
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disulfide-containing monomers in chain-growth polymerization
Marlena
Pięta
 a,
Vishal B.
Purohit
a,
Joanna
Pietrasik
b and
Christopher M.
Plummer
*a
a,
Vishal B.
Purohit
a,
Joanna
Pietrasik
b and
Christopher M.
Plummer
*a
aInternational Centre for Research on Innovative Biobased Materials (ICRI-BioM)—International Research Agenda, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland. E-mail: christopher.plummer@p.lodz.pl
bInstitute of Polymer and Dye Technology, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 1st December 2022
Abstract
Due to the significance of disulfide bonds within modern material and medicinal sciences, much attention has been paid to the synthesis of disulfide-containing polymers. Within this review article, we attempt to provide a comprehensive overview of the diversity of disulfide-containing polymers that can be obtained by the chain-growth polymerization of disulfide-containing monomers. This article covers the synthesis of polymers by free radical polymerization (FRP), i.e., vinyl monomers having side-chains incorporating disulfide bonds, and also the polymerization of disulfide containing heterocyclic monomers by ring-opening polymerization (ROP/rROP/ROMP). In addition, polymerization where disulfide-containing heterocycles undergo a ring-opening process that directly involves the disulfide bond are discussed. The article summarizes the state-of-the-art in polymer synthesis, and also outlines various post-polymerization modifications and biological application studies that demonstrate the importance of disulfide containing macromolecules in polymer science.
 Marlena Pięta | Marlena Pięta completed her Ph.D. in 2018 at Lodz University of Technology (TUL), Poland in the area of organic synthesis, specifically the synthesis of chiral azaheterocycles with cytotoxic activity. In 2019 she worked as researcher in the process chemistry department in Ryvu Therapeutics, Poland. Since 2020 she has been employed as a post-doctorate at TUL. In 2020–2021 she worked on the synthesis of biologically active compounds and profluorescent probes for ROS in a medicinal chemistry group, and from 2022 onwards she is working on the synthesis of new monomers for radical ring-opening polymerization (rROP) in a polymer chemistry group. |
 Vishal B. Purohit | Vishal Purohit completed his Ph.D. in 2017 under the supervision of Dr K. H. Patel and Prof. D. K. Raval at S. P. University (India), where he studied NHC-transition metal catalyzed C–H activation reactions. Between 2017–2020 he was employed as an Assistant Professor at Shri A. N. Patel PG Institute of Science and Research (India). During 2020–2021, he worked as a Postdoc with Prof. Karol Grela at the University of Warsaw (Poland). In November 2021, he joined the group of Dr Christopher Plummer as a Postdoc at Lodz University of Technology (Poland), working on the synthesis of new monomers for ring-opening polymerization. |
 Joanna Pietrasik | Joanna Pietrasik received her Ph.D. in 2003 at Lodz University of Technology (TUL), Poland; next she spent 2 years at Carnegie Mellon University (CMU), USA, in the group of Prof. Matyjaszewski as a postdoc. She obtained her habilitation degree in 2014. She now holds the position of professor at TUL. Her research activity is dedicated to polymer synthesis, inorganic particles, surface modifications, as well as hybrid materials and nanocomposite properties. |
 Christopher M. Plummer | Christopher Plummer completed his Ph.D. in 2016 at RMIT University, Australia in the area of organic synthesis, specifically the synthesis of oxygenated heterocycles. Between 2016–2019 he was employed as a post-doctorate at Sun Yat-sen University (SYSU), China working on the post-modification of polyolefins. In 2019–2021 he worked as a researcher in an industry-funded position at Aix-Marseille University, France, working on the synthesis of new monomers for radical ring-opening polymerization (rROP). Following this, he took up a position as Junior Principal Investigator at Lodz University of Technology (TUL), Poland, and is a presently a group leader in polymer chemistry. |
1. Introduction
Disulfides are generally regarded as dynamic covalent bonds and have a typical dissociation energy of ca. 60 kcal mol−1 (251 kJ mol−1), far exceeding the non-covalent interactions that are present in supramolecular polymers (4–20 kJ mol−1).1 Nevertheless, disulfide bonds can be readily cleaved and replaced with other bonds, as well as reformed on demand. The range of potential cleaving and bond exchanging triggers is extensive, with multiple physical (e.g. light, heat, mechanical force, magnetic field) and chemical triggers (e.g. nucleophiles, reducing agents, radicals). In nature, disulfide bonds occur in abundance as part of the intramolecular cross-linking of peptides and protein backbones. Indeed, the stability of such naturally occurring macromolecules relies upon the participation of disulfide bonds within their secondary and tertiary structures (e.g. α-helix), these structures being essential for physiological activity (Fig. 1A).2 Cysteine-derived disulfide cross-linking is present in proteins of various sizes and functions, from small (10–50 amino acid) peptides such as growth factors and cytokines, to cysteine-rich biomaterials such as keratin-associated proteins and other supramolecular assemblies. Although disulfide linkages are plentiful in cysteine-rich materials, they are also important for the stabilization of surface loops and secondary structure domains that are vital for the physiological activity of proteins (Fig. 1B). Moreover, there also exists many naturally occurring non-peptide disulfides, for example the enzyme cofactor essential for aerobic metabolism α-lipoic acid M1, and asparagusic acid M2 which can be isolated from Asparagus officinalis,3 among others4,5 (Fig. 1C).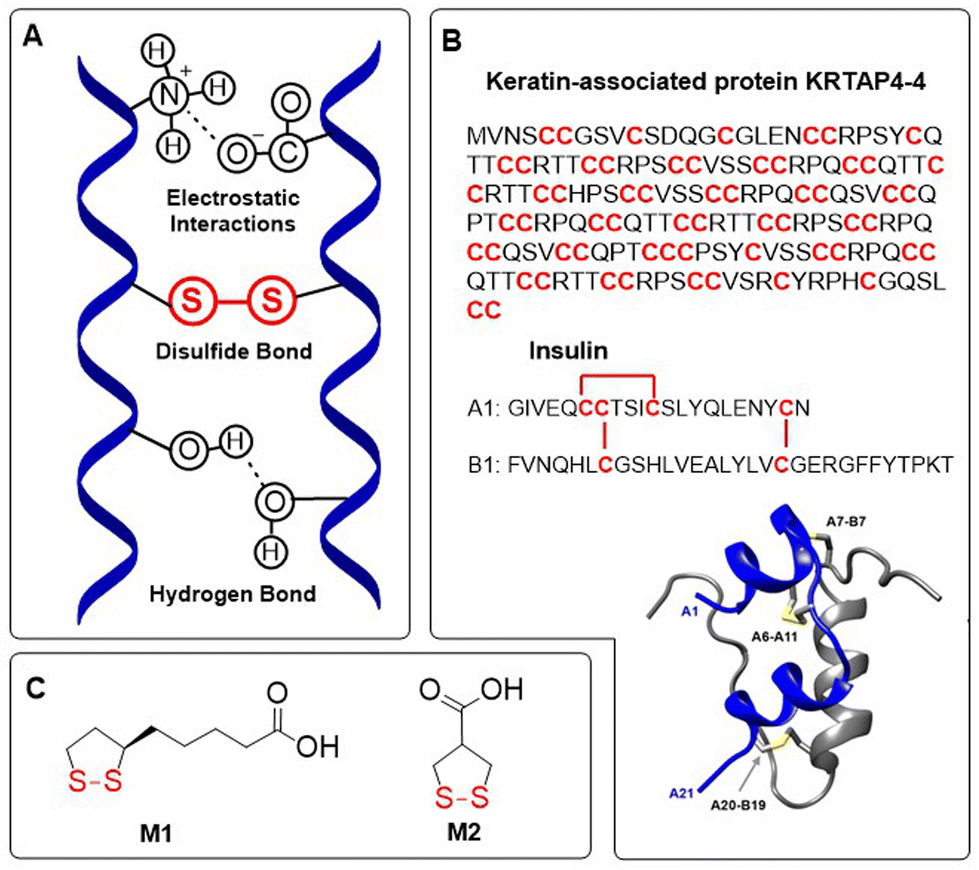 | ||
| Fig. 1 (A) Disulfide cross-linking in proteins influences the formation of secondary and tertiary structures; (B) keratin-associated protein KRTAP4-4 represents a disulfide-rich protein with cysteine constituting 37% of the amino acid sequences, and peptide hormone insulin which consists of two chains bound together by disulfide bonds. Ribbon diagram of insulin showing the location of disulfide bonds, adapted under a Creative Commons licence from van Lierop et al.;11 (C) naturally occurring 1,2-dithiolanes. | ||
The incorporation of disulfide bonds into a polymer backbone can instil polymers with properties that include adhesion, flexural strength, adaptability, degradability, and even self-repair.6 Pendent disulfide functionalities can be exploited to prepare polymers with stimuli-sensitive and dynamic architectures. The reversible polymerization of cyclic disulfides, in particular 1,2-dithiolanes, has been widely exploited in bioconjugation and the design of self-healing materials.7–9 Moreover, polymers displaying disulfide-containing pendant groups demonstrate significant stability in the blood stream and the extracellular environment, while simultaneously displaying reactivity to intracellular antioxidants such as glutathione (GSH). This phenomenon has been widely exploited for the transportation of therapeutic agents attached via disulfide bond to the intracellular environment, readily enabling the circumvention of innate obstacles (e.g. lipophilicity).10
Due to the significance of disulfide bonds in modern polymer chemistry, we present this review article focusing on disulfide-containing polymers synthesized by chain-growth polymerization. The article encompasses disulfides located within both the polymer backbone and the pendant side-chains, specifically those that originate from disulfide-containing monomers. The article is divided into multiple sections. Within the first section the synthesis of polymers by free-radical polymerization (FRP) is reviewed, i.e., vinyl monomers that have side-chains incorporating disulfide bonds. Then, the synthesis of polymers using disulfide-containing cyclic monomers by ring-opening polymerization (rROP/ROP/ROMP) is reviewed. Within these sections, interesting post-modifications, assembly formations and biological studies will be discussed which additionally support and substantiate research at the synthetic level. Following this, polymerization where disulfide-containing monomers undergo a ring-opening process involving the disulfide bond are covered. Finally, a section describing common methods for monomer synthesis is included. The scope of the article is limited to the polymerization of disulfide-containing monomers, and therefore trisulfide-containing monomers and the vulcanization of rubber are outside of the scope and are not covered.
2. Free-radical polymerization
During the process of free-radical polymerization (FRP) a polymer is formed by the successive addition of monomers to an actively propagating radical-containing chain end. In a typical polymerization, an initiator commences the polymerization by its homolytic degradation to form a radical species which then adds to the radical-acceptor moiety (alkene, alkyne, thiocarbonyl, etc.) of a monomer. This mechanistic process generates a new radical, and the propagation process repeats until radical termination. As the propagating chains are all initiated at different times, this leads to non-homogenous polymer samples. As specialist polymer applications require more precisely controlled polymer architectures, various techniques known under the umbrella term “reversible-deactivation radical polymerization” (RDRP) can be applied. In an ideal RDRP process, the molecular weight of the growing polymer chains increases equally with time. The most prominent RDRP techniques are atom transfer radical polymerization (ATRP), reversible addition/fragmentation chain transfer polymerization (RAFT), and nitroxide-mediated polymerization (NMP).2.1 Monomers containing pendant disulfide functionality
The incorporation of disulfide bonds into pendant side-chains of a polymer allows for the fine adjustment of polymer properties by enabling post-polymerization modification. Typical post-modifications include thiol-disulfide exchange with thiolated molecules, reduction followed by utilization of the free thiol group as a nucleophile, and polymer gelation by disulfide bond metathesis. Polymers containing pendant disulfide bonds have therefore attracted attention for the design of targeted drug delivery systems. Selective activation and payload release is enabled by the disulfide group, being tuned to characteristics such as acidity or antioxidant concentration. Thus, in recent years there have been a multitude of reports detailing polymer micelles with covalently linked or encapsulated anti-tumour agents. In this section, polymers synthesized from (2.1.1) pyridyl disulfide monomers, (2.1.2) monomers with disulfide self-immolative linkers, and (2.1.3) bifunctional cross-linking agents are presented.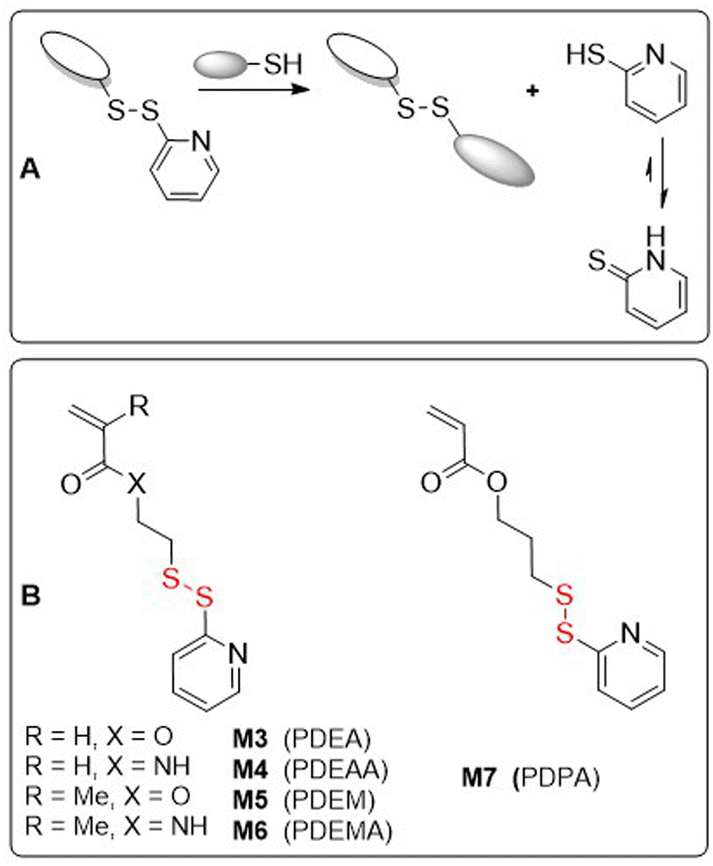 | ||
| Fig. 2 (A) Application of pyridyl disulfide group in a thiol–disulfide exchange giving a new disulfide and unreactive 2-pyridinethione; (B) monomers containing pyridyl disulfide groups. | ||
Monomers containing pyridyl disulfide moieties have been demonstrated to be useful in a range of modern polymer synthesis techniques, including FRP, ATRP and RAFT. In 1998, Ruffner and coworkers, desiring to obtain a polymer with functional pendant groups that were stable under aqueous conditions, copolymerized methacrylamide M6 with N-(2-hydroxypropyl)methacrylamide (HPMA) to fabricate polymers with up to 8.3 mol% M6.13 The authors confirmed that the pyridyl disulfide group was stable in aqueous media at pH ≤8, as well as capable of undergoing rapid thiol–disulfide exchange with l-cysteine or 2-mercaptoethanol in aqueous solution. Poly(HPMA-co-M6) was also conjugated with thiol-modified oligonucleotides and effectively absorbed into HeLa cells via endocytosis.
Hoffman and coworkers later copolymerized M7 with butyl acrylate (BA) and methacrylic acid (MAA) to produce amphiphilic random terpolymers with up to 7 mol% M7 and Mn values of 10.6–124 kg mol−1 (Đ = 1.3–2.3).14 These polymers were readily conjugated with oligopeptides by disulfide bond, and complexed with therapeutic nucleic acids. It was confirmed that poly(M7-co-BA-co-MAA) readily diffused into the cell cytoplasm where it exhibited low cell toxicity. Moreover, it was also demonstrated that disulfide-conjugated drugs can be released in the presence of GSH. Alternative polymers where MAA was replaced with pH sensitive monomers such as ethylacrylic acid (EAA) or propylacrylic acid (PAA) were also reported.15
Thayumanavan and coworkers applied RAFT and ATRP techniques for the fabrication of copolymers of M5 and N-hydroxysuccinimide methacrylate (NHSMA).16 Homopolymerization of M5 under RAFT conditions resulted in an insoluble product. Fortunately, an ATRP-based approach successfully provided polyM5 with an Mn = 6.7 kg mol−1 (Đ = 1.2). Under analogous conditions, poly(NHSMA-co-M5) were obtained with Mn values of 9.7–19.0 kg mol−1 (Đ = 1.3–1.6). Finally, thiol–disulfide exchange reactions using 1-undecanethiol and thiol-modified fluorescent anthracene were performed to study their release profiles using the reducing agent 1,4-dithiothreitol (DTT).
Bulmus and coworkers synthesized polyM5 using RAFT and then subjected it to thiol–disulfide exchange with 3-mercaptopropionic acid, 4-mercaptobutanol, 11-mercaptoundecanol, and reduced L-glutathione (GLT). The obtained modified polymers subsequently self-assembled into spherical nanoparticles in aqueous solution.17PolyM5 was also applied as a macro-RAFT agent for the polymerization of HPMA to provide amphiphilic block copolymers (Fig. 3).18 Treatment of these block polymers with tris(2-carboxyethyl)phosphine (TCEP) led to successful conjugation with maleimide-modified anticancer drug doxorubicin (DOX), with simultaneous cross-linking and self-assembly. The formed nanomicelles were reported to exhibit in vivo stability and released the conjugated DOX at acidic pH.
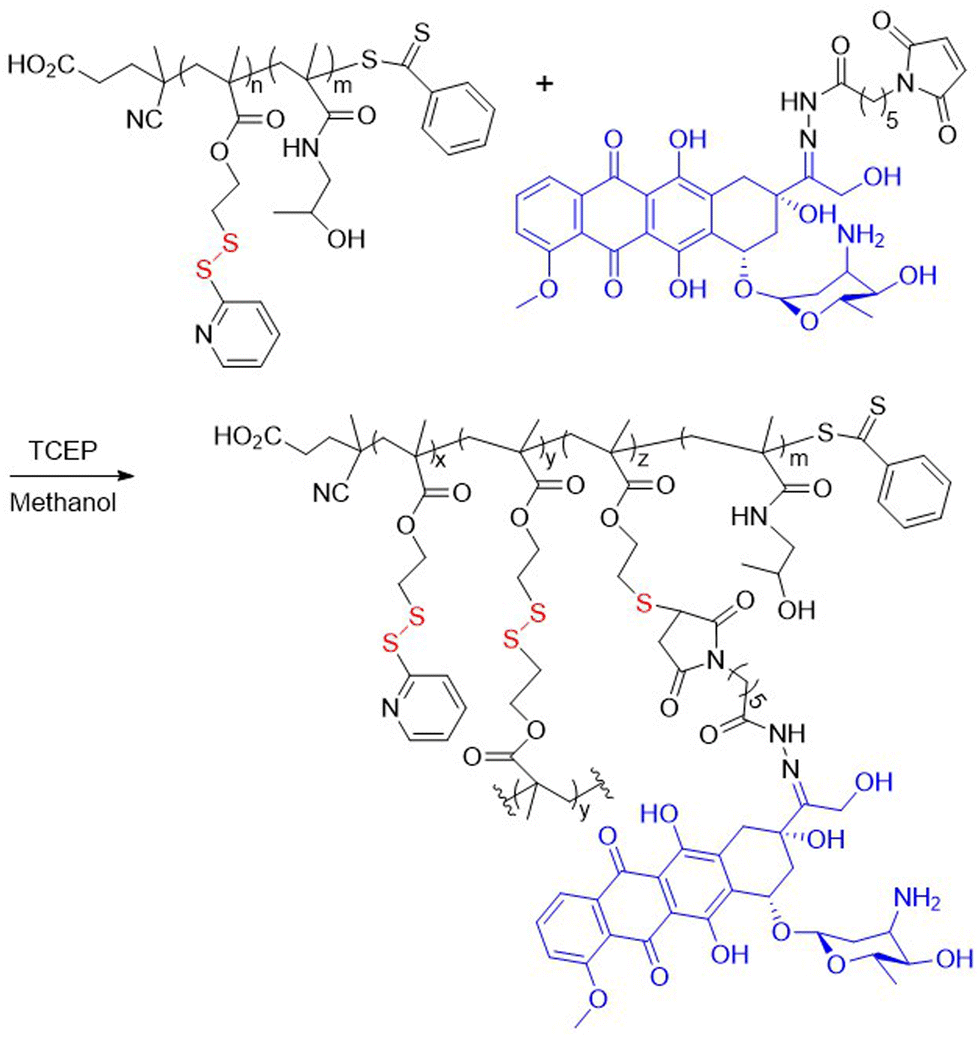 | ||
| Fig. 3 Conjugation of poly(M5-b-HPMA) with maleimide-modified DOX with simultaneous cross-linking. | ||
Later, Thayumanavan and coworkers presented multiple research articles concerning polymers with incorporated pyridyl disulfide moieties.19–32 For example, RAFT copolymerization of M5 and poly(ethylene oxide) monomethacrylate (PEGMA) provided poly(M5-co-PEGMA) which was used to generate well-defined spherical nanogels by its treatment with DTT, with the size dependent on both the monomer ratio and the presence of various salts.19,20,23 Subsequently, the formation of micelles with encapsulated hydrophobic fluorescent dyes and their release profile were studied.19–21 In addition, it was reported that through thiol–disulfide exchange poly(M5-co-PEGMA) could be equipped with protein ligands that could facilitate receptor-dependant cell internalization.22,24,26 Conjugates of poly(M5-co-PEGMA) and caspase-3 proteins were reported able to enter HeLa cell lines, unlike the unconjugated protein.30 Post-modification in which pyridyl disulfide groups were exchanged with various small-molecules provided multistimuli-responsive polymers that were reactive to chemical (redox, pH), biological (protein) and/or physical (light) stimuli.25
Additionally, thiol–disulfide exchange of poly(M5-co-PEGMA) with 2-mercaptoethanol followed by condensation of the resulting alcohol with 4-nitrophenyl chloroformate provided modified polymer containing p-(nitrophenylcarbonate)ethyl disulfide pendants (Fig. 4A). These alternative disulfide-containing pendant groups were reported to be less sterically hindered than pyridyl disulfide groups and therefore allowed for accelerated GSH-induced payload release.31 These polymers were then conjugated with lysine-containing proteins and diamine cross-linkers using amine-carbonate condensation. Poly(M5-co-PEGMA) was also subjected to methylation of the pyridyl nitrogen, with the resulting cationic groups readily undergoing complexation with nucleic acids via electrostatic interactions (Fig. 4B).32 Following subsequent cross-linking, these fabricated nanoassemblies were reported to exhibit lower cytotoxicity compared to classical delivery vehicles, which was attributed to their non-cationic character.
