cut into pieces (3–4 mm). It was then placed in a round mould in an aluminium plate and was pressed at 140 °C for 1 h with 50 bars in a hot press. After pressing, a uniform disk was obtained (Fig. 5).
 | ||
| Fig. 5 (a) Membrane of poly4 prepared from 1 equiv. divinyl sulfone, 0.66 equiv. triethanolamine and 0.10 equiv. DBU; (b) photograph of a cut membrane of poly4 placed in mould; (c) photograph of a membrane of poly4 obtained from cut pieces shown in b after hot press treatment (140 °C, 1 h, 50 bar). | ||
Based on the retro-Michael reaction and its dynamic behaviour, the material can be reprocessed and healed again at elevated temperatures. With 10 mol% DBU, better healing could be observed than that with 5 mol% DBU (Fig. S44†) as the retro oxa-Michael reaction is promoted. Similar effects for different catalyst loadings have been reported for other materials.37 The materials’ thermal and spectroscopic properties do not change upon reprocessing (Td 95% = 220 °C, Tg = 3 °C) (Fig. S42 and S43†). The procedure of cutting poly4 into pieces and reprocessing it in the hot press was in total repeated three times. In all cases, a uniform, self-healed disk was obtained. However, after the third time, new IR bands could be observed (Fig. S45†), pointing to sulfinic acids as already observed by MALDI-TOF MS for linear polymers.
The dynamic properties of poly4 were further investigated by rheometry. Stress relaxation measurements by applying a strain of 1% with a normal force of 1 N were carried out. Thermally activated network rearrangement allowed for the relaxation of the applied stress in the temperature range from 170 to 200 °C resulting in a temperature dependent viscous flow (Fig. 6a) exhibiting a relatively high flow activation energy (Ea) of 173 ± 13.4 kJ mol−1 (Fig. 6b). As discussed by Elling and Dichtel, the observed Arrhenius relationship between temperature and relaxation time is not in contradiction to a dissociative mechanism in the bond breaking process.38 The initial stress relaxation values (G0) differ only marginally between the different temperatures (Fig. S46 and S47) meaning that any decline in relaxation modulus over time is mainly influenced by bond reformation rather than by the thermal properties of the residual material.
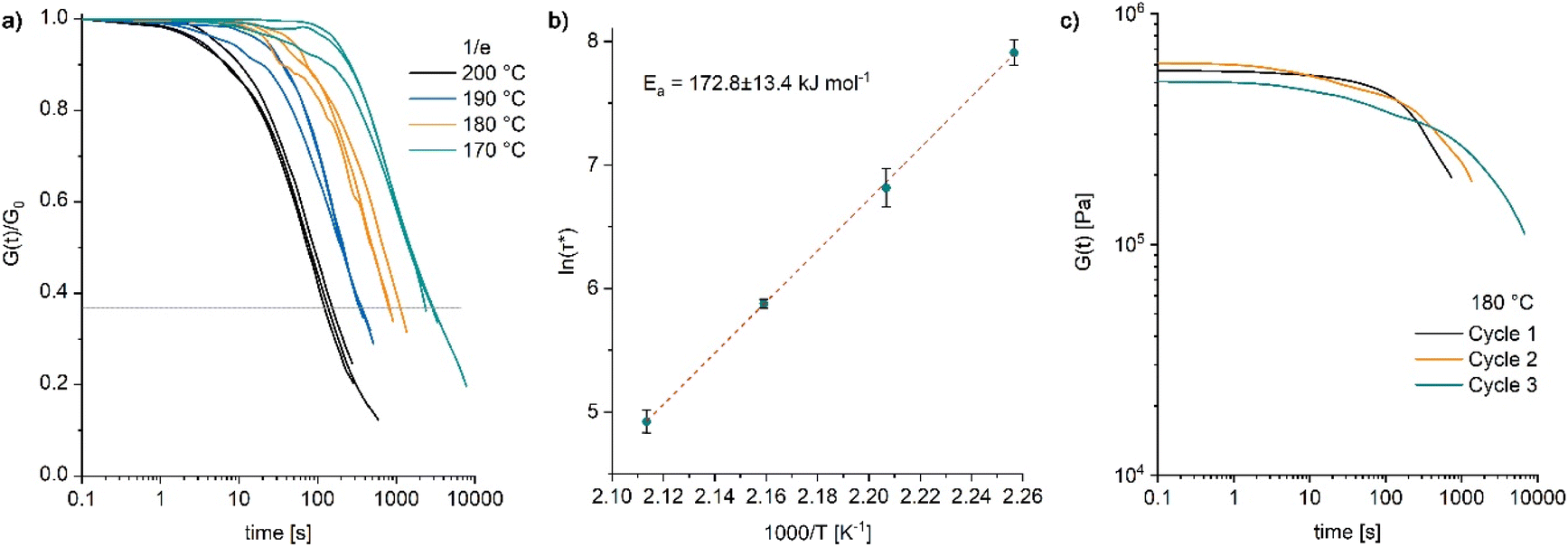 | ||
| Fig. 6 (a) Normalized stress–relaxation curves obtained from poly4 at temperatures ranging from 170 to 200 °C with 1/e (dotted line) used for the determination of the characteristic relaxation time (τ*); (b) fitting of the relaxation times (τ*) vs. temperature data by the Arrhenius equation for poly4; (c) non-normalised stress–relaxation curves obtained from poly4 measured repeatedly at a temperature of 180 °C. | ||