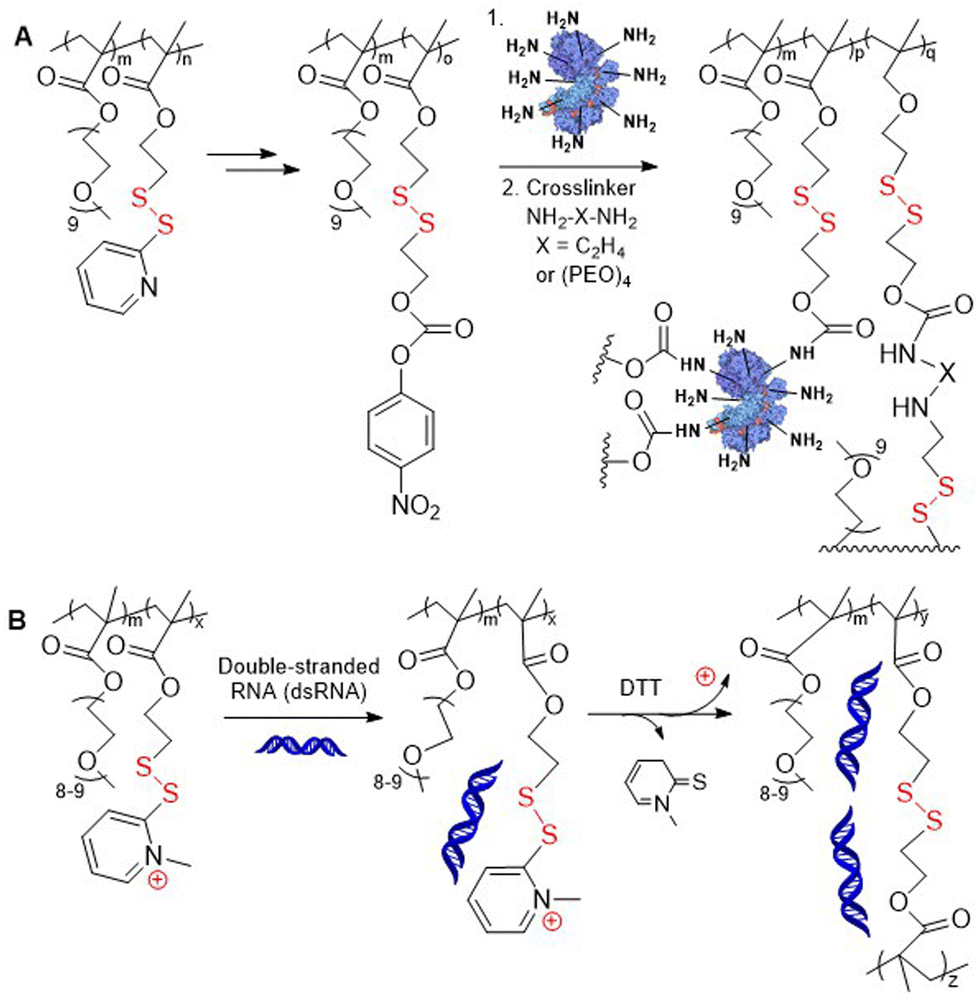 | ||
| Fig. 4 (A) Post-modification of poly(PEGMA-co-M5) for protein conjugation; (B) complexation of poly(PEGMA-co-MeM5) with dsRNA through electrostatic interaction. | ||
Terpolymers obtained by the RAFT copolymerization of M5, PEGMA and 2-(diisopropylamino) ethyl methacrylate (DPAM) were converted into nanogels in an aqueous solution of DTT.27,28 The introduction of diisopropylamine moieties was intended to enable the nanogel to be positively charged at an acidic pH to assist in cellular uptake. In addition, the RAFT copolymerization of M5, PEGMA and glycidyl methacrylate (GMA) provided a polymer which was used to form and study composite supramolecular nanoassemblies.29Poly(PAA-b-DMA-co-M6) (DMA = N,N-dimethylacrylamide) obtained by RAFT polymerization was conjugated to ovalbumin which had been modified by the introduction of a thiol moiety.33 The conjugates were tested as protein-based vaccines and were reportedly able to stimulate the immune system of mice, and to subsequently enhance the rejection of cancer cells.
Xu and coworkers explored M3-based polymers obtained by FRP.34–41 Thus, polyM3, poly(M3-co-PEGMA) and poly(M3-co-PEGMA-b-NiPMA) (NiPMA = N-isopropylmethacrylamide) were fabricated, with the pyridyl disulfide groups then subjected to post-modification, being replaced with various active molecules including camptothecin (CPT),36 Herceptin,36 acetylcysteine,40 diethyldithiocarbamate,41 disulfiram,42 and lactobionic acid.38,41 Some of the modified polymers were also subjected to aqueous self-assembly to give micelles encapsulating anticancer agents such as paclitaxel,34 doxorubicin34,35 or a silicon photosensitiser for photodynamic therapy.37Poly(M3-co-PEGMA) was also obtained by RAFT methodology (Mn = 7.8 kg mol−1, Đ = 1.27) and subjected to nanogel formation in the presence of DTT to encapsulate DOX.42
In 2012, Jackson and Fulton copolymerized M4 with N-ethylacrylamide-2-(4-formylbenzamide) (EFB) or N-(tert-butoxycarbonyl)-propylaminoacrylamide (PAAA) to provide poly(M4-co-EFB-co-DMA) and poly(M4-co-PAAA-co-DMA)via RAFT methodology.43 After removal of the Boc protection, both polymers were cross-linked through imine bond formation. Additionally, during this process a hydrophobic dye was encapsulated. The nanoassembly was further strengthen by disulfide linkage formation resulting from its treatment with DTT. It was reported that the release of the dye readily occurred in the presence of a reducing agent at lowered pH. Similarly, poly(M4-co-EFB-co-DMA) was obtained by Segura-Sánchez et al. and was linked with thiolated chitosan by both imine and disulfide linkage, with acidic and reductive conditions allowing chitosan cleavage.44 Ji and coworkers copolymerized 2-methylene-1,3-dioxepane (MDO) with PEGMA and M5 to provide a terpolymer with an Mn = 24.0 kg mol−1 (Đ = 1.58) (Fig. 5).45,46 The poly(MDO-co-PEGMA-co-M5) was subsequently grafted with DOX and then self-assembled to provide spherical micelles which exhibited strong toxicity against lung carcinoma cells, while blank micelles reported to be non-toxic.
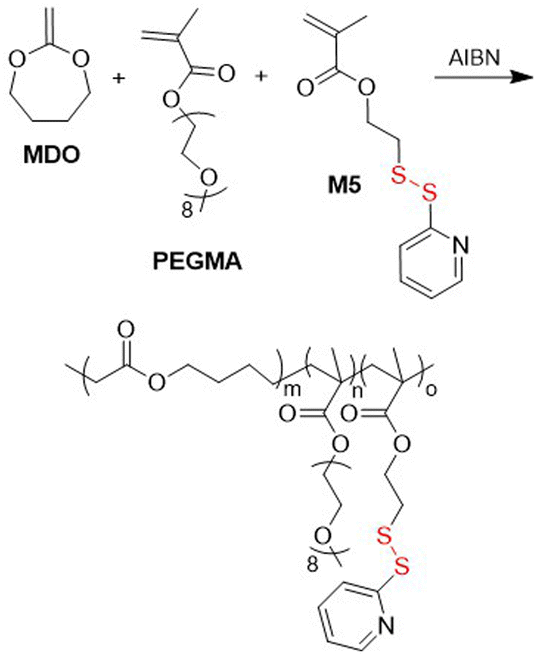 | ||
| Fig. 5 Synthesis of poly(MDO-co-PEGMA-co-M5). | ||
In addition, DOX was encapsulated into a pseudo-diblock polymer of M5 and hyaluronic acid (HA), obtained via ATRP methodology with a subsequent azide click-reaction, which was then self-assembled into micelles and subsequently cross-linked.47 The system exhibited excellent bloodstream stability and high tumor targeting, as well as improved therapeutic efficacy. In addition, RAFT polymerization was utilized to synthesize poly(M6-co-HPMA) with an Mn = 13.0 kg mol−1 (Đ = 1.13) (Fig. 6).48 This macro-chain transfer agent (macro-CTA) was subsequently copolymerized with HPMA and a methacrylamide-functionalized oligolysine monomer, followed by conjugation with an endosomolytic peptide, melittin. The resulting block copolymer was then electrostatically complexed with plasmid DNA to provide a gene delivery system that exhibited enhanced delivery to both HeLa and neuron-like cell lines.
 | ||
| Fig. 6 Gene delivery system of polymer with oligolysine (OL), melittin and plasmid DNA. | ||
Water soluble poly(NiPAA-co-AA-co-M4) with an Mn = 100 kg mol−1 (Đ = 5.1) was conjugated with thiolated single strand DNA, and by subsequent hybridization with the complementary DNA strand, a double strand DNA conjugate was formed which was then applied for aggregation studies.49Poly(M5-b-PEG) (PEG = polyethylene glycol) obtained by RAFT polymerization with an Mn = 11.6 kg mol−1 was stirred with gold nanoparticles in DMF to provide polymer-coated AuNPs with a virtually neutral surface which exhibited good stability under various physiological conditions and reduced non-specific adsorption of biomolecules.50 Oxopentanoate ethyl methacrylate (OPM) and a pyrene-terminated RAFT agent were utilized in synthesis of tri-block terpolymer poly(OPM-b-M3-b-PEG) (Fig. 7).51 This macromolecule was conjugated with O-benzylhydroxylamine (BHA) via imine bond formation with the ketone group of OPM. Then, the modified polymer was attached to graphene through π–π stacking interactions resulting in a polymer/graphene composite, with BHA and 2-pyridinethione (PT) release being investigated.
 | ||
| Fig. 7 Synthesis of polymer/graphene composite from poly(OPM-b-M3-b-PEG). | ||
Applying RAFT polymerization, M6 was incorporated into poly(MCT-co-M6-b-PEGMA-co-DEM), where MCT equals “methacrylated macrocyclic coumarin caged thiol” (Fig. 8A/B).52 An aqueous solution of this polymer was then cross-linked by light irradiation to produce nanoparticles that were able to encapsulate hydrophobic compounds. These nanoparticles were reported to exhibit good stability, performing the controlled release of guest molecules under redox conditions. The mechanism of the photo-triggered cross-linking initiates with the release of free thiol from the photo-responsive MCT moiety which subsequently reacts with pyridyl disulfide functional groups to create disulfide bridges (Fig. 8C).53 By the application of various irradiation conditions, different cross-linking densities were accomplished.
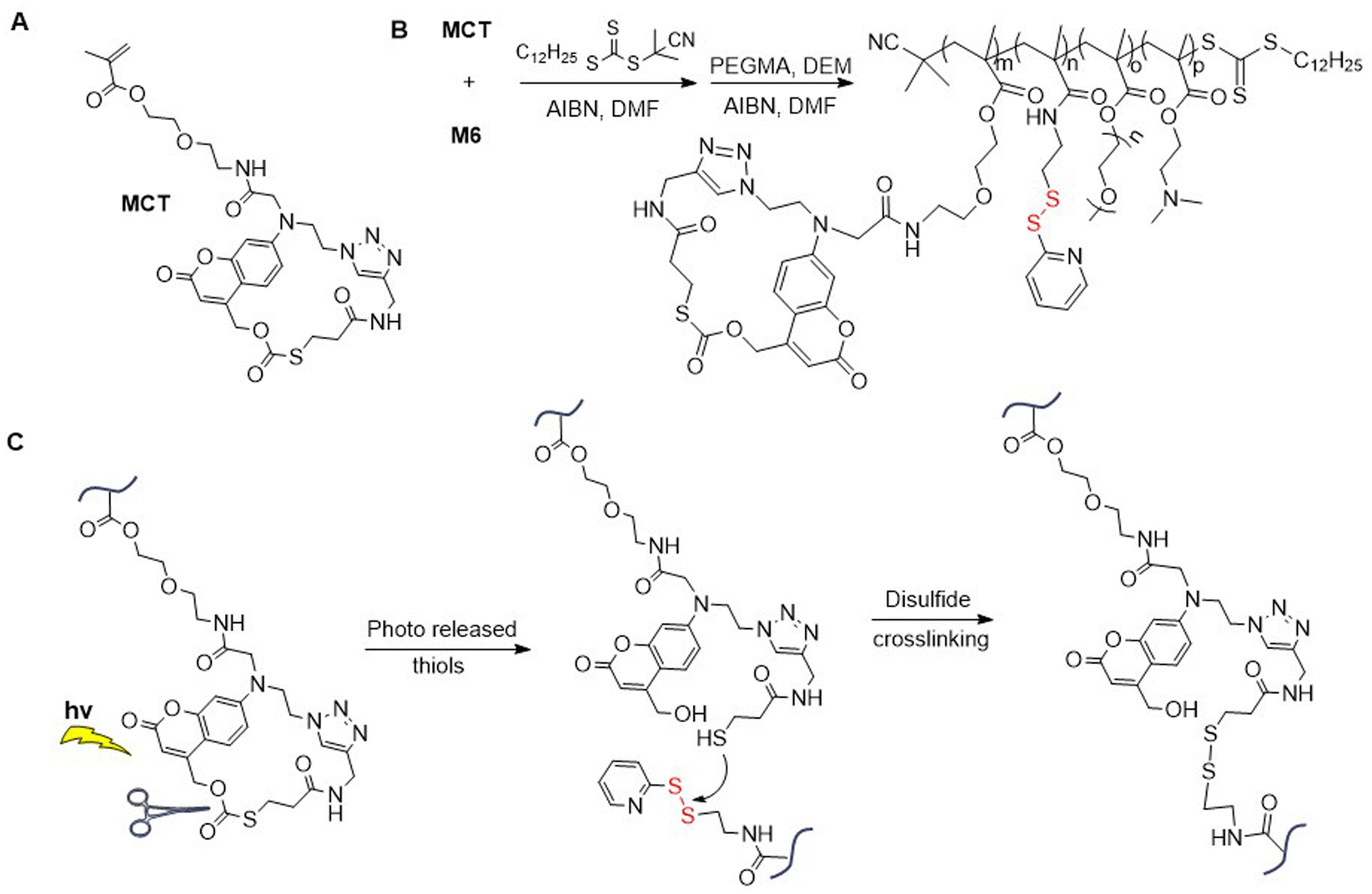 | ||
| Fig. 8 (A) Structure of MCT; (B) synthesis of poly(MCT-co-M6-co-PEG-co-DEM); (C) photo-triggered cross-linking of poly(MCT-co-M6-co-PEGMA-co-DEM). | ||
A series of statistical copolymers involving M5 and N-vinyllactams were prepared via RAFT methodology.54 The authors fabricated polymer film by spin-coating a solution of poly(NVP-co-M5) (NVP = N-vinylpyrrolidone) on solid substrates, which was then functionalized by treatment with an aqueous solution of thiol-containing molecules. It was demonstrated that these films promoted the adhesion and growth of HeLa cell lines, unlike PEG-based polymers. A number of potential copolymer carriers possessing an incorporated pyridyl disulfide moiety were also obtained by RAFT methodologies. Terpolymer poly(PEGMA-co-PDEGMA-co-M5) was synthesized and then conjugated with thiol-functionalized porcine pancreatic lipase to produce thermo-responsive nanogels upon treatment with a meso-2,3-dimercaptosuccinic acid (DMSA) solution.55 Similarly, poly(tBMA-co-M5) (TBM = tert-butylmethacrylate) was conjugated to bovine serum albumin (BSA) to provide covalently connected nanostructures.56 In another study, poly(PDEGMA-b-(PEGMA-co-M5)) was self-assembled above its lower critical solution temperature (LCST) and then cross-linked with reduced BSA.57 After lowering the temperature to below the LCST, proteinosomes containing preserved BSA secondary structure and activity were formed that were capable of internalization into breast cancer cell lines. In addition, poly(DEGMA-co-M5) was coupled with thiol-modified lysozyme to study the cloud point of self-assembled nanostructures, and poly(TEGMA-co-M5) was grafted with various peptides, i.e. bivalirudin, a thrombin inhibitor with the ability to reduce scar formation.58,59
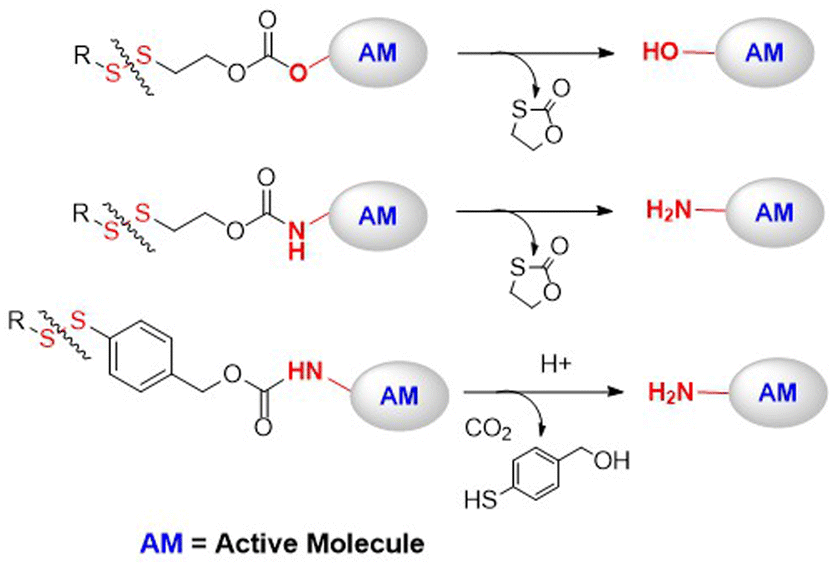 | ||
| Fig. 9 DSILs that were conjugated with polymer carriers. AM = active molecule. | ||
There are a few examples of polymer-conjugates with DSILs obtained via chain-growth polymerization utilizing disulfide-containing monomers. Zelikin and co-workers developed a list of macromolecular prodrugs consisting of DSILs linked to antiviral agents.62–71 Monomers were prepared containing: (a) acrylate or methacrylate, (b) β-DTE carbamate linkers, and (c) bioactive molecules bound through a hydroxyl group (Fig. 10A). Activated monomers were copolymerized by RAFT with HPMA or MAA, with the authors reporting that no degradation of the disulfide bonds was observed during this process.62 Panobinostat (PANO) possessing a secondary amine was also incorporated into a RAFT-derived copolymer (Fig. 10B).66 Despite the low yields of the polymerization, polymers with a PANO content up to 11 mol% were obtained. Macromolecular prodrugs of ribavirin (RBV) and azidothymidine (AZT) were also obtained, being installed to levels of up to 24% by weight.62,63,65
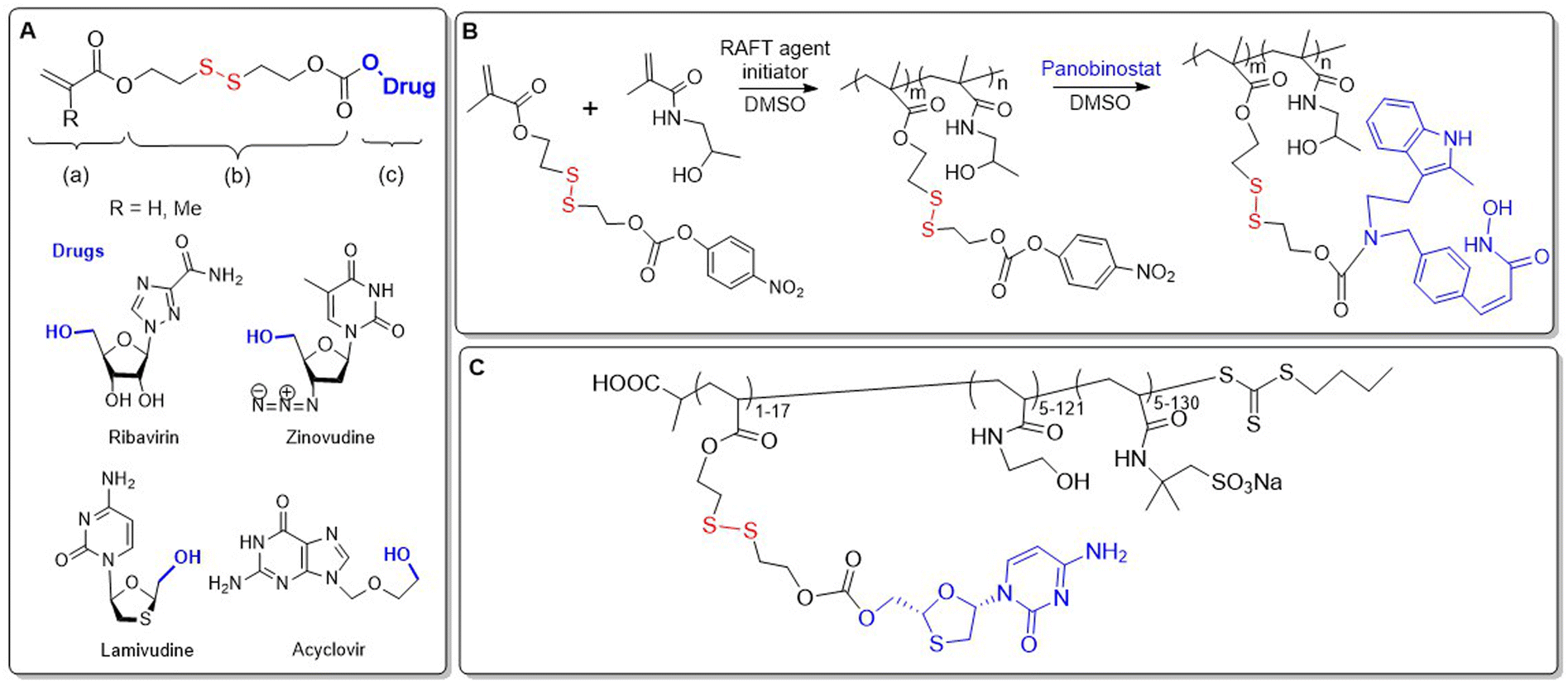 | ||
| Fig. 10 (A) Macromolecular prodrug consisting of DSILs linked to antiviral agents; (B) synthesis of polymer loaded with PANO; (C) terpolymer equipped with 3TC and sodium sulfonate moieties. | ||
All of the reported studies demonstrated a significant advantage for the use of DSILs versus the direct conjugation of the prodrug via ester linkage. Anti-inflammatory activity of RBV prodrugs in cultured macrophages showed a high dependence on both the Mn value and the drug loading percentage.63 To address the problem of HIV drug resistance, polymers containing multiple prodrugs that were active toward numerous viral replication stages were prepared.67,70,72 A terpolymer was thereby fabricated from HPMA and two methacrylate based DSILs, equipped with AZT and lamivudine (3TC) that proved to be stable under extracellular conditions, while readily releasing payloads in environments resembling the cytosol.70,72 Moreover, this polymer exhibited synergistic potency which exceeded the unmodified prodrugs. Since polyanionic side chains are known to increase anti-HIV efficacy, the authors designed statistical copolymers equipped with sulfonate groups. Terpolymers were prepared through the RAFT copolymerization of the DSIL derivative of 3TC with N-hydroxyethyl acrylamide (HEAm) and 2-acrylamido-2-methanepropane sodium sulfonate (AMPS) (Fig. 10C).67 These polymers demonstrated potent reverse transcriptase inhibition, and therefore alternative monomers containing anionic functionalities were copolymerized with ribavirine acrylate and methacrylate DSIL monomers.69
In 2018, Zelikin and coworkers reported albumin–polymer drug conjugates.69–71 Terpolymers consisting of HPMA and AZT/3TC-methacrylate DSIL monomers were obtained with Mn values ranging from 15.9–20.3 kg mol−1 and a drug content of approximately 10 mol%. The polymers were then conjugated with albumin to install ca. 1 protein per polymer chain. It was confirmed that these conjugates provided human T cells with strong protection from HIV infection (Fig. 11A).70 Then, a novel RAFT agent was applied for the copolymerization of HPMA and Acyclovir (ACV) bound through a DSIL to methacrylate, which was then non-covalently associated with albumin (Fig. 11B).71 While the pristine drugs exhibit poor pharmacokinetics, the conjugate displayed high activity against herpes simplex virus type 2 in using mice studies.
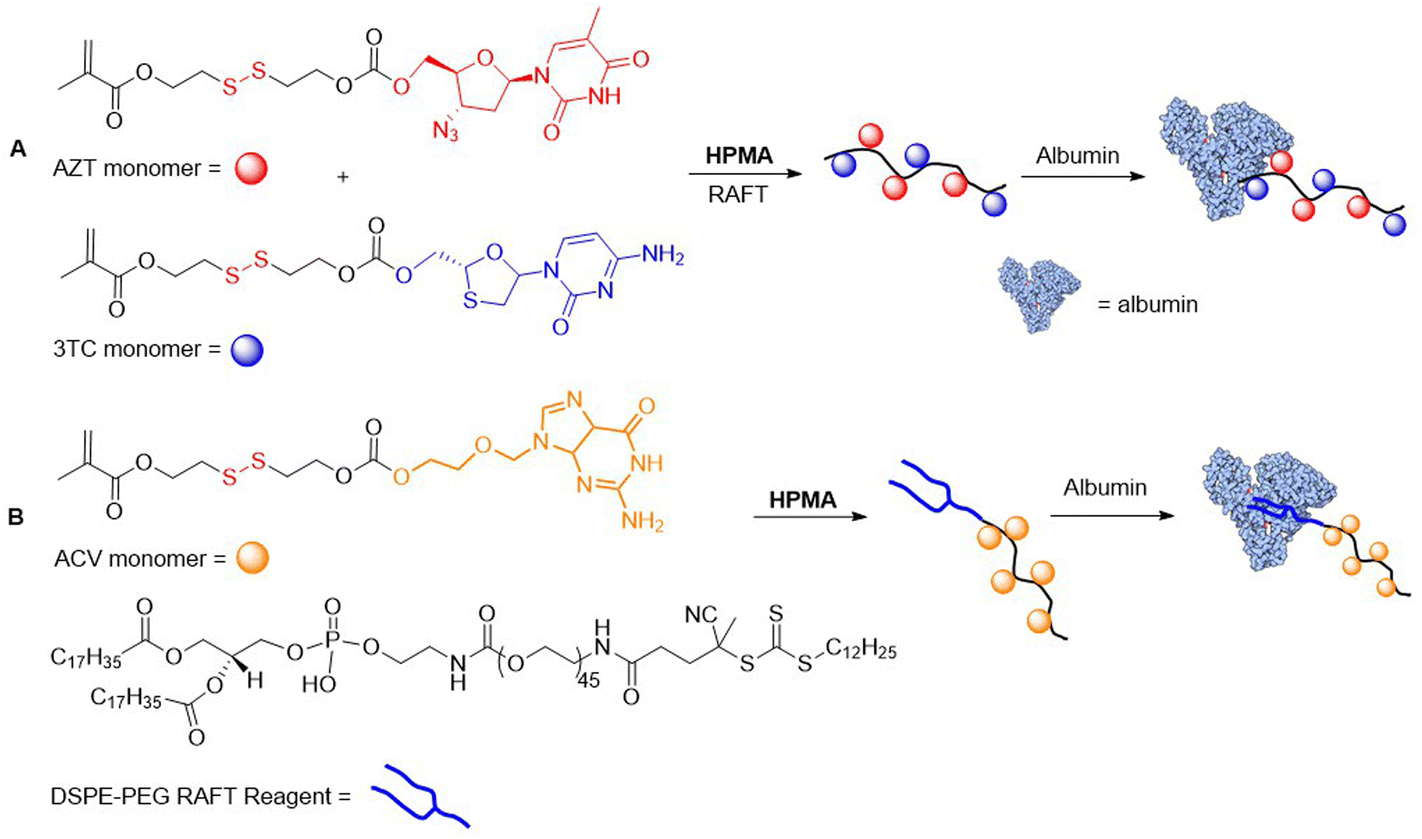 | ||
| Fig. 11 (A) Synthesis of covalently bound albumin–HPMA-based polymer containing AZT/3TC-methacrylate DSILs; (B) synthesis of HPMA-based polymer–ACV conjugates which are non-covalently associated with albumin. | ||
Oupický et al. reported a HPMA-based polymeric prodrug of AMD3465 (a potent HIV entry inhibitor) bound via a β-DTE carbamate linker to methacrylate.73 Radical copolymerization provided a polymer with an AMD3465 content of ca. 22 wt%, which could be effectively released after treatment with GSH. Moreover, due to their cationic character, these polymers were able to form nanosized polyplexes with microRNA and then to deliver it into cells. This dual-function polymer exhibited a stronger inhibition of cancer cell migration in comparison to individual treatments. In addition, camptothecin was bound via DSIL linkage to methacrylate and then subjected to RAFT polymerization with 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) in the presence of PEG113-CPDB (CPDB = 4-(4-cyanopentanoic acid) dithiobenzoate).74 The resulting macro-CTA was reacted with benzyl methacrylate (BzMA) and bis(methacryloyl)cysteamine (BMCy) in a RAFT polymerization, with subsequent self-assembly (Fig. 12). Diffusion of GSH molecules into the core of the nanoparticle enabled degradation of disulfide linkages, followed by reshuffling and camptothecin release.
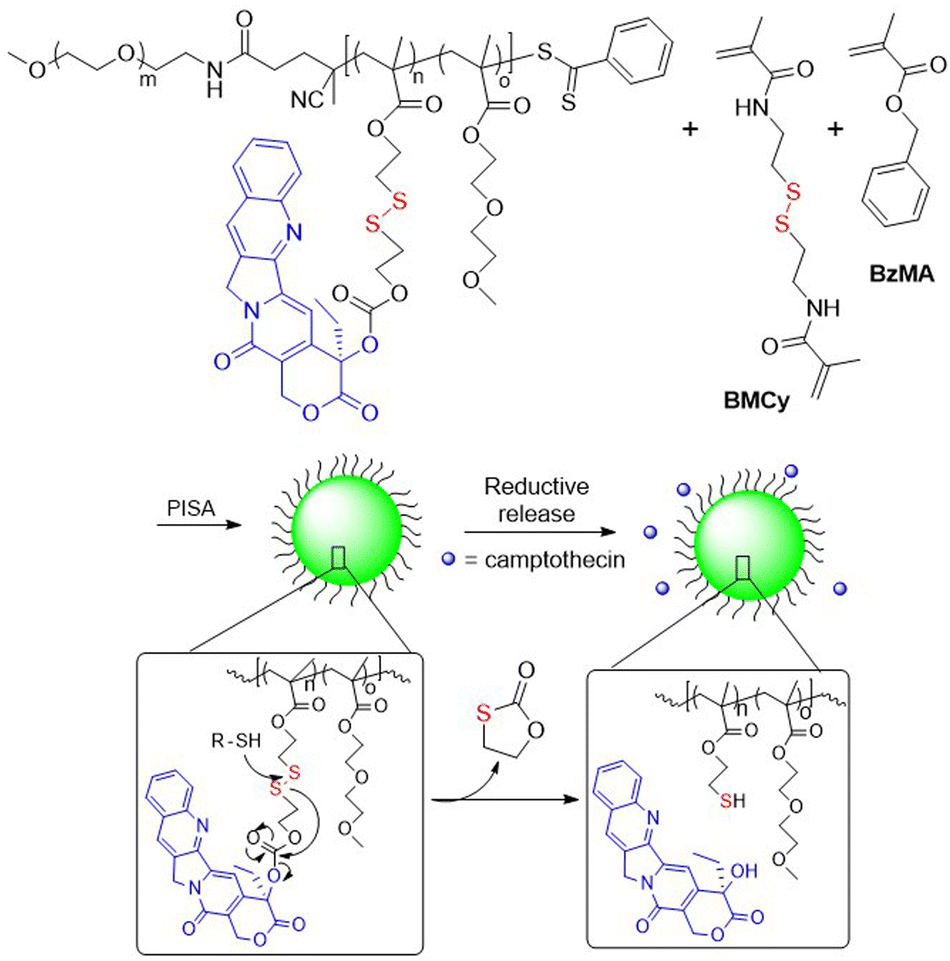 | ||
| Fig. 12 Synthesis of DSIL camptothecin delivery system using RAFT polymerization, with self-assembly and reductive drug release. | ||
Recently, a redox-responsive monomer M8 was reported that displayed robust conversion via 1,6-elimination to release 4-mercaptobenzyl alcohol (Fig. 13A).75 Copolymerization of M8, MMA and pH-responsive monomer BEEMA yielded a terpolymer that could release two corrosion inhibitors in acidic (benzoic acid) or reductive (tryptamine) conditions (Fig. 13B). The corrosion rate of steel coated with poly(MMA-co-BEEMA-co-M8) was reduced 800 times when compared to uncoated steel, and 19 times compared to steel coated with poly(MMA).
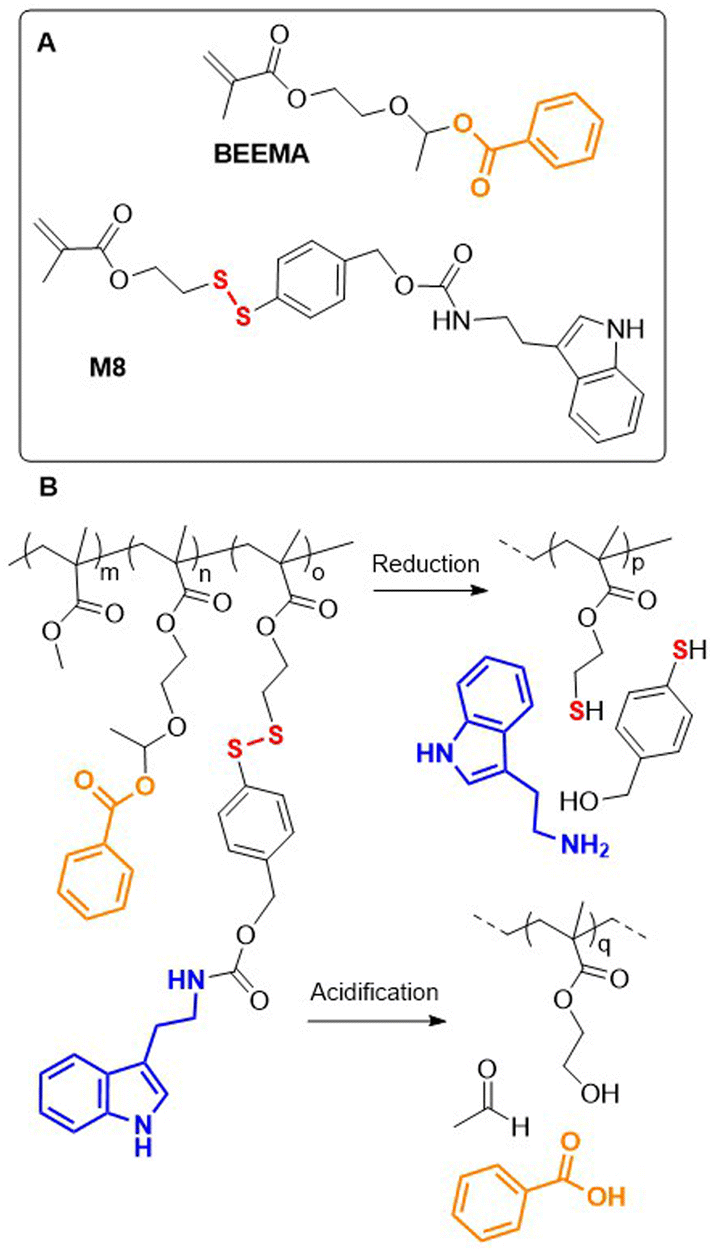 | ||
| Fig. 13 (A) Monomers for synthesis of anti-corrosion coatings; (B) release of benzoic acid or tryptamine from poly(MMA-co-BEEMA-co-M8) in acidic or reductive conditions. | ||
 | ||
| Fig. 14 Monomers with disulfide moiety located in pendant. | ||
2.2 Disulfide-based cross-linking monomers
An additional class of monomers that possess disulfide bonds in the pendant are the bifunctional vinyl monomers used for their cross-linking ability (Fig. 15A). Among these monomers are 2,2′-dithioethanol derivatives: acrylate M11 (DSDA) and methacrylate M13 (DSDMA) and cystamine derivatives: M12 (BACy) and M14 (BMCy) (Fig. 15B). There are reports of the utilization of ATRP and RAFT to synthesize degradable polymers with M11 or M13, focusing on conditions that enable or prevent gelation. This research relies heavily on the application of the Flory–Stockmayer theory which predicts gelation based upon the quantity of cross-linking per polymer chain. The studies refer also to the synthesis of various soluble branched copolymers where monovinyl monomers were polymerized with multivinyl cross-linkers in solution,78 emulsion,79 or suspension,80 and also by controlled radical cross-linking copolymerization.81 Other reports detail the development of branched stimuli-responsive drug delivery systems that are readily degradable under intracellular reducing conditions, where monomers M12 or M14 are typically utilized.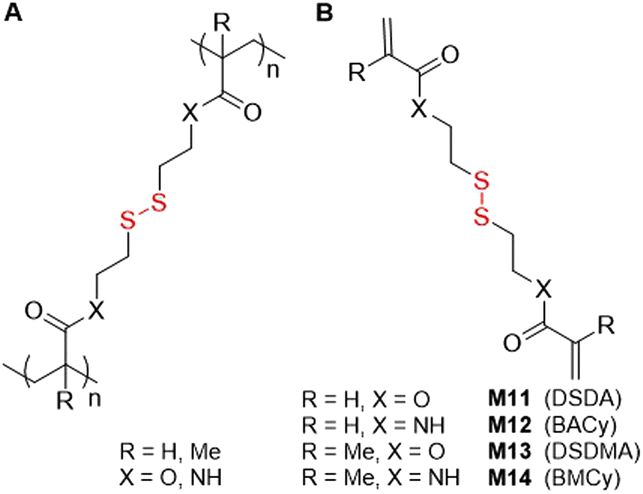 | ||
| Fig. 15 (A) Polymer containing a disulfide cross-linking monomer; (B) structures of disulfide-containing bifunctional monomers. | ||
Matyjaszewski and coworkers applied M13 for the preparation of well-defined degradable copolymers by ATRP.7,82–90 Through copolymerization of MMA with M13 degradable gels were prepared with 3.5 or 1.2 cross-links per chain and an Mn up to 14.9 kg mol−1 (Đ = 1.5–1.6).82,84 Subsequently, the branched gels were explored as macroinitiators for chain extension with styrene via ATRP. In addition, miktoarm star terpolymers of poly(MMA-b-M13-b-BA) were fabricated via an “in–out” methodology (Fig. 16).83 To achieve this, the synthesis of a polyMMA macroinitiator was followed by subsequent chain extension and cross-linking with M13 to provide degradable star copolymers, which were then again extended using BA. Degradation studies revealed that only 19% of the total Br functionality in poly(MMA-b-M13) was initiated during the ATRP polymerization of BA, which was attributed to the high level of core cross-linking. Unfortunately, the occurrence of inter- and intra-star arm–arm couplings was also confirmed.
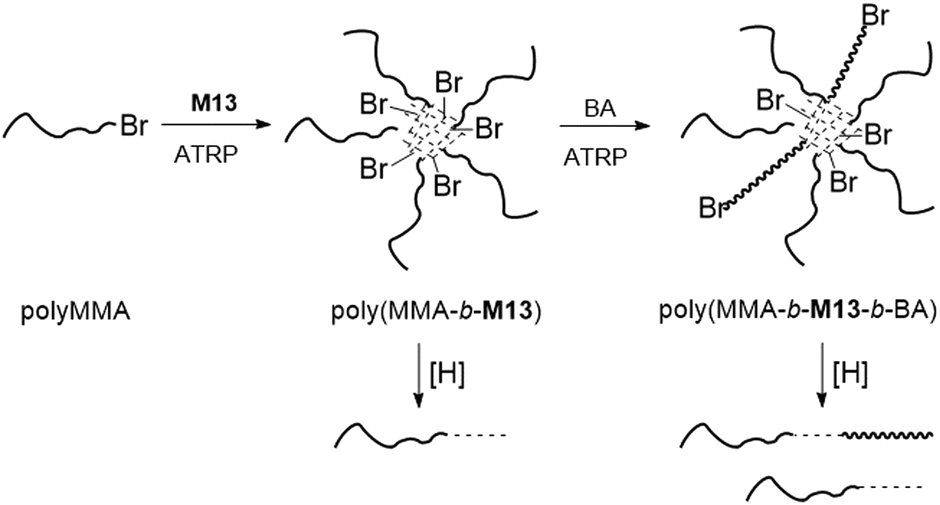 | ||
| Fig. 16 Synthesis of degradable miktoarm polymers from MMA and M13 and BA via ATRP. | ||
Stable, hollow polymer nanocapsules with a cross-linked shell were fabricated by ATRP.85 Firstly, an amphiphilic block copolymer of PEGMA and n-butyl methacrylate (BMA) was obtained and used as macroinitiator in an interfacial miniemulsion copolymerization involving BMA and M13. As a result, stable nanoparticles were obtained, which after degradation with PBu3, yielded polymers with an Mn = 34.2 kg mol−1 (Đ = 2). It should be noted that relatively high dispersity values were obtained compared to the reference polymer without M13 incorporation (Đ = 1.5). Furthermore, a sample of N3-PEGMA-b-PBMA-Cl was prepared, which possessed a halogen end-group to enable the initiation of ATRP, as well as an azide end-group to enable post-modification (Fig. 17A).86 Together with monofunctional PEGMA-b-PBMA-Cl, it was copolymerized with BMA and M13 in a miniemulsion to form nanocapsules with cross-linked shells. Finally, the azide group was reacted with a functionalized dansyl probe in an azide–alkyne cycloaddition, or the polymer was utilized as a macro-ATRP initiator to construct an additional polymer shell (Fig. 17B).