Repeated stress relaxation experiments were conducted with the same sample at 180 °C to study the effect of reprocessing on the viscoelastic behaviour. Between the measurements, the sample was dismantled at room temperature and repositioned in the rheometer. With each processing cycle, a subsequent increase in the relaxation time (Fig. 6c) and no significant drop in the modulus (also evident from the frequency sweep tests, Fig. S48†) were observed. Such a behaviour indicates a loss in reprocessability and is likely caused by an advancing decomposition of the catalyst DBU. Thermal decomposition of DBU under similar conditions has been reported.39
Conclusion
The reversibility of the oxa-Michael reaction can be utilized to build covalent adaptable networks which we herein demonstrated by reprocessing an oxa-Michael derived polymer network made from divinyl sulfone and triethanolamine. In general, the retro oxa-Michael reaction is Brønsted base catalysed and fast at elevated temperatures (>100 °C). This could be shown by alcohol exchange reactions in model studies of oxa-Michael adducts based on sulfones, acrylonitrile and acrylates. In the case of acrylates as Michael acceptors, transesterification is a concurring reaction. Studying the retro oxa-Michael reaction of soluble polymers revealed a molar mass reduction upon heat treatment. It was traced back to a minor irreversible side-reaction, which is most likely caused by the concurring carba-Michael reactions leading to Rauhut–Currier repeat units. Moreover, in the presence of a base, oxa-Michael polymers degrade at lower temperatures and can be depolymerized into small fragments in the presence of alcohols.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development, and the Christian Doppler Research Association (Christian Doppler Laboratory for Organocatalysis in Polymerization) is gratefully acknowledged. We thank Karin Bartl for the TGA and DSC measurements and A. Daniel Boese (University of Graz) for providing access to HPC resources and acknowledge the use of the Somapp Lab, a core facility supported by the Austrian Federal Ministry of Education, Science and Research, the Graz University of Technology, the University of Graz and Anton Paar GmbH.References
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD Publishing, Paris, 2022. DOI:10.1787/de747aef-en.
- B. D. Vogt, K. K. Stokes and S. K. Kumar, Why is Recycling of Postconsumer Plastics so Challenging?, ACS Appl. Polym. Mater., 2021, 3, 4325–4346, DOI:10.1021/acsapm.1c00648.
- C. Jehanno, J. W. Alty, M. Roosen, S. de Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Critical advances and future opportunities in upcycling commodity polymers, Nature, 2022, 603, 803–814, DOI:10.1038/s41586-021-04350-0.
- (a) Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, Thiol Specific and Tracelessly Removable Bioconjugation via Michael Addition to 5-Methylene Pyrrolones, J. Am. Chem. Soc., 2017, 139, 6146–6151, DOI:10.1021/jacs.7b00670; (b) B. Shi and M. F. Greaney, Reversible Michael addition of thiols as a new tool for dynamic combinatorial chemistry, Chem. Commun., 2005, 886–888, 10.1039/b414300k.
- B. Zhang, P. Chakma, M. P. Shulman, J. Ke, Z. A. Digby and D. Konkolewicz, Probing the mechanism of thermally driven thiol-Michael dynamic covalent chemistry, Org. Biomol. Chem., 2018, 16, 2725–2734, 10.1039/c8ob00397a.