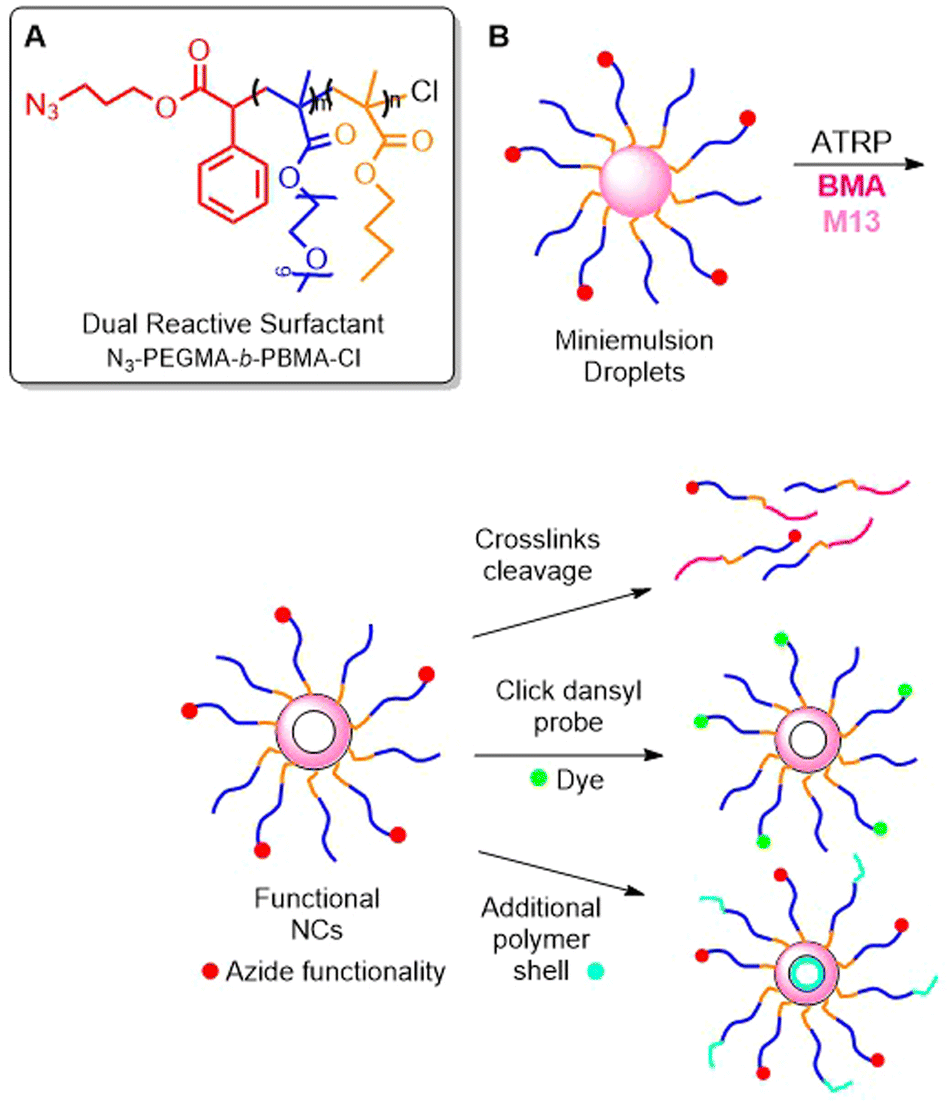 | ||
| Fig. 17 (A) Structure of dual-functional N3-PEGMA-b-PBMA-Cl; (B) scheme of interfacial miniemulsion ATRP of N3-PEGMA-b-PBMA-Cl, PEGMA-b-PBMA-Cl, BMA and M13 and post-modification. | ||
In addition, monomer M13 was applied in the synthesis of a disulfide-containing cross-linked star polymer, prepared via a core-first methodology (Fig. 18).7,87 Firstly, ethylene glycol diacrylate (EGDA) was homopolymerized under high dilution to prevent macroscopic gelation. When the conversion reached 91%, BA was added to produce a poly(EGDA-b-BA) star polymer with an Mn of ca. 375 kg mol−1. This polymer was then subjected to chain extension with cross-linking monomer M13 to introduce disulfide functionalities into the arm ends. Treatment with PBu3 resulted in disulfide bond cleavage to produce individual soluble stars, but under oxidizing conditions the gel could be reformed. In addition, the star polymers in reduced form were deposited on silicon wafer and then oxidized to form an insoluble film which was reported to display self-healing properties.7
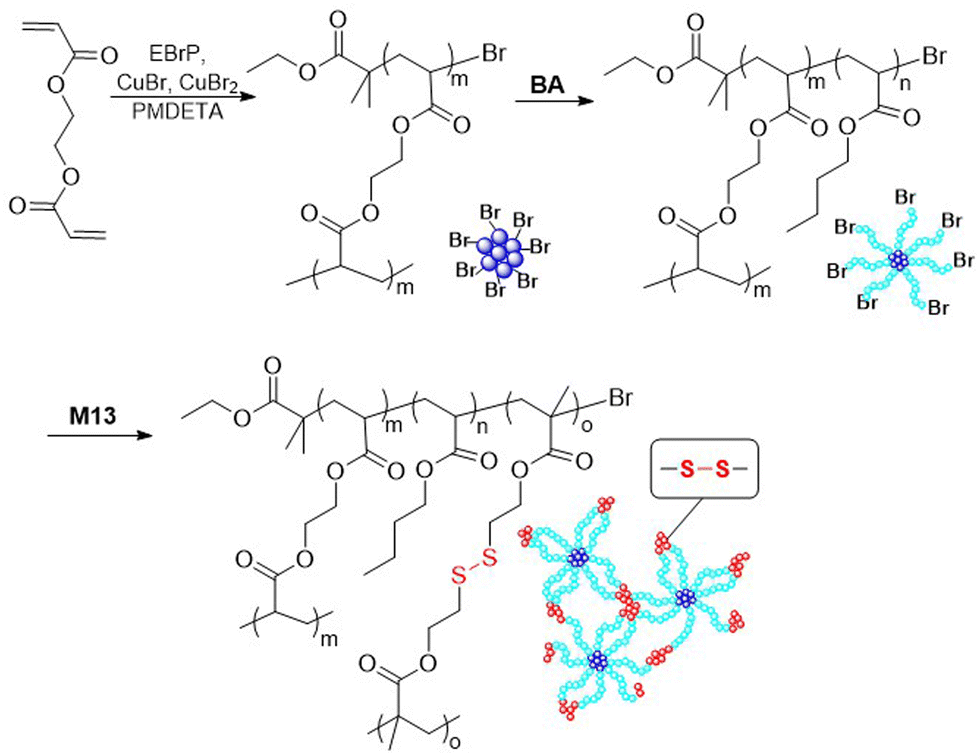 | ||
| Fig. 18 Synthesis of star polymer poly(EGDA-b-BA-b-M13). | ||
Next, 2-ethylhexyl methacrylate (EHMA) was copolymerized by interfacial miniemulsion ATRP with M13 to provide a polymer with Mn = 30.5 kg mol−1 (Đ = 1.6) containing voids within the macroporous structure ranging from 3–15 μm. Polymerization conditions employing a less hydrophobic catalyst resulted in a non-fully degradable copolymer that contained a less uniform cross-linked network, and lower stiffness and yield strength.87 Branched copolymers fabricated by the incorporation of M11 and M13 were also reported by Armes et al.91–97 Firstly, HPMA was copolymerized with M13 by ATRP.91 It was confirmed that high levels of M13 led to macrogelation and an increase in Mn and dispersity. Analysis revealed that the dispersity of the reductively degraded copolymer was analogous to the dispersity of linear polyHPMA obtained under the same conditions in the absence of M13. Thus, it appeared that high molecular weights were caused mostly by M13 branching and not by the chain transfer or termination by combination. Based on these results, highly branched, hydrophobic polymers (Mw = 292 kg mol−1) suitable for electrospinning were synthesized.92
Then, Armes et al. studied the RAFT and ATRP copolymerizations of 2-hydroxypropyl acrylate (HPA) with M13.92 For both techniques it was observed that copolymerization involving the cross-linking agent (M13) was slower than linear homopolymerization. Interestingly, higher levels of cross-links per chain were incorporated by RAFT before macrogelation occurred. This was explained by the occurrence of intramolecular cyclization of the bifunctional vinyl monomer, instead of intermolecular cross-linking. In further studies on the copolymerization of 2-aminoethyl methacrylate (AMA)94 or MMA95–97 with M13 it was confirmed that the level of intermolecular cross-linking versus intramolecular cyclization was highly dependent upon M13 concentration and its molar ratio to the CTA. It was reported that intermolecular branches were formed at all monomer concentrations, but that intramolecular cyclization occurs predominantly at a lower feed ratio of M13 and lower M13/CTA molar ratios. Unusually, it was concluded that initial monomer concentration was more important for the microstructure of the polymer product than the polymerization technique.
Tsarevsky et al. has studied polymerization of M13 with functional vinyl monomers in the presence of efficient chain transfer agents.98,99 Thus, M13 was copolymerized with diethylene glycol methyl ether methacrylate (DEGMEMA),98 or oligo(ethylene oxide) methyl ether methacrylate (OEGMEMA)98 or ethyl cyanoacrylate (ETA) and 2-chloroethyl methacrylate (ClEMa)99 under ATRP conditions with CBr4 to yield degradable hyperbranched polymers with multiple peripheral alkyl bromide groups. To delay gelation with a large M13 input in the feed, the ratio of CTA to initiator was raised to up to 40 (for copolymerization with DEGMEMA or OEGMEMA) or 200 (copolymerization with ECA and CIEMA). By the addition of ca. 5% of crosslinking monomer poly(DEGMEMA-co-M13) was obtained with Mn = 5.1 kDa (Đ = 3.1). Poly(DEGMEMA-co-M13) were used as macroinitiators in a copolymerization with MMA to yield star copolymers with reductively degradable cores.98 Tsarevsky et al. also obtained hyperbranched disulfide-containing polymers avoiding gelation by the copolymerization of MMA and an inimer derived from M13 but containing an ATRP-initiator moiety.100
Paulusse and coworkers obtained poly(DMAEMA-co-M11) (DMAEMA = N,N-dimethylaminoethyl methacrylate) in RAFT polymerization.101 Copolymers with dispersity values <2 were obtained when the process was ended after ca. 10 hours, however, when the polymerization was extended to 12 hours, a substantial increase in dispersity (Đ = ca. 8.0) and Mn values (from 16.5 to 32.0 kg mol−1) were observed, which supported previous observations regarding branching occurring at higher conversions by RAFT processes.93Poly(DMAEMA-co-M11) was also obtained by ATRP methodology, which after capping with 3-morpholinopropylamine (MPA) was subjected to the formation of polyplexes with plasmid DNA that exhibited good transfection capability.102 This feature, as well as their cytotoxicity and interaction with nucleic acids, were affected by the degree of branching and the length of the primary-chain molecules. Moreover, by the same technique a larger “knot” structure of cationic polymer poly(DMAEMA-co-M11-co-PEGMA) with 5.6% branching was produced that exhibited a transfection profile for astrocytes that superseded commercially available reagents.103
To produce new nanohydrogels as potential platforms for drug delivery, M12 and M14 were frequently considered.74,104–109 Wang and coworkers obtained nanohydrogels in a distillation-precipitation FRP of MAA with M12.104 Due to the electrostatic interactions between DOX amine groups and the carboxyl groups incorporated into the nanohydrogels it was possible to load the matrix with up to 42.3 wt% DOX at physiological pH. The DOX-loaded nanohydrogels exhibited pH and redox dual-responsive drug release capabilities, as well as non-toxicity toward normal cell lines, but were reported to have high cytotoxicity to human tumour cells. In addition, M14 and modified hyaluronic acid (HA) were copolymerized to produce nanogels for DOX encapsulation (Fig. 19).105 To enable effective penetration of the blood brain barrier, components such as (a) phenylboronic acid (PBA) which displays affinity for sialic acid, and (b) lactoferrin (Lf) which exhibits affinity to receptor-associated proteins which are highly expressed in glioma cancer cells, were incorporated. Studies on drug release, cytotoxicity, cellular uptake, brain permeability and the biodistribution of these nanoassemblies demonstrated their clear superiority to conventional systems.
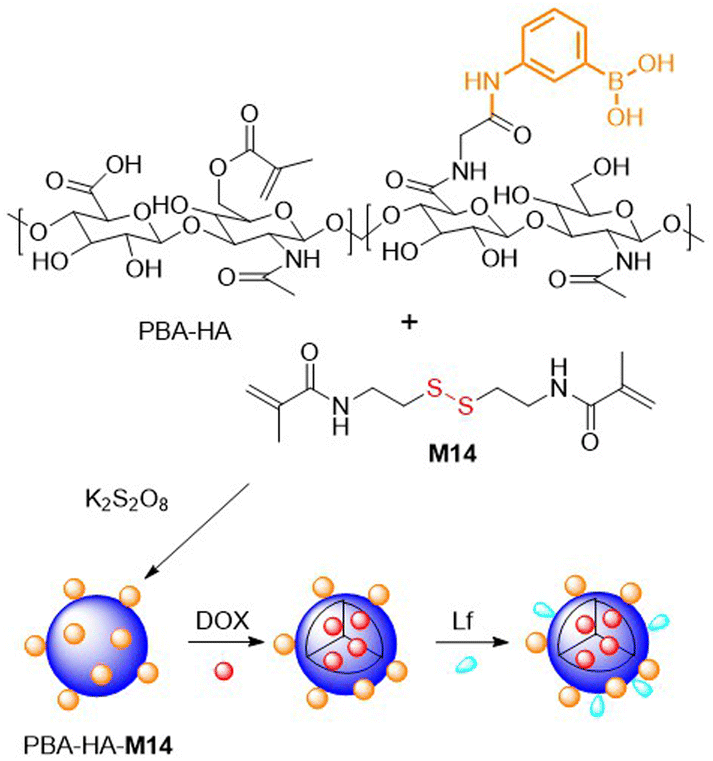 | ||
| Fig. 19 Schematic synthesis of HA and M14 based nanogel loaded with DOX, designed for effective glioma cell penetration. | ||
In 2021, another DOX delivery system was fabricated with M12 and cyclodextrin (CD) nanosponges used as binding components.106 Thus, inverse-emulsion FRP polymerization of AA, M12 and acryloyl-6-ethylenediamine-6-deoxy-β-cyclodextrin (β-CD-NH-ACy) yielded hyper cross-linked polymer (Fig. 20). Further DOX inclusion and complexation with 22.6% drug loading provided spherical-like structures that were responsive to reductive and acidic conditions, effectively releasing DOX at cytosolic GSH levels at a pH of 5.0. These DOX@Nanosponges proved to be cytotoxic against lung cancer cells, in which they were internalized by endocytosis.
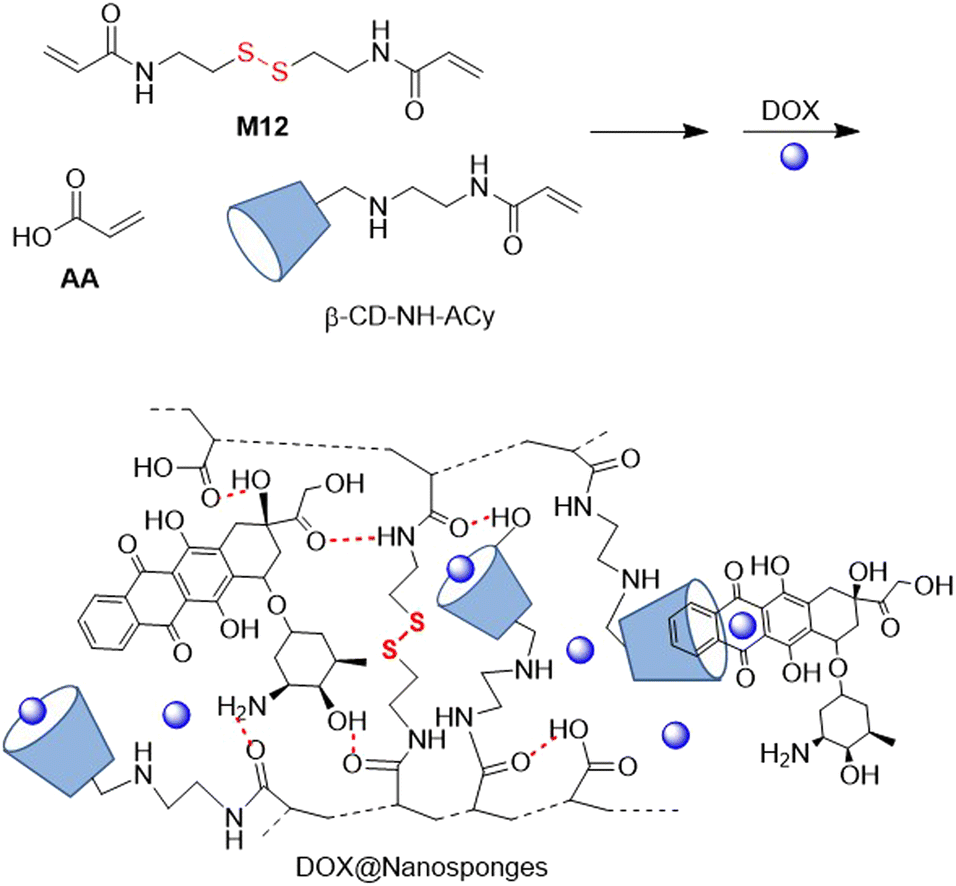 | ||
| Fig. 20 DOX delivery systems with degradable cyclodextrin (CD) nanosponges. | ||
Copolymerization of (2-hydroxyethyl) methacrylate (HEMA) and M12via a FRP precipitation-polymerization in water in the presence of cisplatin provided hydrogels imprinted with the anticancer drug.107 The cisplatin remained complexed with the polymer through hydrogen bonding and was reported to be more effectively released in acidic conditions than at physiological pH. As mentioned in section 2.1.2, M14 (BMCy) has also been applied for the synthesis of polymer drug delivery systems involving camptothecin (CPT).74 Thus, 2,2′-dithiodiethanol methacrylate monomer conjugated with CPT was incorporated to produce block-copolymers PEG-b-poly(MEO2MA-co-CPT)-b-poly(M14-co-BzMA)via RAFT methodology with up to 36.3% of molar drug content (Fig. 12).
Lee and coworkers synthesized copolymers of acrylamide (AM) and M12 as an disulfide moiety-enriched alternative to known copolymers of AM and N,N-methylenebisacrylamide which are exploited for protein resolution during sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).108 The M12-based polyacrylamide hydrogel exhibited a higher swelling ratio and pore size than the traditional hydrogel, with comparable capability for protein separation. In addition, RAFT polymerization was utilized to produce branched polymers from poly(ethylene glycol) methacrylate, M12 and photodegradable monomer 1,3-di(acryloxymethyl)-2-nitrobenzene (DANB).109 After additional hyperbranching in aqueous solution, nanohydrogels were obtained as a result of intermolecular disulfide exchange which exhibited temperature-, photo- and redox-sensitivity.
2.3 Radical ring-opening polymerization (rROP)
Radical ring-opening polymerization (rROP) is a free-radical polymerization technique that involves ring-opening and subsequent propagation involving a cyclic monomer. The harnessing of this technique enables the simple introduction of heteroatom-containing functional groups into a polymer backbone. However, there exists only a few examples within the literature of the utilization of rROP for the synthesis of polymers containing disulfide moieties, being exclusively undertaken using monomer M15 (MTC) (Fig. 21).101,110–112 Hawker and co-workers RAFT copolymerized M15 with MMA to provide statistical copolymers possessing reactive disulfide units within the vinyl backbone and exo-methylidene groups which could be applied for further functionalization.110 At all feed ratios, a good agreement between monomer feed ratio and product composition was observed. Moreover, the treatment of the copolymer with sodium methoxide resulted in the decay of the ester groups without affecting the disulfide bonds. Conversely, the disulfide bonds could be selectively degraded in solutions of hydrazine or tributylphosphine. | ||
| Fig. 21 Synthesis and degradation of poly(MMA-co-M15). | ||
Then, Paulusse et al. copolymerized M15 with HPMA in the presence of a hydrophilic PGMA56 macro-CTA (PGMA = poly(glycerol monomethacrylate) in aqueous solution, with concurrent polymerization-induced self-assembly (PISA) to provide various nanostructures including spheres, worms or vesicles, dependent upon the degree of polymerization (Fig. 22).111 However, only a small contribution of M15 in the terpolymer could be introduced without affecting polymer dispersity and nanoparticle morphology. It was concluded that dispersity values of <1.4 could be obtained with a feed ratio of M15 below 0.5%, and well-defined nanostructures could be formed with an incorporation of M15 below 1%.
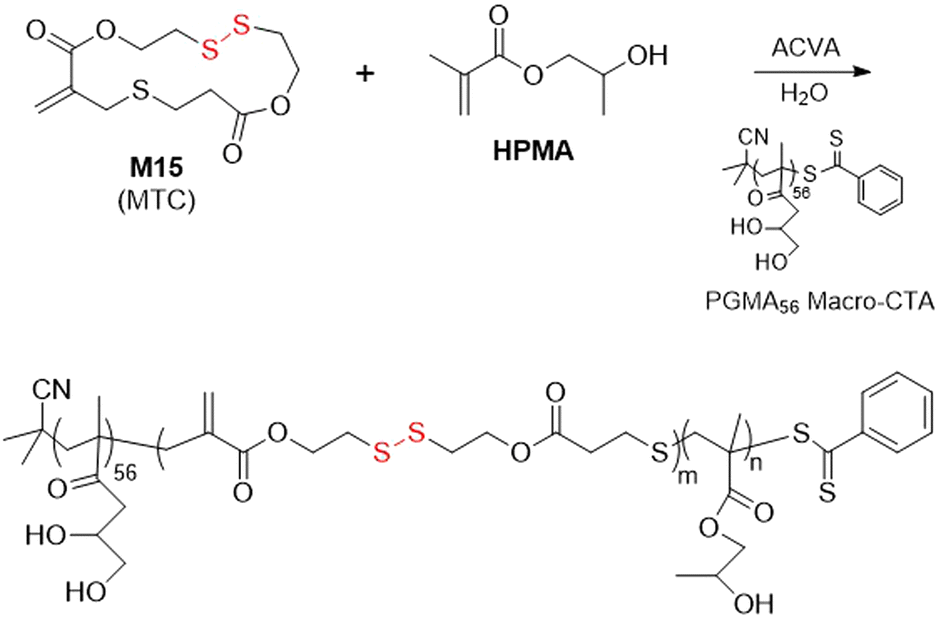 | ||
| Fig. 22 Synthesis of poly(GMA)-b-(M15-co-HPMA). | ||
In addition, monomer M15 was subjected to RAFT copolymerization with DMAEMA and tri(ethyleneglycol) diacrylate (TEGDA) (Fig. 23).101 Polymerization for ca. 5 hours provided a controlled linear chain growth without intermolecular cross-linking, until a critical concentration point was achieved during which cross-linking occurred, this process being readily monitored using SEC by a change from unimodal to bimodal signal. Following degradation, a unimodal peak could again be observed by SEC analysis, implying a statistical incorporation of M15 into the polymer backbone. Moreover, this polymer was end-capped with 3-morpholinopropylamine (MPA) and studied as a gene delivery agent for plasmid DNA, displaying improved transfection efficiency and lowered cytotoxicity in comparison to other systems.102,103
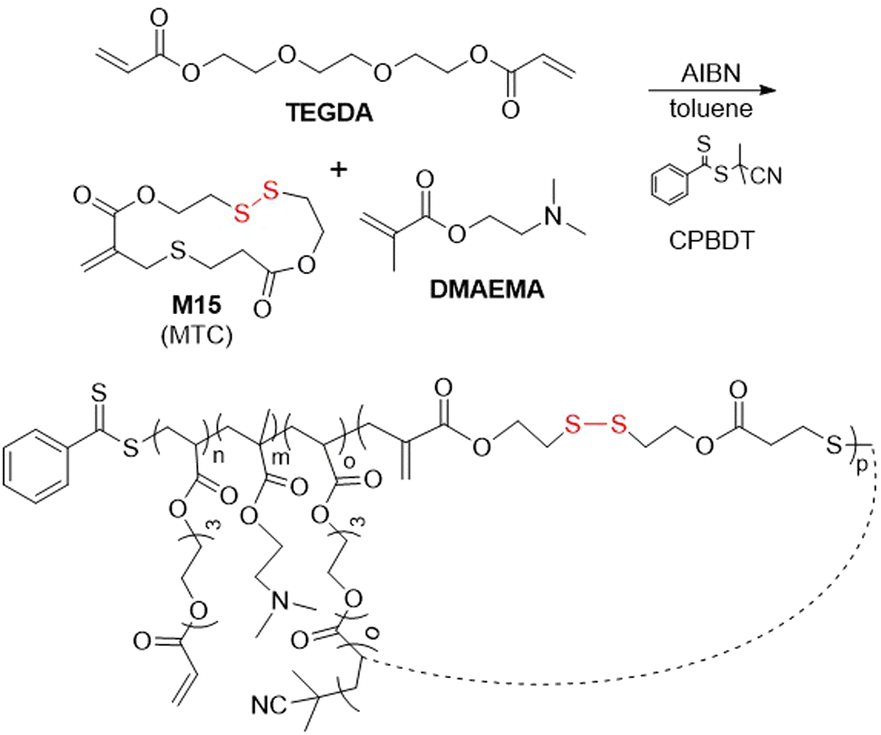 | ||
| Fig. 23 Synthesis of poly(DMAEMA-co-TEGDA-co-M15). | ||
The RAFT polymerization of anticancer drug paclitaxel conjugated to methacrylate (PTXMA), M15 and 2-(diisopropylamino)ethyl methacrylate (DPAEMA) in the presence of a PEGMA macro-CTA provided copolymers with Mn values of 13.6–26.9 kg mol−1 (Đ = 1.4–1.5) (Fig. 24).112 Unfortunately, the final polymer composition of both the M15 and PTXMA was reported to be less than half of the initial feed. By a self-assembly process in water, aggregates having a vesicle shape were obtained. Their anticancer activity was studied in vitro and the polymer carrier PEGMA-b-(DPAEMA-co-PTXMA-co-M15) was demonstrated to have a higher efficacy than the PTXMA monomer.
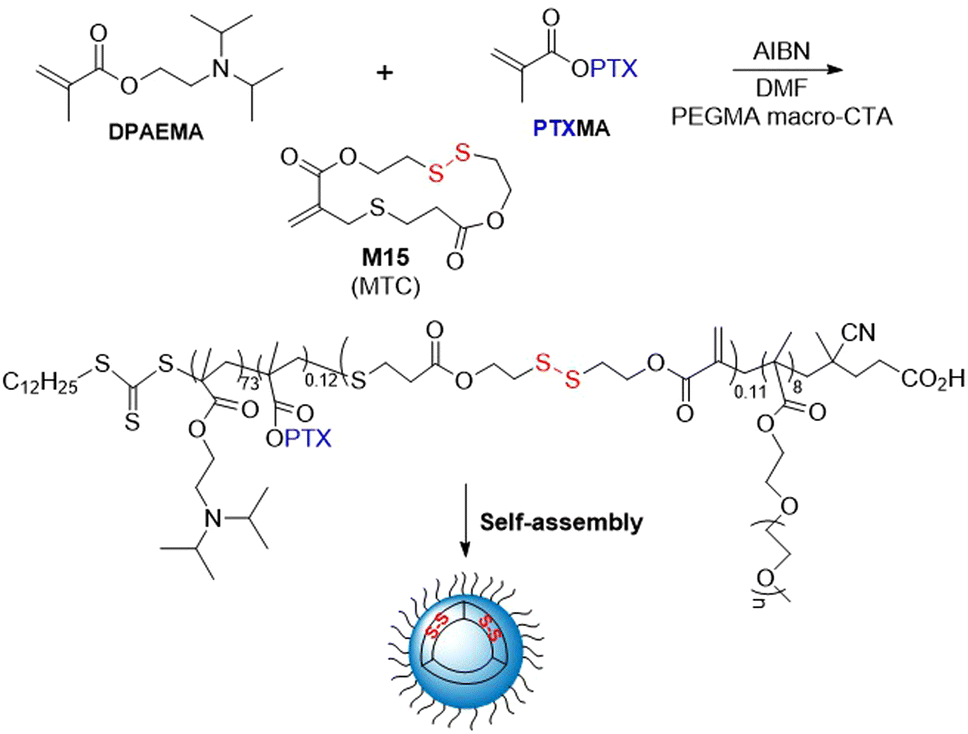 | ||
| Fig. 24 Synthesis and self-assembly of PEGMA-b-(DPAEMA-co-PTXMA-co-M15). | ||
3. Ring-opening polymerization (ROP)
The application of ring-opening polymerization (ROP) to fabricate polymers containing disulfide moieties is generally represented by processes with an anionic character, and can be performed using N-carboxyanhydrides, carbonate, and lactone monomers. The effectiveness of the ROP of lactones and carbonates relies upon the ring-strain of the monomer. For smaller ring heterocycles, the high strain promotes polymerization, while for macrocyclic monomers (≥12 membered) polymerization is frequently more challenging. Moreover, disulfide bonds incorporated into such heterocyclic monomers provide additional limitations in terms of suitable polymerization conditions and catalysts. Nevertheless, there exists multiple reports of effective ROPs of monomers containing disulfide moieties.3.1 Monomers providing pendant disulfides
Three cysteine derived N-carboxyanhydrides (NCAs) with disulfide containing pendants (M16a–c) were subjected to ROP to provide polymers with Mn values ranging from 2.5 to 8.3 kg mol−1 (Đ = 1.13–1.30) (Fig. 25A).113 Copolymerization of M16c with mPEG45-NH2 as a macroinitiator resulted in a block copolymer with an Mn = 11.7 kg mol−1 (Đ = 1.1). It was reported that aqueous solutions of this polymer displayed thermal responsivity, but unusually that the reduction in transmittance during the sol–gel transition was not reversible, which was attributed to disulfide bond exchange. Carbonate M17 and ε-caprolactone (ε-CL) were subjected to ROP in the presence of isopropanol as an initiator and Sn(Oct)2 as a catalyst to provide poly(ε-CL-co-M17) with an Mn = 29.8 kg mol−1 (Đ = 1.33) (Fig. 25B).114 After thiol-exchange with thiolated polyethylene glycol, the resulting biocompatible copolymer was transformed into a doxorubicin-loaded micelle and examined for its cytotoxic activity.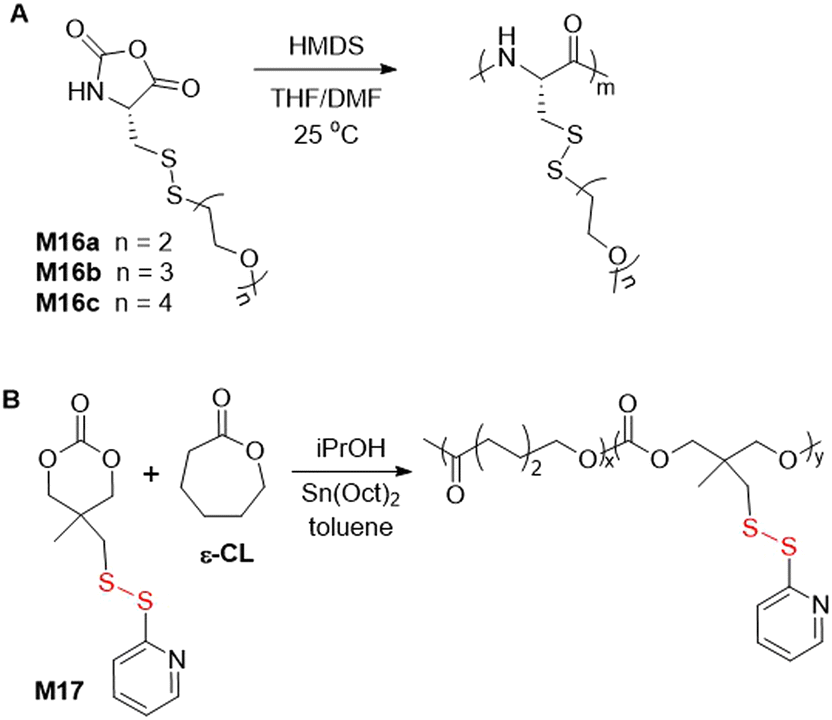 | ||
| Fig. 25 (A) Synthesis of polyM16; (B) synthesis of poly(ε-CL-co-M17). | ||
3.2 Monomers providing backbone disulfides
Lee and coworkers performed the ROP of M18 initiated with alcohol and using diphenylphosphate (DPP) as catalyst (Fig. 26A).115 In the presence of benzyl, propargyl or isopropyl alcohol, homopolymers were obtained with Mn values up to 21.7 kg mol−1, (Đ = ca. 1.05) with conversions of >99%. Moreover, polymerizations of M18 with mPEO-OH (Mn = 2.0 kg mol−1) provided block copolymers. An additional block copolymer was obtained when M18 was added after complete consumption of ε-CL, providing poly(M18-b-ε-CL) Degradation of this polymer was confirmed under reductive conditions, as well as by UV irradiation. Disulfide-containing macrocyclic carbonate M19 was also successfully subjected to ROP initiated by benzyl alcohol in the presence of triazabicyclodecene (TBD) to fabricate polymers with Mn values up to 37.2 kg mol−1 (Đ = 1.28) (Fig. 26B).116,117 The activity of the systems was greatly dependent upon the type of organocatalysts applied. It was also reported that chemically catalysed polymerizations displayed the advantage of shorter reaction times at lower temperatures, as well as improved dispersities, in comparison with enzymatic polymerization.118 | ||
| Fig. 26 (A) Synthesis of polyM18; (B) polymerization of macrocyclic carbonate M19. | ||
4. Ring-opening metathesis polymerization (ROMP)
Ring-opening metathesis polymerization (ROMP) is a chain-growth strategy for the synthesis of polymers containing carbon–carbon double bonds in the backbone. ROMP is derived from research into olefin metathesis first investigated by Y. Chauvin and later extensively elaborated by R. H. Grubbs. The polymerization process for a broad range of cyclic olefins is characterized by a high selectivity and functional group tolerance, but nevertheless the incorporation of disulfide moieties by ROMP remains problematic. The basis of this issue involves the coordination of the disulfide moiety to the transition metal catalyst.119 However, multiple reports of the copolymerization of monomer M20 with cyclooctanes,120 or the ROMP of cysteine-based macrocycles 24a/b appear within the literature.121–123Emrick and coworkers reported the effective copolymerization of M20 with a series of cis-cyclooctane analogues (type M21), although the homopolymerization of M20 was reported ineffective owing to complications involving catalyst coordination (Fig. 27A).120 Copolymerizations of M20 and M21 using Grubbs third generation (G3) catalyst readily provided polymer which was confirmed to contain a random distribution of disulfide moieties. Moreover, a terpolymerization of M20, M21a and phosphoester monomer M22 was performed to provide a polymer with orthogonal degradation properties (Fig. 27B).
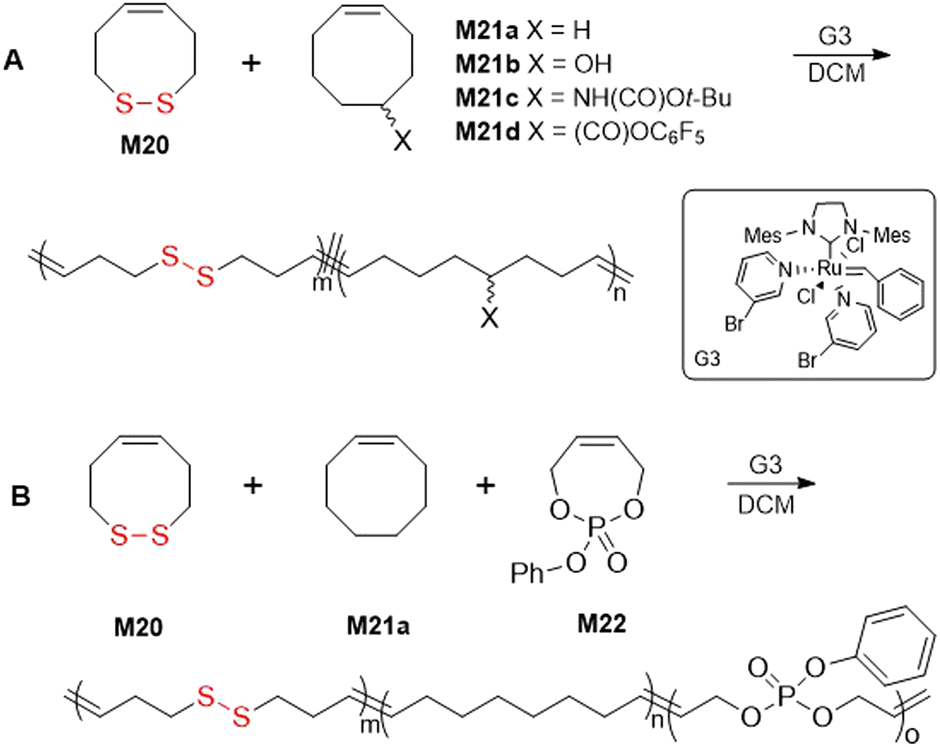 | ||
| Fig. 27 (A) Copolymerization of M20 with cis-cyclooctanes M21a–d; (B) terpolymerization of M20 with M21a and phosphoester M22. | ||
Schlaad and co-workers reported the ROMP of cysteine-based macrocycles M24a/b (Fig. 28).121–123 Polymerization of M23a was attempted with Hoveyda–Grubbs second generation (HG2) catalyst, however only oligomer could be obtained. Monomers M24a/b were therefore prepared by ring-closing metathesis (RCM) of the olefinated Boc-l-cysteine dimers M23a/b. Following this, the ROMP of M24a using G3 catalyst readily provided polyM24a with an Mn = 10.5 kg mol−1 (Đ = 2.2). Since these monomers exhibited low ring-strain, their polymerization was conducted at high concentration in order to obtain favorable entropy. Moreover, it was postulated that the disulfide bonds had little effect on the polymerization owing to the significant distance between the catalyst site and disulfide bond location.
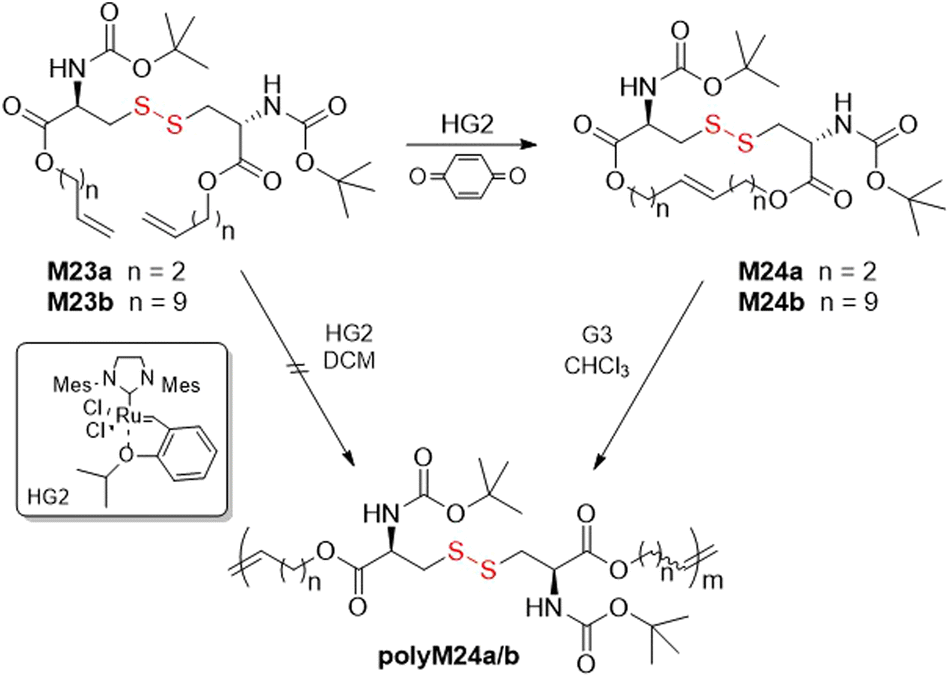 | ||
| Fig. 28 ROMP of macrocycles M24a/b. | ||
5. Miscellaneous
5.1 Radical ring-opening polymerization of the disulfide bond – thermal- and photo-initiated
The first reports regarding the radical ring-opening polymerization of disulfide-containing monomers actually referred to naturally occurring lipoic acid (M1) and were comprised of reactions initiated by heat or irradiation (Fig. 29). In 1955, it was serendipitously discovered by Niu and Reed that M1 could undergo polymerization during the oxidative conditions that were required for its synthesis.124 Later, M1 was purposely polymerized at 65 °C to provide a colourless material which could be decomposed using NaOH to recover monomer.125 Calvin and coworkers simultaneously explored the photopolymerization of 1,2-dithiolane monomers M1 and M25, demonstrating that irradiating these monomers results in diradical formation and subsequent polymerization in neutral solution.126 Conversely, in acidified solution the dithiolane ring was destroyed to yield thiol and sulfenic acid, with no polymerization.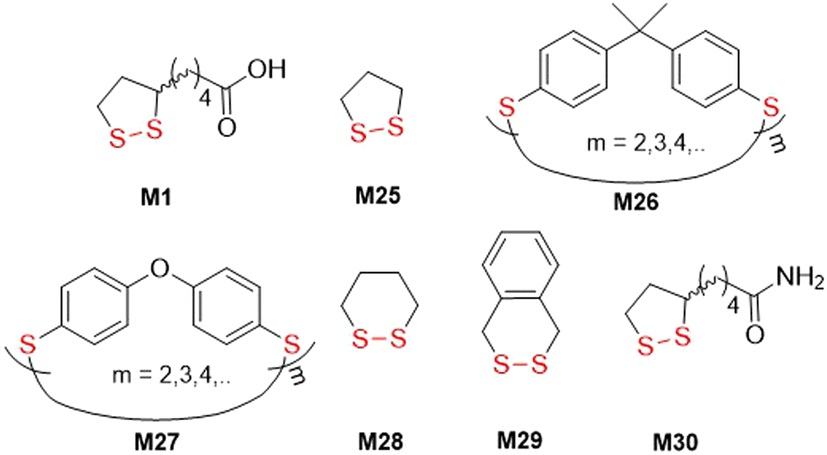 | ||
| Fig. 29 Cyclic disulfide monomers used for thermal- and photo-induced radical ring-opening polymerizations. | ||
Hay and coworkers later reported the thermally-induced radical copolymerization of macrocyclic aromatic disulfides M26 and M27 (both consisting of mixed quantities of oligomer).127–131 It was reported that the melt homopolymerization of oligomeric M26 was possible above its melting point, and was rapid at 200 °C, providing polyM26 with Mn = 88.0 kg mol−1 (Đ = 2.15).128 Moreover, solution polymerization also provided polyM26 with Mn = 64.0 kg mol−1 (Đ = 1.92). For both processes, the Mn and dispersity remained relatively stable for short reaction times, but at high temperatures (>250 °C) or after prolonged reaction time the occurrence of substantial cross-linking was confirmed. Endo and coworkers reported the thermally-induced rROP of M28/M29,132–135M1,136 and the copolymerization of M28 and M1.137,138 It was demonstrated that these monomers can polymerize in bulk without an initiator when held above their melting point, but that cyclic polymers were formed by a back-biting mechanism (Fig. 30A).133,135–137 Furthermore, it was established that these conditions provided entangled polycatenane macrostructures (Fig. 30B). Photoinduced degradation in dilute solution led to a loss in molecular weight, but also the preservation of cyclic character, attributed to the disconnection of the polycatenane structure (Fig. 30C).
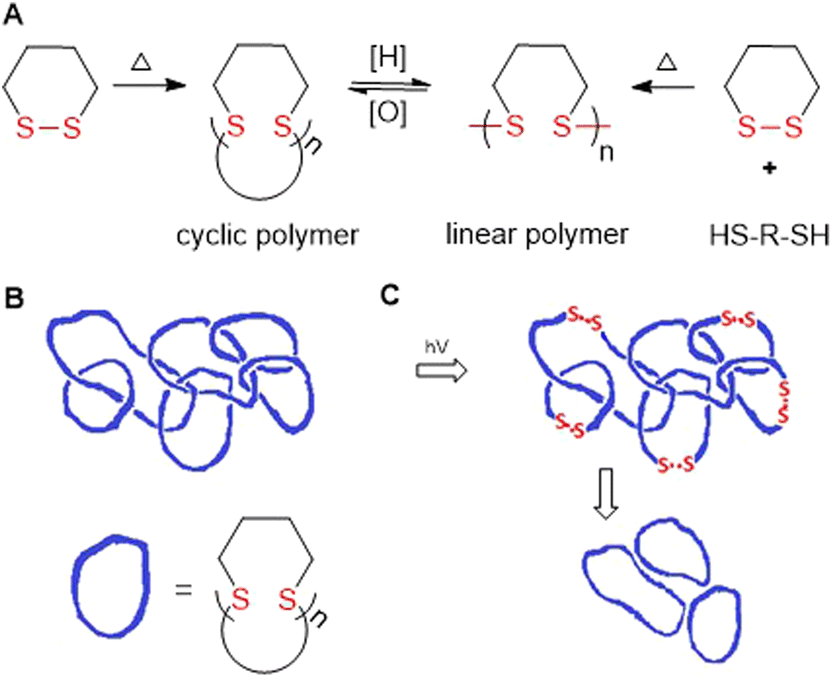 | ||
| Fig. 30 (A) Polymerization of 1,2-dithianes in initiator-free conditions versus thiol-mediated reactions leading to cyclic or linear polymers, respectively; (B) polycatenane structure of poly(disulfide)s; (C) photoinduced degradation leading to non-entangled cyclic polymer formation. | ||
Bulk polymerization of M29 at 90 °C led to cyclic polymer with an Mn = 11.8 kg mol−1 (Đ = 1.95), characterized by higher thermal stability and lower crystallinity than polyM28.135 Likewise, the bulk polymerization of M1 at 90 °C produced a cyclic polymer with Mn = 13.7 kg mol−1 (Đ = 1.5) in 67% yield.136 Interestingly, bulk polymerization in the presence of 6,8-dimercapto-octanoic acid (DHLA) at 120 °C provided a linear polymer with an Mn = 9.1 (Đ = 2.7), but with significantly lower monomer conversion. Bulk copolymerization of M28 and M1 at 80 °C with varying feed ratios led to a series of random sequence copolymers with Mn = 20.1–55.0 kg mol−1 (Đ = 1.45–2.35).137 Due to the difference in ring-strain energies, monomer conversion increased with an increase in M1 feed, which was also more frequently incorporated into the copolymer than M28. Endo also performed thermally-induced copolymerizations of lipoamide M30 and styrene in solution.139 An increase in the feed ratio of M30 resulted in a decrease in conversion, and no polymer was obtained at 50 mol% M30 feed. Differences in the reactivity between the polymeric thiyl and polystyryl radicals during propagation, as well as chain transfer to the amide group, were suspected of causing retardation of the copolymerization. Conversely, if a solution of M30 and styrene was irradiated with UV light at 40 °C, copolymers with enriched M30 relative to the feed were produced.139 Interestingly, irradiation of a solution of styrene produced only traces of polymer, unlike the copolymerization which was presumably initiated by a homolytic fission of M30 to produce initiating diradical species.