- C. F. H. Allen, J. O. Fournier and W. J. Humphlett, The thermal reversibility of the Michael reaction: IV. thiol adducts, Can. J. Chem., 1964, 42, 2616–2620, DOI:10.1139/v64-383.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Dynamic Thiol–Michael Chemistry for Thermoresponsive Rehealable and Malleable Networks, Macromolecules, 2016, 49, 6871–6878, DOI:10.1021/acs.macromol.6b01061.
- P. Chakma, Z. A. Digby, J. Via, M. P. Shulman, J. L. Sparks and D. Konkolewicz, Tuning thermoresponsive network materials through macromolecular architecture and dynamic thiol-Michael chemistry, Polym. Chem., 2018, 9, 4744–4756, 10.1039/C8PY00947C.
- S. P. Daymon and K. M. Miller, Probing the dynamic and rehealing behavior of crosslinked polyester networks containing thermoreversible thiol-Michael bonds, Polymer, 2018, 145, 286–293, DOI:10.1016/j.polymer.2018.05.009.
- P. Chakma, S. V. Wanasinghe, C. N. Morley, S. C. Francesconi, K. Saito, J. L. Sparks and D. Konkolewicz, Heat- and Light-Responsive Materials Through Pairing Dynamic Thiol-Michael and Coumarin Chemistry, Macromol. Rapid Commun., 2021, 42, 2100070, DOI:10.1002/marc.202100070.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Self-Healing Polymer Networks Based on Reversible Michael Addition Reactions, Macromol. Chem. Phys., 2016, 217, 2541–2550, DOI:10.1002/macp.201600353.
- N. van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol-yne Cross-Linking, Angew. Chem., Int. Ed., 2020, 59, 3609–3617, DOI:10.1002/anie.201912902.
- A. Genest, D. Portinha, E. Fleury and F. Ganachaud, The aza-Michael reaction as an alternative strategy to generate advanced silicon-based (macro)molecules and materials, Prog. Polym. Sci., 2017, 72, 61–110, DOI:10.1016/j.progpolymsci.2017.02.002.
- F. C. Whitmore, H. S. Mosher, R. R. Adams, R. B. Taylor, E. C. Chapin, C. Weisel and W. Yanko, Basically Substituted Aliphatic Nitriles and their Catalytic Reduction to Amines, J. Am. Chem. Soc., 1944, 66, 725–731, DOI:10.1021/ja01233a019.
- (a) C. Taplan, M. Guerre and F. E. Du Prez, Covalent Adaptable Networks Using β-Amino Esters as Thermally Reversible Building Blocks, J. Am. Chem. Soc., 2021, 9140–9150, DOI:10.1021/jacs.1c03316; (b) D. Berne, G. Coste, R. Morales-Cerrada, M. Boursier, J. Pinaud, V. Ladmiral and S. Caillol, Taking advantage of β-hydroxy amine enhanced reactivity and functionality for the synthesis of dual covalent adaptable networks, Polym. Chem., 2022, 13, 3806–3814, 10.1039/D2PY00274D.
- R. Baruah, A. Kumar, R. R. Ujjwal, S. Kedia, A. Ranjan and U. Ojha, Recyclable Thermosets Based on Dynamic Amidation and Aza-Michael Addition Chemistry, Macromolecules, 2016, 49, 7814–7824, DOI:10.1021/acs.macromol.6b01807.
- X. Guo, F. Gao, F. Chen, J. Zhong, L. Shen, C. Lin and Y. Lin, Dynamic Enamine-one Bond Based Vitrimer via Amino-yne Click Reaction, ACS Macro Lett., 2021, 10, 1186–1190, DOI:10.1021/acsmacrolett.1c00550.
- S. Xu, Z. Liao, A. Dianat, S.-W. Park, M. A. Addicoat, Y. Fu, D. L. Pastoetter, F. G. Fabozzi, Y. Liu, G. Cuniberti, M. Richter, S. Hecht and X. Feng, Combination of Knoevenagel Polycondensation and Water-Assisted Dynamic Michael-Addition-Elimination for the Synthesis of Vinylene-Linked 2D Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2022, 61, e202202492, DOI:10.1002/anie.202202492.
- C. F. Nising and S. Bräse, The oxa-Michael reaction: from recent developments to applications in natural product synthesis, Chem. Soc. Rev., 2008, 37, 1218–1228, 10.1039/b718357g.