Recently, it was demonstrated that photo-induced polymerization without solvent could also provide interlocked products.140 Therefore, M1 was melted at 120 °C to provide a transparent liquid, which during cooling provided a transparent yellow polymer gel, followed by crystallization of the unreacted M1. Then, by irradiation with UV/visible light, polymerization of the remaining monomers occurred to give a colourless film consisting of interlocked cyclic poly(disulfide)s which could be converted by thermal depolymerization to starting monomer. Macromonomers M31 and M32, consisting of lipoates containing poly(dimethylsiloxane) fragments, were irradiated with UV light to provide bottlebrush polymers in a grafting-through polymerization strategy (Fig. 31A).141 After subjection to thermal depolymerization, ca. 30–40% of the original monomer (M31) could be recovered. Irradiation-induced copolymerization of M33 with cross-linker poly(ethylene glycol)-diacrylate in the presence of a photoinitiator provided material that was used as resin for 3D printing, capable of both thermal and photo-induced reprocessing (Fig. 31B).142
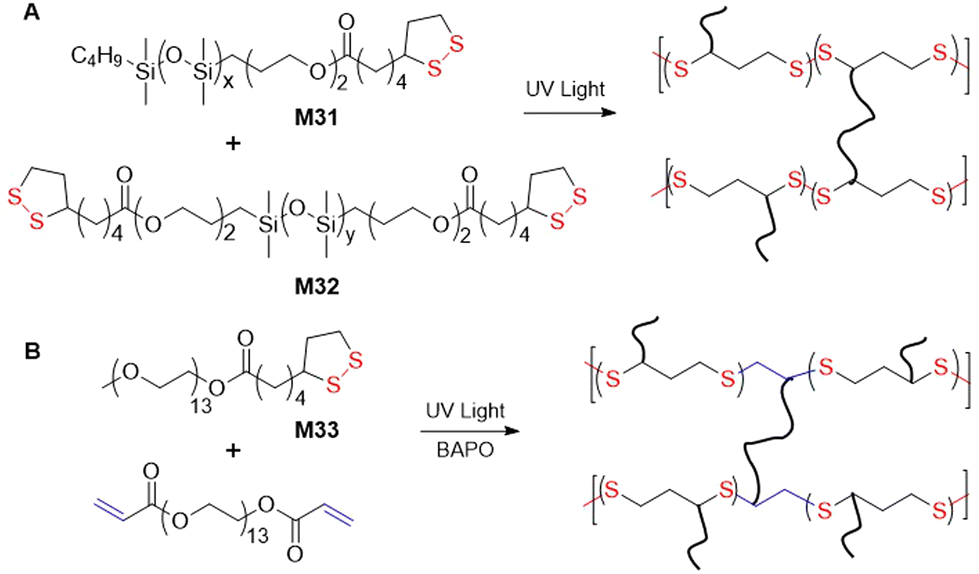 | ||
| Fig. 31 Light-mediated synthesis of dynamic bottlebrush elastomers from lipoic acid-based monomers M31–33. | ||
Orthogonal monomers containing vinyl or acrylate groups linked with lipoic or asparagusic acid moieties can be polymerized to provide linear polymers with intact pendant disulfide units which can then undergo photoinduced polymerization to lead to gelation. For example, M34 was copolymerized with BMA to form a linear polymer with Mn values of 17.0–21.0 kg mol−1 (Đ = 1.36–1.63) (Fig. 32).143 The disulfide bonds within the poly(BMA-co-M34) were then cleaved and recombined using UV irradiation to provide a cross-linked network. In addition, monomer M35 was subjected to cationic polymerization of the alkene bonds to provide linear polymers containing pendant disulfide units in the side chain (Mn = 1.3 kg mol−1, Đ = 2.5).144 Conversely, when M35 was subjected to radical photopolymerization, branched macromolecules were formed due to the ability of the reactive functionalities to undergo both radical polymerization and thiol–ene coupling.
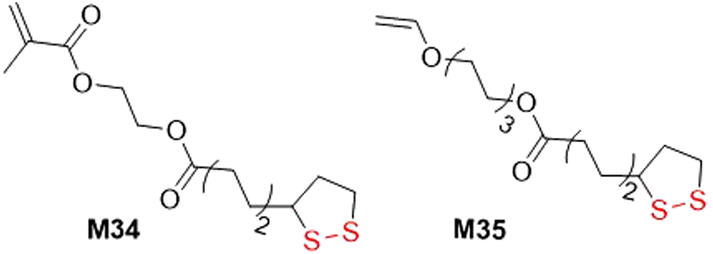 | ||
| Fig. 32 Orthogonal monomers with methacrylate/vinyl and 1,2-dithiolane functionality. | ||
5.2 Radical ring-opening polymerization of the disulfide bond – radically initiated
The polymerization of 1,2-dithiolanes by free-radical initiation has only been modestly reported since it displays several limitations. The radical-initiated homopolymerization of disulfide monomers is reported troublesome, while copolymerization with vinyl comonomers is more efficient, and indeed some reports have been published.145 However, disulfide linkages can only be incorporated into a polymer backbone when two disulfide-containing monomers are added in succession during propagation, and conversely, only monosulfides are installed when a disulfide monomer is preceded or followed by a vinyl monomer, making this methodology less attractive. In 1953, M37 was copolymerized with vinyl acetate (VAc)146 or styrene147 to produce polymers with disulfide linkages in the polymer backbone (Fig. 33). Then, Endo and coworkers subjected M30 to copolymerizations with styrene, acrylonitrile, methyl acrylate, VAc, and MMA in the presence of AIBN, with disulfide monomer feed ratios of 15 mol%.145 Copolymerization was reported to occur in all cases, except for the polymerization with MMA which provided only polyMMA homopolymer, attributed to steric hindrance. For the copolymerization with VAc, the final content of M30 was enriched compared to the feed, with the opposite true for the other monomers.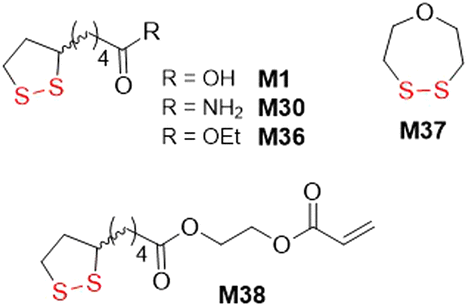 | ||
| Fig. 33 Disulfide monomers in radical ring-opening polymerization induced with a radical initiator. | ||
Tang and Tsarevsky selected specific monomers able to form radicals that were reactive toward 1,2-dithiolane monomers.148 Thereby, copolymerizations of equimolar mixtures of ethyl acrylate (EA) and M1 or M36 to fabricate poly(EA-co-M1) and poly(EA-co-M36), respectively, containing significant quantities of disulfide bonds were performed (Fig. 34).149 Moreover, orthogonal monomer M38 also yielded polymer with disulfide bonds within the polymer backbone. After treatment with DTT, partial degradation was observed alongside an increase in the molecular weight, attributed to thiol–ene reactions between thiol radicals and pendant vinyl groups.
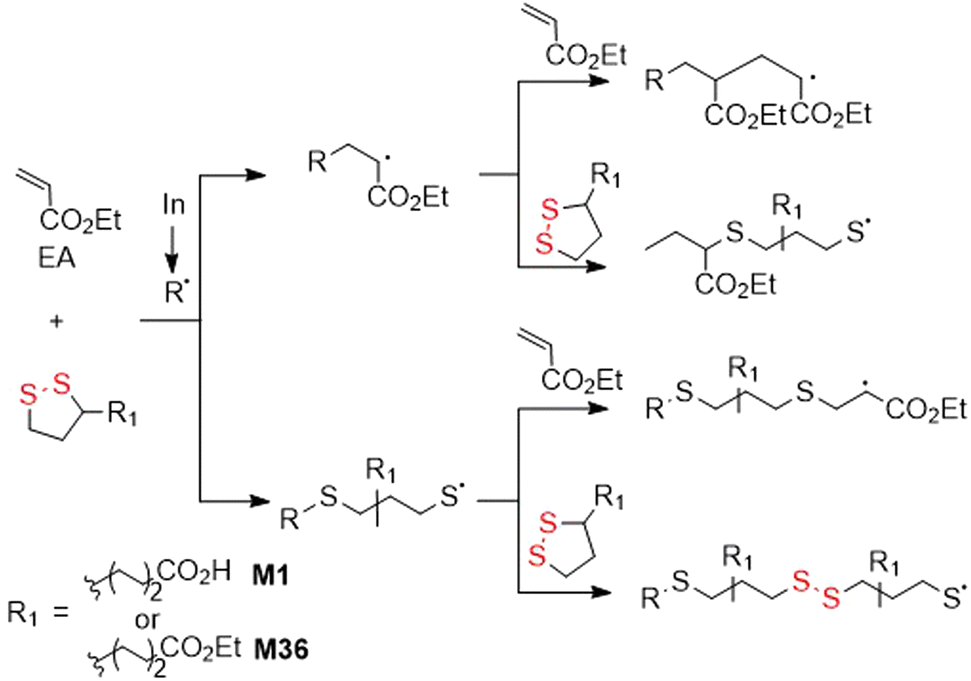 | ||
| Fig. 34 Copolymerization of EA with M1 or M36. | ||
Recently, it was demonstrated that M36 can undergo radical polymerization in bulk or in solution with limited conversion which was lowered further with a rise in temperature or dilution.149 Consequently, it was established that there exists for M36 a monomer–polymer equilibrium with a ceiling temperature of 139 °C.
5.3 Ring-opening polymerization of the disulfide bond – thiolate initiated
The thiolate-initiated anionic ROP of disulfide-containing cyclic monomers is based upon the capability of thiols to act as a nucleophile, breaking the disulfide bond to initiate propagation (Fig. 35). This process can be thermodynamically controlled since the thiolate-disulfide exchange is reversible. All reports within the literature relate to 5- and 6- membered cyclic monomers. Endo and co-workers reported that small amounts of benzyl mercaptan (ca. 0.8 mol%) added to the bulk polymerization of M28 at 80 °C resulting in decreased yields from 84 to 3% when compared to bulk polymerization without thiol initiator.133,134 More versatile studies regarding the incorporation of thiolate initiators were performed using lipoic and asparagusic acid derivatives. In surface-initiated polymerizations, Matile et al. obtained poly(disulfide)s from 1,2-dithiolane monomers M39–M44 (Fig. 35).150,151 In these studies, N-acetyl-l-cysteine methyl ester (Ac-Cys-OMe) and a variety of fluorescent thiols were used as initiators, and iodoacetamides as terminators. Thiol-initiated homopolymerizations were straightforward in terms of control and optimization. Polymerizations of M39 with Ac-Cys-OMe performed in aqueous solution led to polymer with an Mn = 34.3 kg mol−1 (Đ = 1.83) in less than 5 minutes. In addition, the polymerization of M42 and M43 was reported troublesome as these monomers are highly reactive and readily polymerize without an initiator. Cellular uptake studies demonstrated that these cell-penetrating polymers can reach the cytosol of HeLa cells and depolymerize, releasing an active payload.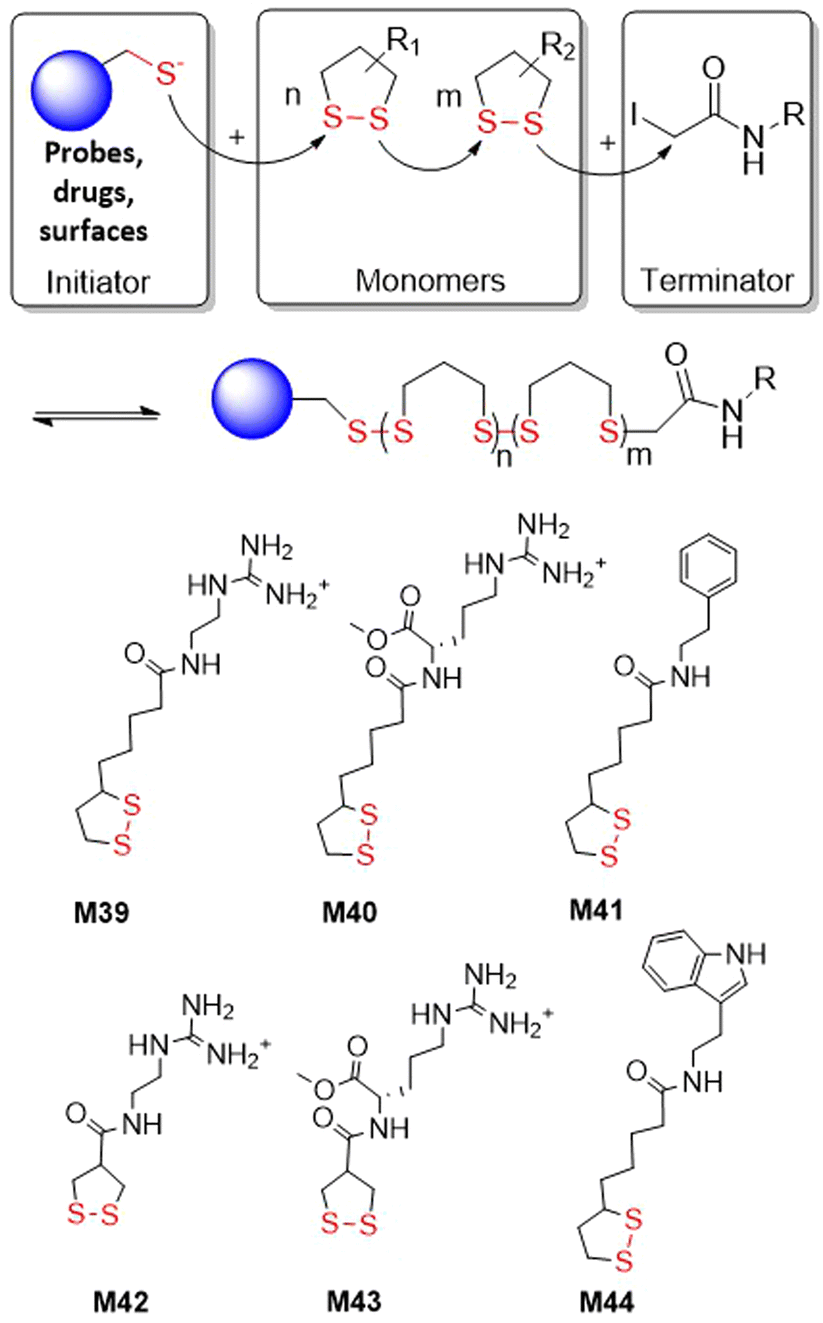 | ||
| Fig. 35 Polymerization from 1,2-dithiolane monomers M39–M44. | ||
Waymouth et al. studied kinetic and thermodynamic differences in the thiol-initiated ring-opening polymerization of 1,2-dithiolanes in regard to the role played by substituents (Fig. 36).152 The methyl ester of methyl substituted asparagusic acid (M45) and methyl lipoate (M46) were compared by benzyl mercaptan-initiated polymerization. It was demonstrated that the polymerization is completely reversible, and that conversion is dictated by a thermodynamic polymerization–depolymerization equilibria. Due to high equilibrium monomer concentration [M]eq values, a high initial dithiolane concentration ([M]0 > [M]eq) was required for ring-opening polymerization to occur. Additionally, it was established that equilibrium constants (Keq) in the ring-opening polymerization of M46 was 3.2 higher than for the same process of M45. Based on the observed rate constant it was determined that the propagation rate of M45 is ca. 4.5 faster, and that the depropagation rate is ca. 14 faster, than M46.
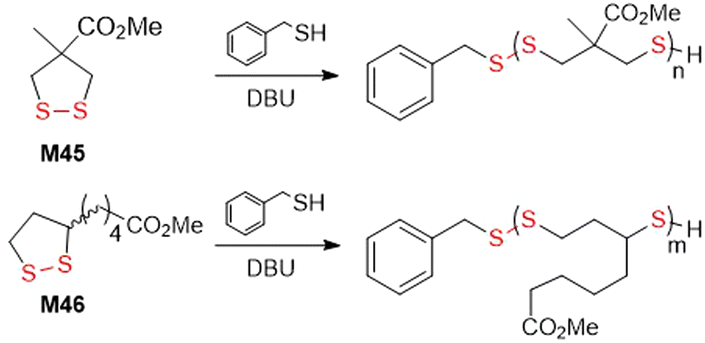 | ||
| Fig. 36 Benzyl mercaptan initiated polymerization of 1,2-dithiolanes M45 an M46. | ||
In 2019, Moore and coworkers reported a topology-controlled polymerization of M46, based upon the structure of the thiol initiator (Fig. 37).153 It was reported that predominantly cyclic products were obtained in polymerizations performed using thiophenol initiators, while prominently linear products were obtained for alkyl thiolate-initiated polymerizations. Using a thiophenol initiator with a monomer to initiator ratios of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively, polymer with an Mn = 22–65 kg mol−1 (Đ = ca. 1.4) could be obtained, while applying a 5000:1 ratio provided polymer with an Mn value as high as 630 kg mol−1 (Đ = 1.27). In polymerizations applying alkyl thiols, products characterized by an Mn of 15–18 kg mol−1 (Đ = ca. 1.4) were produced. Polymerizations performed using (R)-M46 demonstrated a non-regioselectivity of the ring-opening process. Moreover, the kinetics of the polymerization of M46 turned out to be highly dependent upon the choice of base.
:1, respectively, polymer with an Mn = 22–65 kg mol−1 (Đ = ca. 1.4) could be obtained, while applying a 5000:1 ratio provided polymer with an Mn value as high as 630 kg mol−1 (Đ = 1.27). In polymerizations applying alkyl thiols, products characterized by an Mn of 15–18 kg mol−1 (Đ = ca. 1.4) were produced. Polymerizations performed using (R)-M46 demonstrated a non-regioselectivity of the ring-opening process. Moreover, the kinetics of the polymerization of M46 turned out to be highly dependent upon the choice of base.
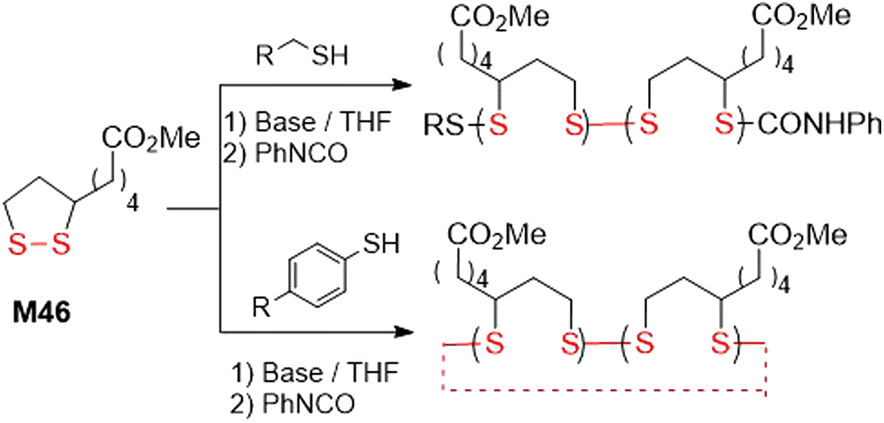 | ||
| Fig. 37 Copolymerization of M46 initiated with aryl and alkyl thiols. | ||
Lu and coworkers performed a rapid and controlled ROP of water-soluble 1,2-dithiolanes M47–M49 initiated by green fluorescence protein (GFP) at temperatures below 0 °C (Fig. 38).154 Low polymerization temperatures were reported necessary to address problems relating to high [M]eq value, side reactions, and a loss of protein function. For cryo-polymerization (from 0 to −30 °C), high initiation efficiency of up to 95% was obtained at pH ≥6.5. Polymers with increasing Mn values were obtained with a decrease in polymerization temperature to −30 °C and a rise in pH value to 7.5–8.5. It was reported that a protein-polymer conjugate of M49 with an Mn = 55 kg mol−1 could be obtained after 90 min at −30 °C.
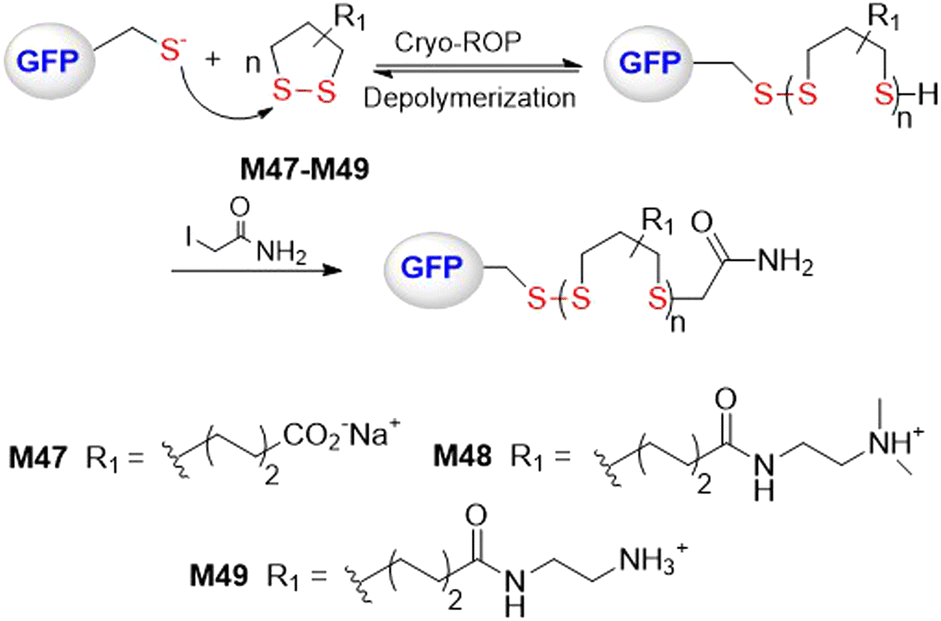 | ||
| Fig. 38 ROP of water-soluble 1,2-dithiolanes M47–M49 initiated by green fluorescence protein (GFP) at low temperatures. | ||
In addition to ROMP (Fig. 28), Schlaad and coworkers also performed the thiolate-initiated polymerization of monomer M24avia the metathesis of its disulfide group (Fig. 39A).122 In a polymerization initiated with methyl thioglycolate and triethylamine, poly(M24a)′ with an Mw = 53–60 kg mol−1 (Đ = 1.8) was obtained. Reaction equilibrium was achieved in less than 5 minutes, with a monomer conversion of ca. 75%. The chemical structures of polyM24a and polyM24a′ were identical (with the exception of the end-group), with a similar distribution of cis/trans isomers. Kinetic investigations confirmed that both ring-opening polymerizations were entropy driven.123 However, while the disulfide metathesis pathway enabled the synthesis of polymers with Mw values up to 180 kg mol−1, olefin metathesis yielded polymers with a maximum Mw value of only ∼70 kg mol−1. In addition, an analogous polymer polyM50, with an amide group in the place of the ester moiety, was obtained from monomer M50 with an Mw = 44 kg mol−1 (Đ = 3.5) and monomer conversion of 87% after 1 h (Fig. 39B).
 | ||
| Fig. 39 (A) Polymerization of L-cysteine derived monomer M24a via disulfide bond metathesis; (B) polymerization of monomer M50 via disulfide bond metathesis. | ||
Polydisulfide-based covalent adaptable liquid crystal networks (CA-LCNs) were obtained from lipoic acid derived monomers M51/M52via thiolate-initiated ROP (Fig. 40).155 Thus, equimolar mixtures of monomers were copolymerized in the presence of 1,6-hexanedithiol and TBD to fabricate self-healing polydisulfide films that displayed reversible shape programmability. In addition, the polymer underwent effective depolymerization into monomer, followed by repolymerization, thereby demonstrating full chemically recyclable behaviour. A thiolate-initiated reversible ROP was also applied for the hydrogelation of polymers containing asparagusic or lipoic acid-derived pendants obtained from monomers M53 and M54 (Fig. 41).156 These cyclic carbonates were subjected to ROP using PEG (14 kg mol−1) as a divalent macroinitiator to fabricate amphiphilic ABA-type triblock polymers with an Mn = 16.9–18.2 kg mol−1 (Đ = 1.13–1.18). These polymers then underwent self-assembly in aqueous solution to form flower-like micelles, with the asparagusic acid-derived hydrogels displaying greater dynamic properties, adaptability, and self-healing than those derived from lipoic acid.
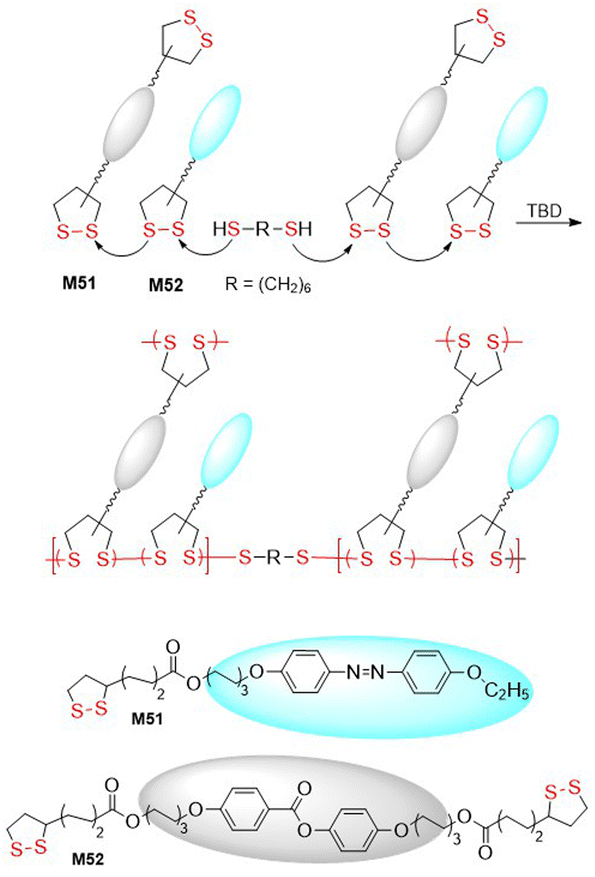 | ||
| Fig. 40 Synthesis of CA-LCNs from monomers M51 and M52. | ||
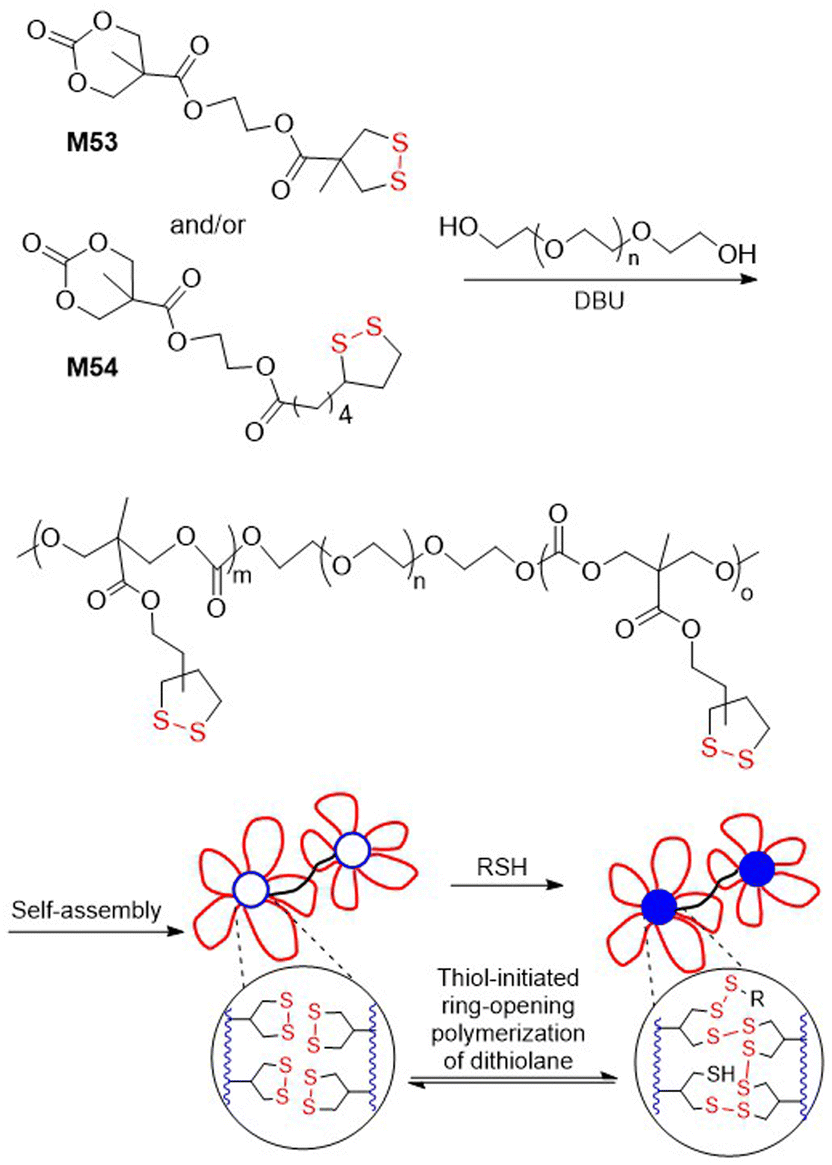 | ||
| Fig. 41 Polymerization of M53 and M54 followed by self-assembly and thiolate-initiated gelation. | ||
6. Common methods for the preparation of disulfide-containing monomers
Disulfide bonds are typically formed by the oxidation of sulfhydryl (-SH) groups (Fig. 42A) or by disulfide-thiol exchange (Fig. 42B). Alternative more specific methods instead apply substrates that contain an equivalent of an “–S+” moiety, for example sulfenyl chlorides or Bunte salts (Fig. 42C).157,158 Typically, the first two methodologies were applied for the synthesis of the monomers within this article. Moreover, the disulfide bond formation was typically performed as the final step in the synthetic pathway, or more rarely, in an earlier step which was followed by simple transformations. | ||
| Fig. 42 Common methods for the synthesis of disulfide-containing molecules. | ||
An overview of several common approaches for the synthesis of disulfide-containing monomers is herewith provided (Fig. 43). An oxidation approach was applied for the synthesis of thioctic acid M1 obtained from compound 55 in the presence of iron chloride,159 or iodine and potassium iodide159,160 (Fig. 43A). Monomer M20 was obtained by the air-oxidation of compound 56 mediated by CsF-impregnated Celite (Fig. 43B).161 Several disulfide-containing monomers were derived from compounds 58 or 60, which are obtained by oxidation of 57 and 59, respectively (Fig. 43C).162,163 Acylation of the resulting diol (58) or diamine (60) with acryloyl 61164,165 and methacryloyl 62105,166 chlorides produces bifunctional vinyl monomers M11–M14, which have been applied for cross-linking during FRP (Fig. 43D). Furthermore, by the reaction of 58 with bis-acid chloride 63, macrocyclic monomer M15 was obtained in 15% yield.110 The application of the same diol (58) in a reaction with diphenylcarbonate 64 in the presence of lipase enzyme provided monomer M19 in 63% yield.118
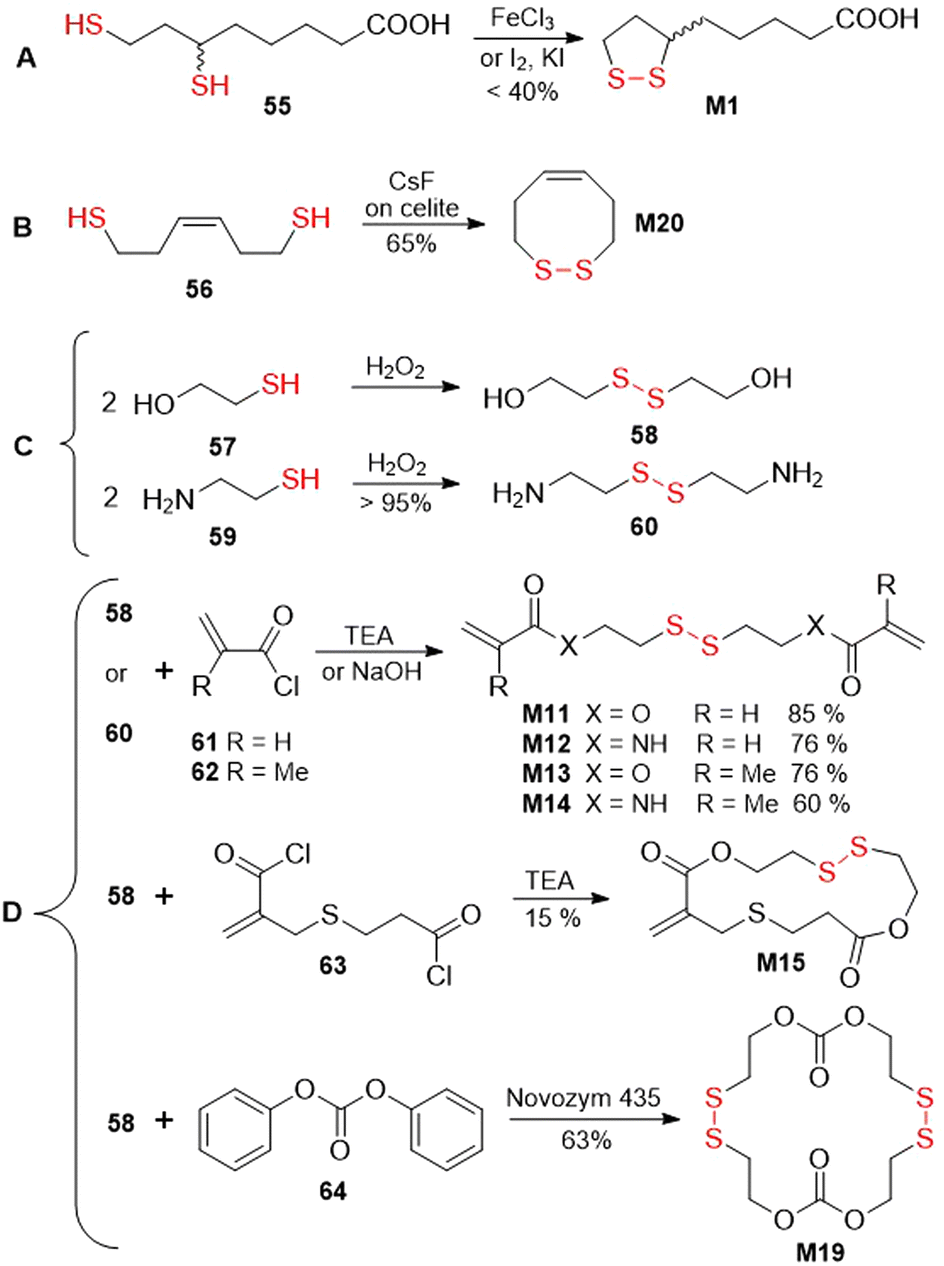 | ||
| Fig. 43 Oxidation approach used in synthesis of disulfide-containing monomers. | ||
Alternatively, a disulfide-thiol exchange approach was applied for the synthesis of pyridyl disulfide functionalized monomers M3–M6 (Fig. 44A). In this way, 2,2′-dipyridyldisulfide 65 was subjected to substitution with 2-hydroxyethanethiol 57 or 2-mercaptoethylamine 59 to provide compounds 66167 or 67,168 respectively, with concurrent release of pyridine-2-thione. These intermediates were then reacted with acryloyl (61)44,49 or methacryloyl (62)16,56 chlorides to efficiently provide monomers M3–M6. Similarly, for the synthesis of monomer M9, compound 68 was subjected to substitution with compound 69 and the resulting intermediate (70) was reacted with 2-hydroxyethanethiol (57) to provide disulfide 71, which was finally acylated with acryloyl chloride (61) to provide monomer M9 (Fig. 44B).76
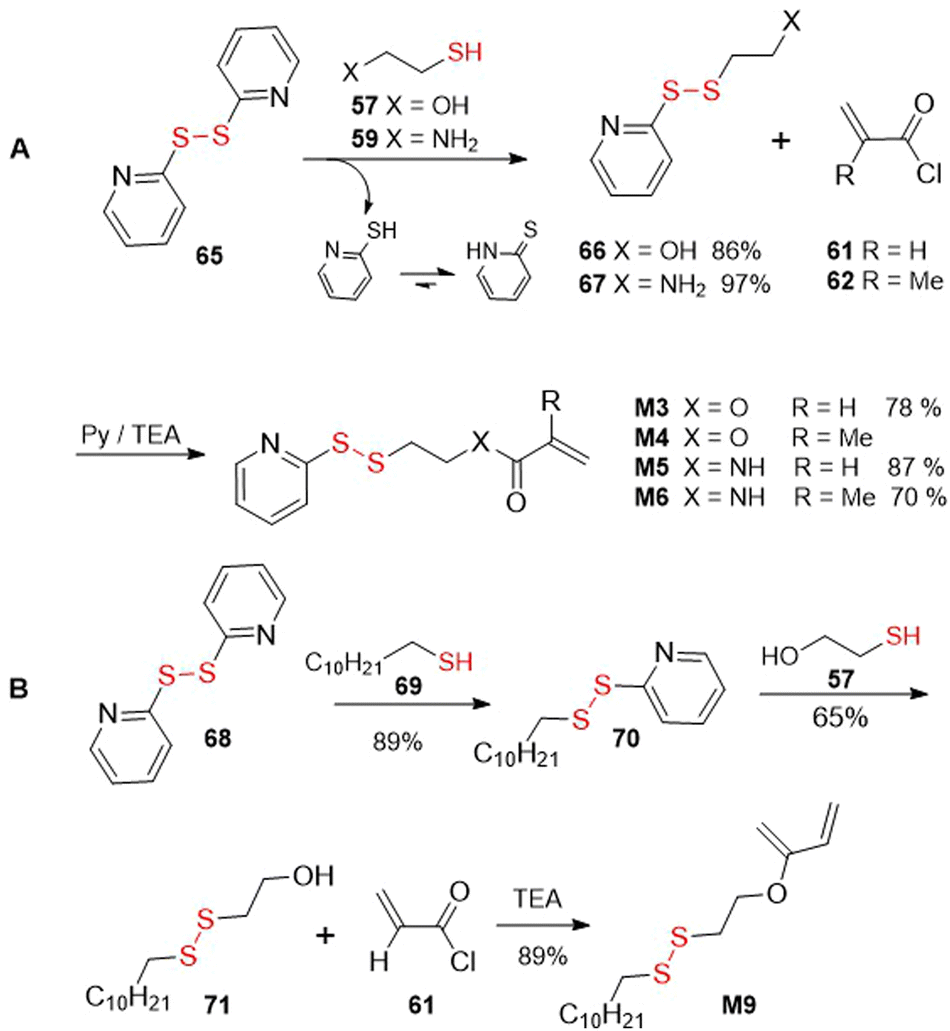 | ||
| Fig. 44 Disulfide-thiol exchange approach in synthesis of disulfide-containing monomers. | ||
The final alternative approach involves the reaction of thiols with various reagents delivering equivalents of “–S+”, leading to asymmetrical disulfides. For example, the thiol group of L-cysteine (73) was ligated with sulfenyl chlorides 72a–c to provide cysteine derivatives 74a–c in good yields, which were then converted into the corresponding N-carboxyanhydrides M16a–c by their reaction with triphosgene (Fig. 45).113
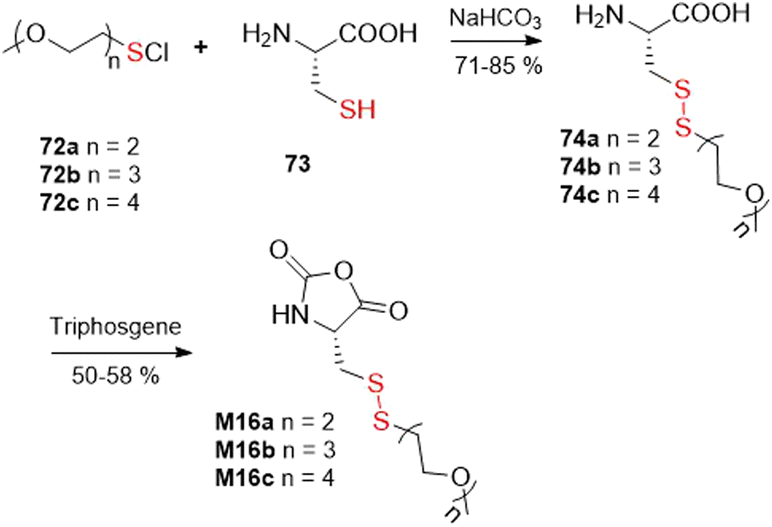 | ||
| Fig. 45 Approach utilizing the equivalent of an “–S+” moiety for the synthesis of disulfide-containing monomers. | ||
7. Conclusion and outlook
Within this review article we have attempted to provide a comprehensive overview of the diversity of disulfide-containing polymers that can be obtained by chain-growth polymerization by the application of disulfide-containing monomers. As the installation of disulfide moieties into polymers furnishes them with highly useful properties that greatly extend their spectrum of applications, it is of great importance to develop new effective ways to introduce disulfide bonds into polymers. The transfer of disulfide moieties from monomer to (co)polymer is possible via a variety of chain-growth polymerization methodologies. These technologies include: (a) radical polymerization by which disulfide bonds can be introduced into pendant groups by FRP, or into the polymer backbone by rROP, (b) ionic ROP performed using cyclic N-carboanhydrides, carbonates and lactones, or (c) the ROMP of cyclic disulfide-containing olefins. Moreover, the polymerization of various compounds by the direct ring-opening of the disulfide-bond by an assortment of triggers including thermal, light, anionic and radicals has provided dynamic materials with high levels of chemical recyclability.Many of the disulfide-containing copolymers discussed herein have been obtained from monomers equipped with active drug molecules, or have been chemically modified with active compounds, and/or conjugated or complexed with proteins and nucleic acid. These polymers have been applied for the fabrication of various nanoarchitectures that have been demonstrated to have increased selectivity, biocompatibility, or transfection ability. The sensitivity of disulfide bonds to reducing conditions has been extensively exploited in research relating to the targeted delivery of active molecules, designed to take advantage of variations in glutathione levels between normal and abnormal cells. In addition, by the application of 1,2-dithiolane monomers, new smart materials with self-healing capabilities, stimuli-responsiveness, adaptiveness, and recyclability have been reported. The effective incorporation of disulfide bonds into a polymer backbone also offers the possibility to obtain new (bio)degradable polymers.
However, the field of disulfide-containing polymers still remains in the initial stages of exploration, and at present, exists predominantly on the bench scale, often facing complications involving tedious monomer synthesis, problematic polymerization, and low monomer incorporation. Nevertheless, the exploration of disulfide-containing polymers remains an extremely active field, and we expect that the future will be a theatre of important innovations regarding the synthesis of new disulfide-containing monomer building blocks that are capable of providing next-generation smart materials that incorporate improved stimuli-responsiveness, degradability, and recyclability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted as a part of the International Research Agendas PLUS programme of the Foundation for Polish Science, co-financed by the European Union under the European Regional Development Fund (MAB PLUS/2019/11).References
- A. G. Gaydon, Dissociation energies and spectra of biatomic molecules, Chapman and Hall, London, 2nd edn, 1953 Search PubMed.
- D. Fass and C. Thorpe, Chem. Rev., 2018, 118, 1169–1198 CrossRef CAS PubMed.
- S. C. Mitchell and R. H. Waring, Phytochemistry, 2014, 97, 5–10 CrossRef CAS PubMed.
- L. Teuber, Sulfur Rep., 1990, 9, 257–333 CrossRef CAS.
- C.-S. Jiang, W. E. G. Müller, H. C. Schröder and Y.-W. Guo, Chem. Rev., 2011, 112, 2179–2207 CrossRef.