- (a) X. Xiong, C. Ovens, A. W. Pilling, J. W. Ward and D. J. Dixon, Highly stereoselective oxy-michael additions to beta,gamma-unsaturated alpha-keto esters: rapid enantioselective synthesis of 3-hydroxybutenolides, Org. Lett., 2008, 10, 565–567, DOI:10.1021/ol702693m; (b) D. H. Wang, R. N. McKenzie, P. R. Buskohl, R. A. Vaia and L.-S. Tan, Hygromorphic Polymers: Synthesis, Retro-Michael Reaction, and Humidity-Driven Actuation of Ester–Sulfonyl Polyimides and Thermally Derived Copolyimides, Macromolecules, 2016, 49, 3286–3299, DOI:10.1021/acs.macromol.6b00250.
- I. C. Stewart, R. G. Bergman and F. D. Toste, Phosphine-catalyzed hydration and hydroalkoxylation of activated olefins: use of a strong nucleophile to generate a strong base, J. Am. Chem. Soc., 2003, 125, 8696–8697, DOI:10.1021/ja035232n.
- J. E. Baldwin, R. C. Thomas, L. I. Kruse and L. Silberman, Rules for ring closure: ring formation by conjugate addition of oxygen nucleophiles, J. Org. Chem., 1977, 24, 3846–3852, DOI:10.1021/jo00444a011.
- K. Ratzenböck, M. M. U. Din, S. M. Fischer, E. Žagar, D. Pahovnik, A. D. Boese, D. Rettenwander and C. Slugovc, Water as a monomer: synthesis of an aliphatic polyethersulfone from divinyl sulfone and water, Chem. Sci., 2022, 13, 6920–6928, 10.1039/D2SC02124B.
- (a) J. Kaur, R. Krishnan, B. Ramalingam and S. Jana, Hydroxyethyl sulfone based reactive coalescing agents for low-VOC waterborne coatings, RSC Adv., 2020, 10, 17171–17179, 10.1039/D0RA00753F; (b) D. J. Berrisford, P. A. Lovell, N. R. Suliman and A. Whiting, Latent reactive groups unveiled through equilibrium dynamics and exemplified in crosslinking during film formation from aqueous polymer colloids, Chem. Commun., 2005, 5904–5906, 10.1039/b512073j.
- A. Polykarpov, A. Palsule and A. Deshpande, Compositions of dicarbonyl substitued-1-alkene, methods to make them, polymers made from them and methodes to make the polymer, US 2022/0112151, 2022 Search PubMed.
- K. Ratzenböck, D. Pahovnik and C. Slugovc, Step-growth polymerisation of alkyl acrylates via concomitant oxa-Michael and transesterification reactions, Polym. Chem., 2020, 11, 7476–7480, 10.1039/d0py01271h.
- S. M. Fischer, S. Renner, A. D. Boese and C. Slugovc, Electron-rich triarylphosphines as nucleophilic catalysts for oxa-Michael reactions, Beilstein J. Org. Chem., 2021, 17, 1689–1697, DOI:10.3762/bjoc.17.117.
- (a) S. Strasser and C. Slugovc, Nucleophile-mediated oxa-Michael addition reactions of divinyl sulfone – a thiol-free option for step-growth polymerisations, Catal. Sci. Technol., 2015, 5, 5091–5094, 10.1039/C5CY01527H; (b) S. Thiyagarajan, V. Krishnakumar and C. Gunanathan, KOtBu-Catalyzed Michael Addition Reactions Under Mild and Solvent-Free Conditions, Chem. – Asian J., 2020, 15, 518–523, DOI:10.1002/asia.201901647; (c) S. M. Fischer, P. Kaschnitz and C. Slugovc, Tris(2,4,6-trimethoxyphenyl)phosphine - a Lewis base able to compete with phosphazene bases in catalysing oxa-Michael reactions, Catal. Sci. Technol., 2022, 12, 6204–6212, 10.1039/D2CY01335E.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, On the Basicity of Organic Bases in Different Media, Eur. J. Org. Chem., 2019, 6735–6748, DOI:10.1002/ejoc.201900956.
- (a) A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases, Org. Process Res. Dev., 2019, 23, 1860–1871, DOI:10.1021/acs.oprd.9b00187; (b) C. A. Lewis Jr. and R. Wolfenden, The Nonenzymatic Decomposition of Guanidines and Amidines, J. Am. Chem. Soc., 2014, 136, 130–136, DOI:10.1021/ja411927k.
- (a) H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Phosphazene-catalyzed oxa-Michael addition click polymerization, Polym. Chem., 2018, 9, 4716–4723, 10.1039/C8PY01089G; (b) Q. Jiang, Y. Zhang, Y. Du, M. Tang, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, Preparation of hyperbranched polymers by oxa-Michael addition polymerization, Polym. Chem., 2020, 11, 1298–1306, 10.1039/C9PY01686D; (c) S. Matsuoka, S. Namera and M. Suzuki, Oxa-Michael addition polymerization of acrylates catalyzed by N-heterocyclic carbenes, Polym. Chem., 2015, 6, 294–301, 10.1039/C4PY01184H; (d) H. Yao, Y. Song, W. Huang, L. Jiang, Q. Jiang, X. Xue, B. Jiang and H. Yang, Preparing Degradable Polymers with Promising Mechanical Properties by Hydrogen Transfer Polymerization, Macromolecules, 2022, 55, 8283–8291, DOI:10.1021/acs.macromol.2c01305.
- S. Strasser, C. Wappl and C. Slugovc, Solvent-free macrocyclisation by nucleophile-mediated oxa-Michael addition polymerisation of divinyl sulfone and alcohols, Polym. Chem., 2017, 8, 1797–1804, 10.1039/C7PY00152E.
- Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Organohalide-catalyzed dehydrative O-alkylation between alcohols: a facile etherification method for aliphatic ether synthesis, Green Chem., 2015, 17, 2774–2779, 10.1039/C5GC00284B.
- N. Ziegenbalg, R. Lohwasser, G. D'Andola, T. Adermann and J. C. Brendel, Oxa-Michael polyaddition of vinylsulfonylethanol for aliphatic polyethersulfones, Polym. Chem., 2021, 12, 4337–4346, 10.1039/D1PY00256B.
- (a) T. Sasaki and H. Yaguchi, Photoinduced unzipping depolymerization of poly(olefin sulfone)s possessing photobase generator and base amplifier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 602–613, DOI:10.1002/pola.23179; (b) T. Sasaki, T. Kondo, M. Noro, K. Saida, H. Yaguchi and Y. Naka, Photoinduced depolymerization in poly(olefin sulfone) films comprised of volatile monomers doped with a photobase generator, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1462–1468, DOI:10.1002/pola.25898; (c) X. Guo, X. Liu, H. Su, S. Shan and Q. Jia, Chemical fixation of SO2 using norbornene with different side-chain groups and control of the physical properties of poly(olefin sulfone), Polym. Int., 2017, 66, 1530–1537, DOI:10.1002/pi.5410; (d) O. P. Lee, H. Lopez Hernandez and J. S. Moore, Tunable Thermal Degradation of Poly(vinyl butyl carbonate sulfone)s via Side-Chain Branching, ACS Macro Lett., 2015, 4, 665–668, DOI:10.1021/acsmacrolett.5b00234.
- (a) E. Kiran, J. K. Gillham and E. Gipstein, Pyrolysis–molecular weight chromatography–vapor-phase infrared spectrophotometry: An on-line system for analysis of polymers. III. Thermal decomposition of polysulfones and polystyrene, J. Appl. Polym. Sci., 1977, 21, 1159–1176, DOI:10.1002/app.1977.070210501; (b) E. Wellisch, E. Gipstein and O. J. Sweeting, Thermal decomposition of polysulfones, J. Appl. Polym. Sci., 1964, 8, 1623–1631, DOI:10.1002/app.1964.070080412; (c) Y.-L. Zhao, W. H. Jones, F. Monnat, F. Wudl and K. N. Houk, Mechanisms of Thermal Decompositions of Polysulfones: A DFT and CBS-QB3 Study, Macromolecules, 2005, 38, 10279–10285, DOI:10.1021/ma051503y.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets, J. Am. Chem. Soc., 2012, 134, 7664–7667, DOI:10.1021/ja302894k.
- B. R. Elling and W. R. Dichtel, Reprocessable Cross-Linked Polymer Networks: Are Associative Exchange Mechanisms Desirable?, ACS Cent. Sci., 2020, 6, 1488–1496, DOI:10.1021/acscentsci.0c00567.
- D. Edinger, H. Weber, E. Žagar, D. Pahovnik and C. Slugovc, Melt Polymerization of Acrylamide Initiated by Nucleophiles: A Route toward Highly Branched and Amorphous Polyamide 3, ACS Appl. Polym. Mater., 2021, 3, 2018–2026, DOI:10.1021/acsapm.1c00084.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01345b |
| This journal is © The Royal Society of Chemistry 2023 |