- K. Kishore and K. Ganesh, Adv. Polym. Sci., 1995, 121, 81–121 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97 RSC.
- B. van Lierop, S. C. Ong, A. Belgi, C. Delaine, S. Andrikopoulos, N. L. Haworth, J. G. Menting, M. C. Lawrence, A. J. Robinson and B. E. Forbes, Sci. Rep., 2017, 7, 17239 CrossRef.
- B. Sui, C. Cheng and P. Xu, Adv. Ther., 2019, 2, 1900062 CrossRef CAS.
- L. Wang, J. Kristensen and D. E. Ruffner, Bioconjugate Chem., 1998, 9, 749–757 CrossRef CAS PubMed.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105–120 CrossRef CAS PubMed.
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2005, 101, 47–58 CrossRef CAS.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS PubMed.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106–3113 CrossRef CAS PubMed.
- J. H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS.
- J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS.
- S. Jiwpanich, J. H. Ryu, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 10683–10685 CrossRef CAS.
- D. C. González-Toro, J. H. Ryu, R. T. Chacko, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2012, 134, 6964–6967 CrossRef.
- L. Li, J.-H. Ryu and S. Thayumanavan, Langmuir, 2012, 29, 50–55 CrossRef PubMed.
- N. M. Matsumoto, D. C. González-Toro, R. T. Chacko, H. D. Maynard and S. Thayumanavan, Polym. Chem., 2013, 4, 2464–2469 RSC.
- X. Liu, D. Hu, Z. Jiang, J. Zhuang, Y. Xu, X. Guo and S. Thayumanavan, Macromolecules, 2016, 49, 6186–6192 CrossRef CAS.
- J.-H. Ryu, S. Bickerton, J. Zhuang and S. Thayumanavan, Biomacromolecules, 2012, 13, 1515–1522 CrossRef CAS.
- L. Li, K. Raghupathi, C. Yuan and S. Thayumanavan, Chem. Sci., 2013, 4, 3654–3660 RSC.
- L. Li and S. Thayumanavan, Langmuir, 2014, 30, 12384–12390 CrossRef CAS PubMed.
- C. Yuan, K. Raghupathi, B. C. Popere, J. Ventura, L. Dai and S. Thayumanavan, Chem. Sci., 2014, 5, 229–234 RSC.
- J. Ventura, S. J. Eron, D. C. González-Toro, K. Raghupathi, F. Wang, J. A. Hardy and S. Thayumanavan, Biomacromolecules, 2015, 16, 3161–3171 CrossRef CAS PubMed.
- K. Dutta, D. Hu, B. Zhao, A. E. Ribbe, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 5676–5679 CrossRef CAS PubMed.
- Z. Jiang, W. Cui, P. Prasad, M. A. Touve, N. C. Gianneschi, J. Mager and S. Thayumanavan, Biomacromolecules, 2018, 20, 435–442 CrossRef.
- E. F. Crownover, A. J. Convertine and P. S. Stayton, Polym. Chem., 2011, 2, 1499–1504 RSC.
- K. C. R. Bahadur and P. Xu, Adv. Mater., 2012, 24, 6479–6483 CrossRef CAS PubMed.
- K. C. R. Bahadur, B. Thapa and P. Xu, Mol. Pharmacol., 2012, 9, 2719–2729 CrossRef.
- K. C. R. Bahadur, V. Chandrashekaran, B. Cheng, H. Chen, M. M. O. Peña, J. Zhang, J. Montgomery and P. Xu, Mol. Pharmacol., 2014, 11, 1897–1905 CrossRef.
- H. He, A. W. Cattran, T. Nguyen, A. L. Nieminen and P. Xu, Biomaterials, 2014, 35, 9546–9553 CrossRef CAS.
- B. Cheng and P. Xu, Toxins, 2020, 12, 582–594 CrossRef CAS PubMed.
- H. He, D. Altomare, U. Ozer, H. Xu, K. Creek, H. Chen and P. Xu, Biomater. Sci., 2015, 4, 115–120 RSC.
- E. Markoutsa and P. Xu, Mol. Pharmacol., 2017, 14, 1591–1600 CrossRef CAS.
- H. He, E. Markoutsa, J. Li and P. Xu, Acta Biomater., 2018, 68, 113–124 CrossRef CAS.
- H. Asadi and S. Khoee, Int. J. Pharmacol., 2016, 511, 424–435 CrossRef CAS PubMed.
- A. W. Jackson and D. A. Fulton, Macromolecules, 2012, 45, 2699–2708 CrossRef CAS.
- H. Palacio, F. Otálvaro, L. F. Giraldo, G. Ponchel and F. Segura-Sánchez, Chem. Pharm. Bull., 2017, 65, 1132–1143 CrossRef CAS.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal and J. Ji, Polym. Chem., 2014, 5, 4061–4068 RSC.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal and J. Ji, Macromol. Chem. Phys., 2014, 215, 1848–1854 CrossRef CAS.
- H. S. Han, K. Y. Choi, H. Ko, J. Jeon, G. Saravanakumar, Y. D. Suh, D. S. Lee and J. H. Park, J. Controlled Release, 2015, 200, 158–166 CrossRef CAS PubMed.
- J. G. Schellinger, J. A. Pahang, R. N. Johnson, D. S. H. Chu, D. L. Sellers, D. O. Maris, A. J. Convertine, P. S. Stayton, P. J. Horner and S. H. Pun, Biomaterials, 2013, 34, 2318–2326 CrossRef CAS.
- Y. Sugawara, T. Tamaki, H. Ohashi and T. Yamaguchi, Soft Matter, 2013, 9, 3331–3340 RSC.
- H. Chen, H. Zou, H. J. Paholak, M. Ito, W. Qian, Y. Che and D. Sun, Polym. Chem., 2014, 5, 2768–2773 RSC.
- Z. Song, Y. Xu, W. Yang, L. Cui, J. Zhang and J. Liu, Eur. Polym. J., 2015, 69, 559–572 CrossRef CAS.
- Y. Yang, Y. Li, Q. Lin, C. Bao and L. Zhu, ACS Macro Lett., 2016, 5, 301–305 CrossRef CAS PubMed.
- Z. Liu, Q. Lin, Y. Sun, T. Liu, C. Bao, F. Li and L. Zhu, Adv. Mater., 2014, 26, 3912–3917 CrossRef CAS PubMed.
- H. Peng, K. Rübsam, X. Huang, F. Jakob, M. Karperien, U. Schwaneberg and A. Pich, Macromolecules, 2016, 49, 7141–7154 CrossRef CAS.
- X. Ji, J. Liu, L. Liu and H. Zhao, Colloids Surf., B, 2016, 148, 41–48 CrossRef CAS PubMed.
- Y. Ju, C. Xing, D. Wu, Y. Wu, L. Wang and H. Zhao, Chem. – Eur. J., 2017, 23, 3366–3374 CrossRef CAS.
- L. Zhang, D. Zhang, Y. Yang and Y. Zhang, Langmuir, 2021, 37, 3950–3959 CrossRef CAS PubMed.
- X. Ji, L. Liu and H. Zhao, Polym. Chem., 2017, 8, 2815–2823 RSC.
- J. Lee, T. Zhao, D. J. Peeler, D. C. Lee, T. J. Pichon, D. Li, K. M. Weigandt, P. J. Horner, L. D. Pozzo, D. L. Sellers and S. H. Pun, Soft Matter, 2020, 16, 3762–3768 RSC.
- Z. Deng, J. Hu, S. Liu, Z. Deng, J. Hu and S. Liu, Macromol. Rapid Commun., 2020, 41, 1900531 CrossRef CAS.
- R. Zhang, T. Nie, Y. Fang, H. Huang and J. Wu, Biomacromolecules, 2022, 23, 1–19 CrossRef PubMed.
- A. Kock, K. Zuwala, A. A. A. Smith, P. Ruiz-Sanchis, B. M. Wohl, M. Tolstrup and A. N. Zelikin, Chem Commun., 2014, 50, 14498–14500 RSC.
- A. A. A. Smith, B. M. Wohl, M. B. L. Kryger, N. Hedemann, C. Guerrero-Sanchez, A. Postma and A. N. Zelikin, Adv. Healthcare Mater., 2014, 3, 1404–1407 CrossRef CAS.
- C. F. Riber, A. A. A. Smith and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 1887–1890 CrossRef CAS.
- P. Ruiz-Sanchis, B. M. Wohl, A. A. A. Smith, K. Zuwala, J. Melchjorsen, M. Tolstrup and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 65–68 CrossRef CAS.
- K. Zuwala, A. A. A. Smith, M. Tolstrup and A. N. Zelikin, Chem. Sci., 2016, 7, 2353–2358 RSC.
- M. Danial, A. H. F. Andersen, K. Zuwala, S. Cosson, C. F. Riber, A. A. A. Smith, M. Tolstrup, G. Moad, A. N. Zelikin and A. Postma, Mol. Pharmacol., 2016, 13, 2397–2410 CrossRef CAS.
- C. F. Riber, T. M. Hinton, P. Gajda, K. Zuwala, M. Tolstrup, C. Stewart and A. N. Zelikin, Mol. Pharmacol., 2017, 14, 234–241 CrossRef CAS.
- K. Zuwala, C. F. Riber, K. B. Løvschall, A. H. F. Andersen, L. Sørensen, P. Gajda, M. Tolstrup and A. N. Zelikin, J. Controlled Release, 2018, 275, 53–66 CrossRef CAS PubMed.
- A. H. F. Andersen, C. F. Riber, K. Zuwala, M. Tolstrup, F. Dagnæs-Hansen, P. W. Denton and A. N. Zelikin, ACS Macro Lett., 2018, 7, 587–591 CrossRef CAS PubMed.
- C. K. Frich, F. Krüger, R. Walther, C. Domar, A. H. F. Andersen, A. Tvilum, F. Dagnæs-Hansen, P. W. Denton, M. Tolstrup, S. R. Paludan, J. Münch and A. N. Zelikin, J. Controlled Release, 2019, 294, 298–310 CrossRef CAS.
- M. Danial, S. Telwatte, D. Tyssen, S. Cosson, G. Tachedjian, G. Moad and A. Postma, Polym. Chem., 2016, 7, 7477–7487 RSC.
- Z.-H. Peng, Y. Xie, Y. Wang, J. Li and D. Oupický, Mol. Pharmacol., 2017, 14, 1395–1404 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2016, 17, 2992–2999 CrossRef CAS.
- P. Srikamut, T. Phakkeeree, F. Seidi, S. Iamsaard and D. Crespy, ACS Appl. Polym. Mater., 2021, 3, 5425–5433 CrossRef CAS.
- J. H. Ryu, R. Roy, J. Ventura and S. Thayumanavan, Langmuir, 2010, 26, 7086–7092 CrossRef CAS PubMed.
- Q. Zhang, S. Aleksanian, S. M. Noh and J. K. Oh, Polym. Chem., 2013, 4, 351–359 RSC.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 1455–1460 CrossRef CAS.
- S. Durie, K. Jerabek, C. Mason and D. C. Sherrington, Macromolecules, 2002, 35, 9665–9672 CrossRef CAS.
- M. Chisholm, N. Hudson, N. Kirtley, F. Vilela and D. C. Sherrington, Macromolecules, 2009, 42, 7745–7752 CrossRef CAS.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965–977 RSC.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995–6004 CrossRef CAS.
- N. V. Tsarevsky, K. Min, N. M. Jahed, H. Gao and K. Matyjaszewski, ACS Symp. Ser., 2006, 939, 184–200 CrossRef CAS.
- W. Li, K. Matyjaszewski, K. Albrecht and M. Möller, Macromolecules, 2009, 42, 8228–8233 CrossRef CAS.
- W. Li, J. A. Yoon and K. Matyjaszewski, J. Am. Chem. Soc., 2010, 132, 7823–7825 CrossRef CAS PubMed.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- M. Lamson, Y. Epshtein-Assor, M. S. Silverstein and K. Matyjaszewski, Polymer, 2013, 54, 4480–4485 CrossRef CAS.
- V. Metri, A. Louhichi, J. Yan, G. P. Baeza, K. Matyjaszewski, D. Vlassopoulos and W. J. Briels, Macromolecules, 2018, 51, 2872–2886 CrossRef CAS.
- Y. Wang and K. Matyjaszewski, React. Funct. Polym., 2022, 170, 105104 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- L. Wang, C. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566–1570 CrossRef CAS.
- C.-D. Vo, J. Rosselgong, S. P. Armes and N. C. Billingham, Macromolecules, 2007, 40, 7119–7125 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2009, 42, 939–945 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- D. L. Popescu and N. V. Tsarevsky, Aust. J. Chem., 2012, 65, 28–34 CrossRef CAS.
- H. Tang and N. v. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3683–3693 CrossRef CAS.
- N. V. Tsarevsky, J. Huang and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2021, 59, 675–684 Search PubMed.
- Y. Gao, V. I. Böhmer, D. Zhou, T. Zhao, W. Wang and J. M. J. Paulusse, J. Controlled Release, 2016, 244, 375–383 CrossRef CAS.
- T. Zhao, H. Zhang, B. Newland, A. Aied, D. Zhou and W. Wang, Angew. Chem., 2014, 126, 6209–6214 CrossRef.
- B. Newland, A. Aied, A. v Pinoncely, Y. Zheng, T. Zhao, H. Zhang, R. Niemeier, E. Dowd, A. Pandit and W. Wang, Nanoscale, 2014, 6, 7526–7533 RSC.
- Y. J. Pan, Y. Y. Chen, D. R. Wang, C. Wei, J. Guo, D. R. Lu, C. C. Chu and C. C. Wang, Biomaterials, 2012, 33, 6570–6579 CrossRef CAS.
- M. Zhang, S. Asghar, C. Tian, Z. Hu, Q. Ping, Z. Chen, F. Shao and Y. Xiao, Carbohydr. Polym., 2021, 253, 117194 CrossRef CAS PubMed.
- Y. Dai, Q. Li, S. Zhang, S. Shi, Y. Li, X. Zhao, L. Zhou, X. Wang, Y. Zhu and W. Li, J. Drug Delivery Sci. Technol., 2021, 64, 102650 CrossRef CAS.
- M. H. Loghmani, A. F. Shojaie and S. A. Hosseini, J. Ind. Eng. Chem., 2021, 96, 98–108 CrossRef CAS.
- Y. S. Lin, Y. L. Huang, W. F. Lee and C. H. Lin, J. Chin. Chem. Soc., 2014, 61, 945–952 CrossRef CAS.
- Q. B. Chen and Y. Z. You, Chem. Lett., 2015, 44, 677–679 CrossRef CAS.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS.
- L. P. D. Ratcliffe, C. Couchon, S. P. Armes and J. M. J. Paulusse, Biomacromolecules, 2016, 17, 2277–2283 CrossRef CAS PubMed.
- J. Du, B. Choi, Y. Liu, A. Feng and S. H. Thang, Polym. Chem., 2019, 10, 1291–1298 RSC.
- Y. Ma, X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2014, 47, 4684–4689 CrossRef CAS.
- W. Chen, Y. Zou, J. Jia, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2013, 46, 699–707 CrossRef CAS.
- S. Kim, K. I. Wittek and Y. Lee, Chem. Sci., 2020, 11, 4882–4886 RSC.
- B. Yan, B. Liang, J. Hou, C. Wei, Y. Xiao, M. Lang and F. Huang, Eur. Polym. J., 2020, 123, 109452 CrossRef CAS.
- C. Wei, C. Lian, B. Yan, Y. Xiao, M. Lang and H. Liu, Polym. Chem., 2020, 11, 744–751 RSC.
- C. Wei, Y. Zhang, B. Yan, Z. Du and M. Lang, Chem. – Eur. J., 2018, 24, 789–792 CrossRef CAS PubMed.
- V. B. Purohit, M. Pięta, J. Pietrasik and C. M. Plummer, Polym. Chem., 2022, 13, 4858–4878 RSC.
- C. C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- F. N. Behrendt and H. Schlaad, Polym. Chem., 2017, 8, 366–369 RSC.
- F. N. Behrendt and H. Schlaad, Macromol. Rapid Commun., 2018, 39, 1700735 CrossRef.
- F. N. Behrendt, A. Hess, M. Lehmann, B. Schmidt and H. Schlaad, Polym. Chem., 2019, 10, 1636–1641 RSC.
- L. J. Reed and C. Niu, J. Am. Chem. Soc., 1955, 77, 416–419 CrossRef CAS.
- R. C. Thomas and L. J. Reed, J. Am. Chem. Soc., 1956, 78, 6148–6149 CrossRef CAS.
- J. A. Barltrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76, 4348–4367 CrossRef CAS.
- Y. Ding and A. S. Hay, Macromolecules, 1996, 29, 6386–6392 CrossRef CAS.
- Y. Ding and A. S. Hay, Polymer, 1997, 38, 2239–2244 CrossRef CAS.
- Z. A. Liang, Y. Z. Meng, L. Li, X. S. Du and A. S. Hay, Macromolecules, 2004, 37, 5837–5840 CrossRef CAS.
- Y. Z. Meng, Z. A. Liang, Y. X. Lu and A. S. Hay, Polymer, 2005, 46, 11117–11124 CrossRef CAS.
- J. A. Chandrasiri and C. A. Wilkie, Polym. Degrad. Stab., 1994, 46, 275–284 CrossRef CAS.
- R. Arakawa, T. Watanabe, T. Fukuo and K. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4403–4406 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143–3150 CrossRef CAS.
- K. Endo, T. Shiroi and N. Murata, Polym. J., 2005, 37, 512–516 CrossRef CAS.
- H. Ishida, A. Kisanuki and K. Endo, Polym. J., 2009, 41, 110–117 CrossRef CAS.
- A. Kisanuki, Y. Kimpara, Y. Oikado, N. Kado, M. Matsumoto and K. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5247–5253 CrossRef CAS.
- K. Endo and T. Yamanaka, Macromolecules, 2006, 39, 4038–4043 CrossRef CAS.
- T. Yamanaka and K. Endo, Polym. J., 2007, 39, 1360–1364 CrossRef CAS.
- Y. Nambu, M. H. Acar, T. Suzuki and T. Endo, Makromol. Chem., 1988, 189, 495–500 CrossRef CAS.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, M. Chen and D.-H. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 44860–44867 CrossRef CAS.
- C. Choi, J. L. Self, Y. Okayama, A. E. Levi, M. Gerst, J. C. Speros, C. J. Hawker, J. R. de Alaniz and C. M. Bates, J. Am. Chem. Soc., 2021, 143, 9866–9871 CrossRef CAS PubMed.
- C. Choi, Y. Okayama, P. T. Morris, L. L. Robinson, M. Gerst, J. C. Speros, C. J. Hawker, J. Read de Alaniz and C. M. Bates, Adv. Funct. Mater., 2022, 32, 2200883 CrossRef CAS.
- S. Maes, V. Scholiers and F. E. du Prez, Macromol. Chem. Phys., 2021, 2100445 Search PubMed.
- S. Brännström, M. Johansson and E. Malmström, Biomacromolecules, 2019, 20, 1308–1316 CrossRef PubMed.
- T. Suzuki, Y. Nambu and T. Endo, Macromolecules, 1990, 75, 1579–1582 CrossRef.
- W. H. Stockmayer, R. O. Howard and J. T. Clarke, J. Am. Chem. Soc., 1953, 75, 1756–1757 CrossRef CAS.
- A. V. Tobolsky and B. Baysal, J. Am. Chem. Soc., 1953, 7, 1757 CrossRef.
- H. Tang and N. V. Tsarevsky, Polym. Chem., 2015, 6, 6936–6945 RSC.
- M. Raeisi and N. V. Tsarevsky, J. Polym. Sci., 2021, 59, 675–684 CrossRef CAS.
- E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS.
- G. Gasparini, E. K. Bang, G. Molinard, D. v. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed.
- J. Lu, H. Wang, Z. Tian, Y. Hou and H. Lu, J. Am. Chem. Soc., 2020, 142, 1217–1221 CrossRef CAS.
- S. Huang, Y. Shen, H. K. Bisoyi, Y. Tao, Z. Liu, M. Wang, H. Yang and Q. Li, J. Am. Chem. Soc., 2021, 143, 12543–12551 CrossRef CAS PubMed.
- K. Margulis, X. Zhang, L. Joubert, K. Bruening, C. J. Tassone, R. N. Zare and R. M. Waymouth, Angew. Chem., 2017, 129, 16575–16580 CrossRef.
- D. Witt, Synthesis, 2008, 2491–2509 CrossRef CAS.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS PubMed.
- M. W. Bullock, J. A. Brockman, E. L. Patterson, J. V. Pierce, M. H. von Saltza, F. Sanders and E. L. R. Stokstad, J. Am. Chem. Soc., 1954, 76, 1828–1832 CrossRef CAS.
- Q. F. Soper, W. E. Buting, J. E. Cochran Jr. and A. Pohland, J. Am. Chem. Soc., 1954, 76, 4109–4112 CrossRef CAS.
- E. L. Ruggles and R. J. Hondal, Tetrahedron Lett., 2006, 47, 4281–4284 CrossRef CAS.
- V. Kesavan, D. Bonnet-Delpon and J. P. Bégué, Synthesis, 2000, 223–225 CrossRef CAS.
- A. H. Nathan and M. T. Bogert, J. Am. Chem. Soc., 1941, 63, 2361–2366 CrossRef CAS.
- T. Lovato, V. Taresco, A. Alazzo, C. Sansone, S. Stolnik, C. Alexander and C. Conte, J. Mater. Chem. B, 2018, 6, 6550–6558 RSC.
- Y. S. Lin, Y. L. Huang, W. F. Lee and C. H. Lin, J. Chin. Chem. Soc., 2014, 61, 945–952 CrossRef CAS.
- C. Miao, F. Li, Y. Zuo, R. Wang and Y. Xiong, RSC Adv., 2016, 6, 3013–3019 RSC.
- L. R. Jones, E. A. Goun, R. Shinde, J. B. Rothbard, C. H. Contag and P. A. Wender, J. Am. Chem. Soc., 2006, 128, 6526–6527 CrossRef CAS PubMed.
- F. Gauthier, A. Malher, J. J. Vasseur, C. Dupouy and F. Debart, Eur. J. Org. Chem., 2019, 5636–5645 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |