
(Bipyridine bisphenolate)-aluminum/onium salt pair: a highly active binary catalyst for ring-opening polymerization of lactide with improved thermostability and protic tolerance†
Mingqian
Wang
,
Zhiqiang
Ding
,
Bin
Wang
 * and
Yuesheng
Li
* and
Yuesheng
Li
Tianjin Key Laboratory of Composite & Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300350, China. E-mail: binwang@tju.edu.cn
First published on 16th November 2022
Abstract
Developing aluminum-based catalysts with high thermostability and good protic tolerance in the ring-opening polymerization (ROP) of lactide (LA) remains a great challenge, because of the decomposition of the active species under these conditions. In this contribution, a series of bipyridine bisphenolate aluminum (BpyBph-Al) complexes were synthesized and used to catalyze the ROP of LA with the assistance of an onium salt as a cocatalyst in epoxide. We disclosed the relationship between the structures of (BpyBph)Al and their catalytic performances, and thoroughly investigated the effects of polymerization conditions (including polymerization temperature, catalyst/cocatalyst and monomer/catalyst feed ratios) on LA polymerization behaviors. The (BpyBph)Al/onium binary catalytic system is of particular interest as it exhibits high thermostability and excellent tolerance to a wide range of protonic impurities, compared with the (salen)Al/onium salt. These features allowed a rapid LA polymerization under bulk and melt conditions with a very high feed ratio of [LA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[catalyst pair]:[expoxide] = 5000:1:2. Finally, the chain initiation mechanism was further clarified by in situ NMR experiments. It was shown that a six-coordinated bis(alkoxide) aluminum complex will be generated by the (BpyBph)Al/onium salt in the presence of epoxide, which can be reversibly converted to a five coordinated (BpyBph)Al alkoxide and an alkoxide salt. The cooperative effects between the (BpyBph)Al alkoxide and the alkoxide salt were mainly responsible for rapid polymerization.
:[catalyst pair]:[expoxide] = 5000:1:2. Finally, the chain initiation mechanism was further clarified by in situ NMR experiments. It was shown that a six-coordinated bis(alkoxide) aluminum complex will be generated by the (BpyBph)Al/onium salt in the presence of epoxide, which can be reversibly converted to a five coordinated (BpyBph)Al alkoxide and an alkoxide salt. The cooperative effects between the (BpyBph)Al alkoxide and the alkoxide salt were mainly responsible for rapid polymerization.
Introduction
Synthetic aliphatic polyesters, such as polylactide (PLA), poly(ε-carprolactone) (PCL) and polyvalerolactone (PVL) are recognized as an important alternative to petroleum-based polyolefins, thanks to their excellent biocompatibility, biodegradability and sustainability.1–8 Ring-opening polymerization (ROP) is currently the most common method for the preparation of PLA, PCL and their copolymers both in academia and industry. The catalyst plays important role in determining the polymerization rate and polymer microstructure. One important objective of contemporary research is to develop catalysts that exhibit sufficient thermal stability under industrially relevant conditions, and can achieve fast polymerization rate and high molar mass polyester at a low catalyst loading. Many catalytic systems, including organometallics,9–19 organocatalysts20–25 and Lewis pairs,26–33 have been developed over the past few decades. Organometallics are one class of versatile catalytic systems for ROP, because their catalytic performance can be easily modulated by tuning the Lewis acidity of the metal centre and modifying the electronic or steric effects of the ligand backbone.34–36 Besides, organometallics can achieve a high degree of control over the stereoregularity of the polymer chain, compared with organocatalysts and Lewis pairs.9,37–45Salen-metal complexes have prevailed in the ROP of lactone, because the steric and electronic effects of the ligand can be easily modulated by chemical modification. In addition, the ONNO tetradentate structure in the salen-type ligand reduces the possibility of ligand exchange, which is beneficial for improving the stability of the catalyst.19,46–55 These features are important advantages for the development of a catalyst system that can be fine-tuned and potentially employed on an industrial scale. The pioneering studies in 1996 by Spassky reported that the (salen)Al complex with a large sterically hindered o-naphthalene bridged group could catalyze the ROP of LA to produce PLA with a high molar mass and narrow dispersity.56,57 Since then, a series of (salen)Al with structural diversity have been synthesized and used for the ROP of LA and other lactones.37,58–69 In most cases, a (salen)Al complex with an alkyl group in the axial position is used as a precursor. This precursor will react with alcohol (generally termed as an initiator) to form (salen)Al alkoxide in situ, which acts as the real active species and can initiate the ROP of LA via the coordination–insertion mechanism.66 The (salen)Al alkyl precursor is easily decomposed and finally deactivated, since the alkyl–Al bond is highly sensitive to moisture.
Metal chloride complexes are more stable than their metal alkyl counterparts, and thus may be a potential catalyst for ROP. However, there are only a few examples of the ROP of LA by metal chloride complexes. In 2001, Chisholm et al. demonstrated that aluminum chloride complexes are inactive in the ROP of LA, but they can lead to poly(propylene oxide)-b-PLA upon addition of propylene oxide.70 Although aluminum chloride complexes exhibited much lower catalytic activity than single-site LnAlOR catalysts, this fascinating work suggested that aluminum chloride will be a promising catalyst for LA polymerization. Clegg et al. found that dimeric aluminum chloride complexes can be activated by the addition of propylene oxide or cyclohexene oxide to afford efficient initiators for the ROP of LA.71 The active species was supposed to be a chloroalkoxide formed by the nucleophilic ring-opening of a coordinated epoxide. In 2017, Pang and co-workers developed air-stable and environmentally friendly salen-iron chloride for the ROP of LA and CL.72 In order to overcome the low initiation efficiency, propylene oxide was used as a solvent to activate the ring-opening reaction in situ. In particular, (salen)Fe chloride can promote the stereoselective polymerization of rac-LA via a chain-end control mechanism. Subsequently, Thomas et al. disclosed a catalyst pair consisting of a (salen)Al chloride and an onium salt.73 This binary system is of particular interest as it is the very first example of an aluminum-based catalyst active for LA polymerization at room temperature. Nonetheless, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in salen-type ligands are sensitive to protonic impurities (such as water, alcohol and organic acid), and catalytic decrease or even complete deactivation will be observed at high temperatures and low catalyst concentration, especially in bulk polymerization, for most of the salen-Al complexes.74
N bonds in salen-type ligands are sensitive to protonic impurities (such as water, alcohol and organic acid), and catalytic decrease or even complete deactivation will be observed at high temperatures and low catalyst concentration, especially in bulk polymerization, for most of the salen-Al complexes.74
Recently, we synthesized a series of bipyridine bisphenolate aluminum (BpyBph-Al) complexes and used these complexes to catalyze maleic anhydride/epoxide copolymerization for the synthesis of unsaturated polyesters.75–77 Compared with a typical (salen)Al/onium salt and Lewis pairs, the (BpyBph)Al/onium salt exhibited significantly improved tolerance to protonic impurities. Thus, the catalytic activity of the (BpyBph)Al/onium salt can be maintained at a low catalyst loading and high concentration of the chain transfer agent (100 equiv. relative to the catalyst). Increasing the catalyst's thermostability and compatibility with protonic impurities allows polymerizations at decreased catalyst concentrations, which is highly desirable to reduce cost, minimize the catalyst residue, and access high molar mass materials.78
Based on these results, the (BpyBph)Al/onium salt binary catalysts were further used to catalyze the ROP of LA in this work (Scheme 1). We disclosed the relationship between the structures of (BpyBph)Al and their catalytic performances, and investigated the effects of polymerization conditions (including polymerization temperature, catalyst/cocatalyst and monomer/catalyst feed ratios) on LA polymerization behaviors. Compared with the (salen)AlCl/onium salt, the (BpyBph)Al/onium salt exhibited superior performance in LA polymerization. The catalytic activity of (BpyBph)AlCl/PPNCl is about 4–8 fold higher than those of (salen)AlCl/PPNCl at 180 °C. Besides, the (BpyBph)Al complexes showed excellent tolerance to a wide range of protonic impurities, compared with (salen)Al complexes. These features allowed a rapid LA polymerization under bulk and melt conditions. Finally, the chain initiation mechanism was clarified by in situ NMR experiments.
 | ||
| Scheme 1 Ring-opening polymerization of LA catalyzed by the (BpyBph)Al/onium salt binary catalyst. | ||
Experimental section
General materials and methods
Unless otherwise stated, all manipulations involving air or water sensitive compounds were conducted under a nitrogen atmosphere in an Etelux Lab2000 glove box or using the standard Schlenk line technique. The epoxides were purchased from Acros, dried over calcium hydride for 48 h, distilled, and stored under a nitrogen atmosphere before use. The (BpyBph)Al complexes were synthesized according to the reported methods.79ε-Caprolactone was dried over CaH2 for 48 hours, distilled and stored under a nitrogen atmosphere. L-Lactide (LLA) was recrystallized twice from dried and degassed toluene, sublimed and vacuum dried for 48 h and stored in a glove box. Bis(triphenylphosphine)ammonium chloride ([PPN]Cl) was recrystallized from dichloromethane/diethyl ether solution, pounded into a powder and dried under vacuum at 60 °C. Bis(triphenylphosphine) ammonium salts of different anions, including [PPN]OAc, [PPN]TFA, and [PPN]Br via [PPN]Cl at 60 °C with sodium salts of the corresponding ions (sodium acetate/triphenylphosphine and sodium fluoroacetate/sodium bromide) obtained by an ion exchange reaction.801H and 13C {1H} NMR spectra were recorded on a Bruker Avance III spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR) at room temperature in CDCl3. The molar mass and dispersity (Đ) of copolymers were determined by size exclusion chromatography (SEC) at 40 °C with a flow rate of 1.0 mL min−1, using THF as the eluent on an Agilent PL-SEC 50 instrument coupled with a refractive index (RI) detector with respect to polystyrene (PS) standards. The columns included a Plgel guard 50 × 7.5 mm column, a PLgel mixed-B 300 × 7.5 mm column and a PLgel mixed-C 300 × 7.5 mm column. Samples for the SEC test were filtered through a 0.22 μm PTFE filter. The matrix-assisted laser desorption/ionization time-of-flight mass spectra of typical polymer samples are performed on a Bruker Autoflex III mass spectrometer in linear, positive ion mode, using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propylidene] malononitrile (DCTB) and 2,5-dihdyroxybenzoic acid (DHB) as a matrix, and THF as a solvent.
General procedures for the ROP of LA
In an Etelux glovebox, the aluminum complex (0.01 mmol), PPNCl (0.01 mmol, 1 equiv.) and LA (1 mmol, 100 equiv.) were added in an oven-dried Schlenk tube equipped with a magnetic stirrer. Then, epoxide (0.5 mL) was further added to the Schlenk tube. The tube was sealed and removed from the glovebox and placed in an oil-bath. After the reaction at 80 °C for the prescribed time, the polymerization was quenched with 0.02 mL of Et2O/HCl and a crude aliquot was withdrawn from the system for determining the monomer conversion by 1H NMR spectroscopy. The reaction mixture was diluted with approximately 5 mL of dichloromethane and precipitated into 50 mL of methanol with vigorous stirring. The crude polymer was collected after filtration and dried under vacuum at 40 °C overnight. The experimental procedure for evaluating the tolerance of Al complexes was similar, but an extra chain transfer agent was mixed with the catalyst before the addition of epoxide.NMR scale reaction of complexes 4 and 8 with quaternary [PPN] onium salts with/without butadiene monoxide (BO)
In a glovebox, complex 4 (5.7 mg, 10 μmol) was dissolved in chloroform-d with the addition of PPNOOCCF3 (10 μmol) without or with the addition of anhydrous and degassed BO (2.6 μL, 30 μmol) in oven dried J. Young NMR tubes, respectively. Complex 8 (5.7 mg, 10 μmol) was handled by PPNCl (10 μmol) according to the same process. The tubes were monitored by in situ NMR at room temperature. The first BO insertion took place rather fast in minutes while the excess of unreacted BO remained for hours.Results and discussion
Ring-opening polymerization of LA by BpyBph-Al/onium binary catalysts
The ROP of LA was selected as a model reaction to evaluate the catalytic performance of the Al complex/onium salt catalyst pair (Table 1). Both the typical (salen)Al complexes 1–2 and (BpyBph)Al complexes 3–8 were used as the Lewis acid, and PPNCl acted as the Lewis base. It is noted that the ROP of LA cannot be initiated by the Al complex/PPNCl binary catalyst in the absence of epoxide. However, all the polymerizations could proceed smoothly by using propylene oxide as a solvent. Thus, epoxide was used as a solvent to generate active species as much as possible and overcome the low initiation efficiency. LA was consumed almost completely by the 1/PPNCl pair within 0.5 h at 80 °C with a feed ratio of [LA]:[Al]:[PPNCl] = 100:1:1, and the TOF was high up to 192 h−1 (entry 1, Table 1). Combining PPNCl with complex 2 with a rigid benzene bridge, we observed a relatively slow polymerization, in which 80% of LA was consumed in 0.5 h (entry 2, Table 1). 3/PPNCl exhibited medium catalytic activity (TOF = 170 h−1) between 1/PPNCl and 2/PPNCl pairs and the distortion of the ONNO-Al coordination plane suggested the flexibility of the bridging linkage that increased in the order: 2 < 3 < 1. Obviously, the catalytic activity of the Al complex/PPNCl pair increased with an increase in the flexibility of the ligand backbone in the Al complex. This result is highly consistent with the observation in the ROP of LA catalyzed by a single component (salen)Al alkoxide complex, in which the polymerization rate will be retarded significantly by using a catalyst with a rigid framework.82
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Conv.b (%) |
TOFc (h−1) |
M
n,theod (kDa) |
M
n,expe (kDa) |
Đe |
|
a Unless otherwise stated, the ROPs were carried out in 0.5 mL of propylene oxide with 10 mmol Al complex at 80 °C for 0.5 h, [LA]0 = 2.0 M, [LA]:[Al]:[PPNCl] = 100:1:1.
b Monomer conversion was determined by 1H NMR.
c TOF (turnover frequency) = mol of anhydride consumed × mol. of catalyst × h−1.
d The theoretical number-averaged molar mass Mn,theo was calculated by Mn,theo = [LA]/[I] × % conv. (LA) × M(LA).
e The experimental number-averaged molar mass Mn,exp and dispersity (Đ) were determined by SEC. The Mn,exp value obtained by SEC was further corrected by a correcting factor of 0.58.81
f The polymerization was conducted at room temperature.
g In the absence of the PPNCl cocatalyst, polymerization time = 8 h.
h [LA]:[Al]:[PPNCl] = 100:1:0.5, polymerization time = 0.5 h.
i [LA]:[Al]:[PPNCl] = 100:1:2, polymerization time = 0.25 h.
j [LA]:[Al]:[PPNCl] = 100:1:5, polymerization time = 0.25 h.
|
||||||
| 1 | 1/PPNCl | 96 | 192 | 13.8 | 6.7 | 1.22 |
| 2 | 2/PPNCl | 80 | 160 | 11.5 | 5.2 | 1.26 |
| 3 | 3/PPNCl | 85 | 170 | 12.3 | 6.3 | 1.24 |
| 4 | 4/PPNCl | 82 | 164 | 11.8 | 5.8 | 1.28 |
| 5 | 5/PPNCl | 90 | 180 | 13.0 | 5.9 | 1.30 |
| 6 | 6/PPNCl | 72 | 144 | 10.4 | 5.0 | 1.25 |
| 7 | 7/PPNCl | 75 | 150 | 10.8 | 5.2 | 1.27 |
| 8 | 8/PPNCl | 78 | 156 | 11.2 | 5.0 | 1.21 |
| 9f | 5/PPNCl | 80 | 16 | 11.5 | 5.5 | 1.22 |
| 10g | 5/— | 15 | 1.9 | 2.2 | — | — |
| 11h | 5/PPNCl | 36 | 72 | 5.2 | 2.1 | 1.22 |
| 12i | 5/PPNCl | 81 | 324 | 11.7 | 4.6 | 1.35 |
| 13j | 5/PPNCl | 92 | 638 | 13.3 | 1.8 | 1.57 |

The electronic effect and steric hindrance in Al complexes significantly affect the polymerization rate as well. Substituting t-Bu groups (complex 3) with more electron-donating –OCH3 groups (complex 4) resulted in a slight decrease in catalytic activity (entry 4, Table 1), while introducing electron-withdrawing fluorine atoms in the para-position (complex 5) increase the catalytic activity to some extent (entry 5, Table 1). Here, we suppose that the electron-withdrawing group will increase the Lewis acidity of the metal center, which facilitates its coordination with LA and activation of the monomer. 6/PPNCl was proved to be less active than 3/PPNCl (TOF: 170 vs. 144 h−1) (entry 6, Table 1). The bulky steric hindrance around aluminum also will hinder the coordination of the active aluminum complex to the next coming LA monomer. Substituting the axial –Cl group in complex 4 with OAc− (complex 7) and TFA− (complex 8) did not affect the polymerization rate (entries 7 and 8, Table 1), and we observed very similar catalytic activity for these three complexes in the presence of PPNCl. We envisioned that the active species formed in situ by 4/PPNCl, 7/PPNCl and 8/PPNCl in the presence of epoxide exhibited similar activity toward lactide (see below). The ROP of LA catalyzed by 5/PPNCl could proceed smoothly at room temperature (entry 9, Table 1), but the polymerization rate dramatically decreased. The resultant PLAs exhibited monomodal dispersity.
The LA polymerization follows first-order kinetics, as evidenced by the good linearity between ln[LA]0/[LA]t and reaction time (Fig. S1†). Notably, the experimental Mn,exp values for the resultant polyester samples are approximately half of the theoretical values (Mn,theo), suggesting the formation of two polymeric chains per metal center. This is in contrast to the LA copolymerization catalyzed by (salen)Al alkoxide, where only one polymer chain is initiated per metal center.
The effect of the [Al]:[PPNCl] feed ratio on LA polymerization was further investigated, considering that the amount of PPNCl will be crucial to generate a potent active species. We observed a slow polymerization by using complex 5 alone and without PPNCl as a cocatalyst. Only 15% of LA was converted by complex 5 after 8 h (entry 10, Table 1). Introducing 0.5 equiv. of PPNCl led to a more rapid polymerization with a LA conversion of 36% in 0.5 h, affording PLA with monomodal dispersity and Mn,exp approximately half of the theoretical value (entry 11, Table 1). The polymerization would be accelerated by further increasing the ratio of [PPNCl]:[Al] to 2:1 and 5:1 (entries 12 and 13, Table 1) with LA conversions of 81% and 92% in 0.25 h, respectively. However, the resultant PLAs exhibited much lower Mn,exp than Mn,theo/2 and broader dispersity. It was envisioned that a high quantity of PPNCl favored substantial chain transfer reactions. In agreement with previous observations with the (porphyrin)AlCl complex, the presence of excess [PPN]Cl can cause a displacement of the alkoxide anion by the chloride, generating the new onium salt [PPN]OR and ultimately leading to the formation of the corresponding bis(alkoxide) complexes. As the polymerizations are conducted in propylene oxide, excess PPNCl can lead to the formation of several [PPN]OR species, finally resulting in PLA with a lower molar mass.
To further highlight the synergistic effect between the BpyBph-Al complex and PPNCl, we further monitored the LA polymerization catalyzed by PPNCl alone at 80 °C with a feed ratio of [LA]:[PPNCl] = 100:1 (Fig. S2†). There is an induction period for PPNCl catalyzed LA polymerization. We did not detect LA consumption within 90 min, and a low LA conversion was observed at 120 min. PPNCl exhibited a very different catalytic behavior from the BpyBph-Al/PPNCl binary system, supporting that (BphBpy)AlCl and PPNCl cooperatively catalyze LA polymerization. PPNCl mainly served as a Lewis base to initiate the ring-opening reaction of epoxide that was activated by (BphBpy)AlCl (see below).
Evaluation of thermostability and tolerance to protic impurities for a binary catalyst
LA polymerization is generally conducted at high temperatures in industry. Meanwhile, a high monomer/catalyst feed ratio is required to access PLA with a high molar mass, whereas the dosage of the trace impurities will also be increased at a high monomer loading. Most of the reported catalysts exhibited decreased catalytic activity or even complete deactivation at high temperatures or high monomer feed ratios owing to the decomposition of the active species.9–19 Based on these considerations, we further evaluated the thermostability and protic impurity tolerance for salen-Al/PPNCl and (BpyBph)-Al/PPNCl binary catalysts with a [LA]:[catalyst pair] feed ratio of 500:1 (Tables 2 and 3).
| Entry | Cat. | T (°C) | t (h) | Conv.b (%) |
TOFc (h−1) |
M
n,theod (kDa) |
M
n,expe (kDa) |
Đe |
|---|---|---|---|---|---|---|---|---|
|
a Unless otherwise stated, the ROPs were carried out in 2.5 mL of propylene oxide with 10 mmol Al complex, [LA]0 = 2.0 M, [LA]:[Al]:[PPNCl] = 500:1:1.
b Monomer conversion was determined by 1H NMR.
c TOF (turnover frequency) = mol. of anhydride consumed × mol. of catalyst × h−1.
d The theoretical number-averaged molar mass Mn,theo was calculated by Mn,theo = [LA]/[I] × % conv. (LA) × M(LA).
e The experimental number-averaged molar mass Mn,exp and dispersity (Đ) were determined by SEC. The Mn,exp value obtained by SEC was further corrected by a correcting factor of 0.58.81
f The polymerization was conducted under bulk and melt conditions with a feed ratio of [LA]:[5]:[PPNCl]:[1,2-epoxyhexane] = 2000:1:1:2.
g The polymerization was conducted under bulk and melt conditions with a feed ratio of [LA]:[5]:[PPNCl]:[1,2-epoxyhexane] = 5000:1:1:2.
|
||||||||
| 1 | 1/PPNCl | 80 | 5 | 93 | 93 | 67.0 | 19.1 | 1.28 |
| 2 | 2/PPNCl | 80 | 5 | 84 | 84 | 60.5 | 15.7 | 1.27 |
| 3 | 5/PPNCl | 80 | 5 | 88 | 88 | 63.4 | 18.0 | 1.25 |
| 4 | 1/PPNCl | 130 | 3 | 67 | 112 | 48.2 | 9.8 | 1.44 |
| 5 | 2/PPNCl | 130 | 3 | 75 | 125 | 54.0 | 10.6 | 1.42 |
| 6 | 5/PPNCl | 130 | 3 | 85 | 142 | 61.2 | 14.2 | 1.44 |
| 7 | 1/PPNCl | 180 | 2 | 12 | 30 | 0.8 | — | — |
| 8 | 2/PPNCl | 180 | 2 | 26 | 65 | 18.7 | — | — |
| 9 | 5/PPNCl | 180 | 2 | 98 | 245 | 70.6 | 13.8 | 1.53 |
| 10f | 5/PPNCl | 200 | 6 | 93 | 310 | 267.8 | 72.5 | 1.28 |
| 11g | 5/PPNCl | 200 | 12 | 87 | 363 | 626.4 | 115.1 | 1.31 |
| Entry | Cat. | T (°C) | Protic agent | Equiv. to [Al-PPNCl] | Conv.b (%) |
TOFc (h−1) |
M
n,expd (kDa) |
Đd |
|---|---|---|---|---|---|---|---|---|
|
a Unless otherwise stated, the ROPs were carried out in 2.5 mL of propylene oxide with 10 mmol Al complex at 80 °C for 5 h, [LA]0 = 2.0 M, [LA]:[Al]:[PPNCl] = 500:1:1.
b Monomer conversion was determined by 1H NMR.
c TOF (turnover frequency) = mol. of anhydride consumed × mol. of catalyst × h−1.
d The experimental number-averaged molar mass Mn,exp and dispersity (Đ) were determined by SEC. The Mn,exp value obtained by SEC was further corrected by a correcting factor of 0.58.81
|
||||||||
| 1 | 5/PPNCl | 80 | BnOH | 10 | 92 | 92 | 5.2 | 1.26 |
| 2 | 5/PPNCl | 80 | BnOH | 20 | 91 | 91 | 3.0 | 1.24 |
| 3 | 5/PPNCl | 80 | BnOH | 50 | 89 | 89 | 1.5 | 1.28 |
| 4 | 5/PPNCl | 80 | BnOH | 100 | 85 | 85 | — | — |
| 5 | 5/PPNCl | 80 | iPrOH | 50 | 95 | 95 | 1.3 | 1.21 |
| 6 | 5/PPNCl | 80 | H2O | 50 | 86 | 86 | 1.0 | 1.18 |
| 7 | 5/PPNCl | 80 | Maleic acid | 50 | 85 | 85 | 1.1 | 1.19 |
| 8 | 1/PPNCl | 80 | BnOH | 50 | 45 | 45 | 0.7 | 1.20 |
| 9 | 1/PPNCl | 80 | BnOH | 100 | 15 | 15 | — | — |
| 10 | 1/PPNCl | 80 | Maleic acid | 50 | 35 | 35 | 0.6 | 1.30 |
| 11 | 2/PPNCl | 80 | BnOH | 50 | 57 | 57 | 0.9 | 1.23 |
| 12 | 2/PPNCl | 80 | BnOH | 100 | 25 | 25 | — | — |
| 13 | 2/PPNCl | 80 | Maleic acid | 50 | 40 | 40 | 0.5 | 1.26 |
The catalytic activities of 1/PPNCl, 2/PPNCl and 5/PPNCl increased to some extent by elevating the polymerization temperature from 80 to 130 °C (entries 1–6, Table 2). However, the activities of 1/PPNCl and 2/PPNCl sharply decreased when further increasing the temperature to 180 °C (entries 7 and 8, Table 2). The LA conversion after 2 h was 12% and 26%, respectively for 1/PPNCl and 2/PPNCl at 180 °C. This result may be ascribed to the decomposition of active species derived from (salen)Al/PPNCl, which can be observed in the ring-opening copolymerization of cyclic anhydride and epoxide at high temperatures.83 By contrast, we observed more rapid polymerization by using 5/PPNCl at 180 °C, and LA was consumed almost completely in 2 h (entry 9, Table 2). These results clearly indicated that the 5/PPNCl pair exhibited much higher thermostability than 1/PPNCl and 2/PPNCl. Here, we supposed that the bipyridine (bpy) bridging group with an extended π-conjugate may be responsible for the high thermostability of the (BpyBph)Al complex. 5/PPNCl could efficiently promote the ROP of LA even under bulk and melt conditions by using 1,2-epoxyhexane with a high boiling point as a co-initiator (entries 10 and 11, Table 2). It was noted that the molar mass of PLA decreased, and the dispersity became broad as the polymerization temperature increased, indicative of serious side reactions (such as transesterification and chain transfer reactions) at high temperatures.
Compared with 1/PPNCl and 2/PPNCl pairs, 5/PPNCl shows good tolerance to protic agents, such as benzyl alcohol (BnOH), isopropanol (iPrOH), water (H2O) and maleic acid. We first chose BnOH to mimic protic impurity and investigated the variation in the catalytic activity of 5/PPNCl under different BnOH loading dosages. We did not observe an obvious decay in catalytic activity by gradually increasing the [BnOH]:[catalyst pair] feed ratio from 10:1 to 100:1 (entries 1–4, Table 3). However, the molar mass of the resultant PLA decreased with an increase of the [BnOH]:[catalyst pair] feed ratio, suggesting that BnOH could serve as a chain transfer agent. The ROPs were further conducted using 5/PPNCl in the presence of 50 equiv. of iPrOH, H2O and maleic acid, respectively (entries 5–7, Table 3). All the copolymerization could proceed smoothly without obvious decay of catalytic activity. These results clearly indicated that 5/PPNCl exhibits good tolerance to a wide range of protic reagents. By contrast, the activity for 1/PPNCl (from 93 to 45 h−1) and 2/PPNCl (from 84 to 57 h−1) substantially decreased in the presence of 50 equiv. of BnOH (entries 8 and 11, Table 3). We observed a further decrease of activity for these two catalysts by increasing the [BnOH]:[catalyst pair] feed ratio to 100:1 (entries 9 and 12). Introducing 50 equiv. of maleic acid as a protic impurity also resulted in a decline in activity for 1/PPNCl and 2/PPNCl, respectively (entries 10 and 13).
Investigation into the chain initiation mechanism
In previous work, Thomas et al. have shown that the origin of the unprecedented activity of the (salen)Al/PPNCl system in LA polymerization is very likely to be caused by the formation of highly active alkoxide species with the help of density theory calculation.73 Here, we conducted in situ NMR experiments to investigate the chain initiation mechanism. We began by measuring the 19F NMR spectra of two different catalyst/cocatalyst combinations, 4/PPNTFA and 8/PPNCl (Fig. S3 and S4†), to elucidate the interaction between the Al complex and the cocatalyst. When PPNTFA was mixed with 4, the peak for TFA− shifted upfield from −74.64 to −76.30 ppm (Fig. S3†); whereas combining 8 with PPNCl led to a small downfield shift of the TFA− peak, from −76.58 to −76.13 ppm (Fig. S4†). In addition, both 4/PPNTFA and 8/PPNCl showed only one peak in their 19F NMR spectra. These results suggest that the six-coordinate (BpyBph)Al(CF3COO−)Cl− anion (INT1 in Scheme 2) was formed via coordination between the nucleophilic anion of PPNX and the five-coordinate Al complex. | ||
| Scheme 2 Proposed mechanism of chain initiation by the (BpyBPh)AlCl/PPNCl catalyst pair according to in situ1H and 19F NMR spectroscopy. | ||
When 3 equiv. of BO was added to 4/PPNTFA or 8/PPNCl, only 1 equiv. of BO underwent a ring opening, and the rest of the BO was still intact even after 24 h (Fig. 1a, Fig. S5, S6 and Tables S1, S2†). 1H and 19F NMR spectroscopy indicated the formation of a five-coordinated Al alkoxide complex INT3 and free PPNTFA (Fig. S3 and S5†). Notably, the six-coordinate complex INT2 was not detected on the NMR time scale. We deduced that the stronger nucleophilicity of TFA− in INT1 resulted in tight coordination with the Al center and that the first equiv. of BO underwent ring opening by the departing Cl−. Once INT2 formed, TFA− dissociated rapidly, and INT3 formed spontaneously, because TFA− is a better leaving group than alkoxide. In the presence of a larger excess of BO (5 equiv. relative to 4/PPNTFA), coordination of INT3 with BO and subsequent nucleophilic attack by TFA− afforded the six-coordinate bis(alkoxide) Al complex INT5 (Fig. 1b). Additionally, the upfield shift of the TFA− peak implied that TFA− was involved in the nucleophilic attack on BO (Fig. S3D†). Notably, the ring opening of PO during chain initiation occurred on both sides of the Al-ONNO coordination plane, and the six-coordinated bis(alkoxide) Al complex INT5 was finally generated (Scheme 2).
 | ||
| Fig. 1 Chemical shifts and integration areas for peaks in the upfield region of the 1H NMR spectra of INT3 and INT5, which formed after 4/PPNTFA was combined with (a) 3 equiv. of BO and (b) 5 equiv. of BO, respectively. The integration area of the signal for the –OCH3 groups of the ligand was used as a reference, and the structures of the intermediates could be determined from the integration areas of the alkoxide groups. | ||
We hypothesized that there will be an association/dissociation equilibrium between a six-coordinated bis(alkoxide) Al complex and a mixture consisting of five-coordinated (BpyBph)Al alkoxide and [PPN]+ alkoxide (PPNOR). During chain propagation, the competitive coordination of the LA monomer with an aluminum center can facilitate the dissociation of the six-coordinated bis(alkoxide) Al complex. The five-coordinated (BpyBph)Al alkoxide alone exhibits extremely low catalytic activity toward LA polymerization, and thus it mainly acts as a Lewis acid and activates the LA monomer by coordination in the vacant axial position. The PPNOR can act as a Lewis base and induce the ring-opening of the activated LA via nucleophilic attack. The cooperative effects between (BpyBph)Al alkoxide and PPNOR are mainly responsible for the rapid polymerization. Once the activated LA is ring-opened by PPNOR, a new six-coordinated bis(alkoxide) Al complex will be generated that can be converted to five-coordinated (BpyBph)Al alkoxide and PPNOR again. Then, the next LA monomer is inserted into the polymer chain with the assistance of the (BpyBph)Al alkoxide/PPNOR pair. The ROP of LA proceeds via coordination anionic polymerization by repeating the above reaction cycle.
It is assumed that each individual alkoxide in the active six-coordinated bis(alkoxide) Al complex could initiate one polymer chain. Finally, two polymer chains were initiated by per metal catalyst. This point could be supported by the experimental results that Mn,exp is approximately Mn/2 (see Table 1). To find more evidence for this mechanism, the PLA sample obtained from 8/PPNCl was further analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. We could observe two populations in the mass spectra, which were attributed to linear PLA with chloroalkoxide and a trifluoroacetic alkoxide chain end, respectively (Fig. 2). In addition, the chloroalkoxide and trifluoracetic alkoxide chain ends could be further confirmed by 1H, 13C and DEPT(135°) NMR spectra (Fig. S7–S9†). These results strongly indicated that the six-coordinated bis(alkoxide) Al complex was the real active species for LA polymerization. Each of the alkoxide groups, which was derived from the axial group in the Al complex and the nucleophilic anion in the PPN salt, respectively, could initiate one polymer chain. The MALDI-TOF MS data also suggested that propylene oxide is only ring-opened to form active species but will not be inserted into the active polymer chain during propagation.
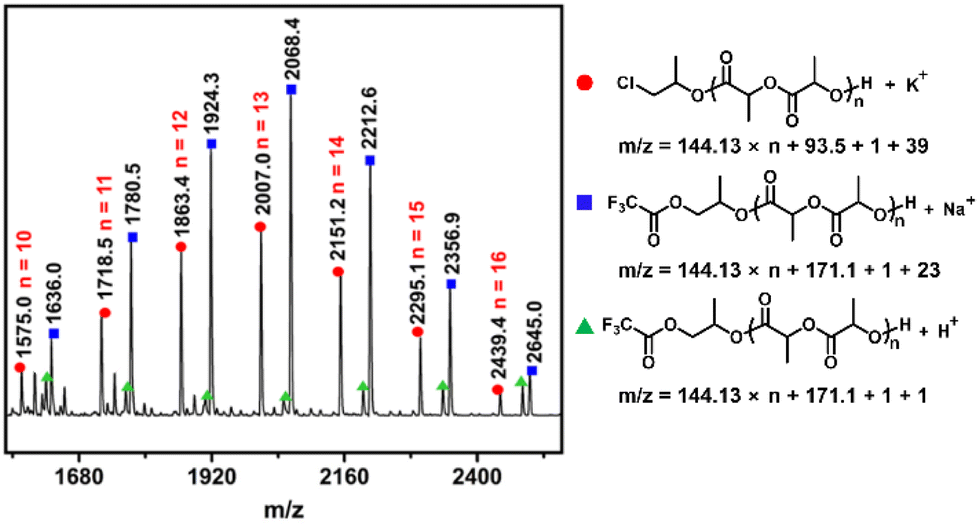 | ||
| Fig. 2 MALDI-TOF mass spectrum of polylactide (polymerization conditions: catalyst 8/PPNCl, temperature 80 °C, [LA]/[Al]/[PPNCl] = 100/1/1, [LA] = 2 M, the solvent is 1 mL of PO, the polymerization is 5 min, the LA conversion rate is 38%). | ||
As mentioned above, the (BpyBph)Al chloride complex could promote LA polymerization in the presence of propylene oxide without an onium salt as a cocatalyst, resulting in a very slow polymerization with a LA conversion of 15% after 8 h at 80 °C. The chain initiation mechanism was thus further investigated by an in situ NMR experiment. We did observe the formation of five-coordinated neutral Al alkoxide species after mixing complex 4 alone with BO (Fig. S10†). This five-coordinated Al alkoxide cannot be converted to the six-coordinated bis(alkoxide) Al complex in the presence of 5 equiv. of BO without the PPNCl cocatalyst. Thus, the five-coordinated neutral Al alkoxide was the active species that can initiate the ROP of LA via the coordination–insertion mechanism (Scheme 3). Such a “co-initiation mechanism” by epoxides was also found in the LA polymerization catalyzed by aluminum-, titanium- and iron chloride systems.71,72
 | ||
| Scheme 3 Coordination–insertion mechanism for chain propagation initiated by (BpyBph)AlCl without the PPNCl cocatalyst. | ||
Conclusions
In summary, a (BpyBph)Al/PPNCl binary system was proved to be a promising catalyst for the ROP of LA. The catalytic activity could be improved by introducing electron-withdrawing groups in the para-position of phenolate or elevating the reaction temperature. Increasing the [PPNCl]:[Al] feed ratio not only accelerates the polymerization, but also leads to a decrease in the molecular weight of the resultant PLA owing to the formation of more active species at a high PPNCl concentration. Compared with the (salen)AlCl/onium salt, the (BpyBph)Al/onium salt exhibited superior performance in LA polymerization, in view of the increased catalytic activity at high temperatures and good compatibility with a wide range of protonic impurities. Thus, lactide polymerization could be conducted under bulk and melt conditions with a high [LA]:[catalyst pair] feed ratio of up to 5000:1.
In situ NMR experiments and end group analysis by MALDI-TOF MS revealed that a highly active six-coordinated bis(alkoxide) aluminum complex was formed by reaction of the (BpyBph)Al/onium salt with epoxide, and it will be transformed into a neutral five-coordinated aluminum alkoxide and a PPN+ alkoxide. The five-coordinated aluminum alkoxide (Lewis acid) and PPN+ alkoxide (Lewis base) cooperatively catalyze the ROP of LA, finally leading to very rapid LA polymerization. Each individual alkoxide in the active six-coordinated bis(alkoxide) Al complex could initiate one polymer chain, and thus two polymer chains were initiated per metal catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 52222302 and 51973156). The authors also highly appreciate Dr Xiao-Lu Chen for the initial experiments.References
- W. Amass, A. Amass and B. Tighe, Polym. Int., 1998, 47, 89–144 CrossRef CAS.
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1997, 30, 3470–3476 CrossRef CAS.
- M. D. Dasaratha, K. Vema, R. Jayakumar and C. Vamsadhara, Int. J. Pharm., 2003, 268, 23–29 CrossRef.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- Y. Hori, Y. Gonda, Y. Takahashi and T. Hagiwara, Macromolecules, 1996, 29, 804–806 CrossRef CAS.
- X. Pang, X. Zhuang, Z. Tang and X. Chen, Biotechnol. J., 2010, 5, 1125–1136 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS PubMed.
- D. Li, B. Gao and Q. Duan, New J. Chem., 2019, 43, 6943–6950 RSC.
- J. A. Stewart, P. McKeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- S. Kaler and M. D. Jones, Dalton Trans., 2022, 51, 1241–1256 RSC.
- S. Chen, Z. Jia, L. Cao, Y. Li, X. Han, X. Pan and J. Wu, Macromolecules, 2021, 54, 9027–9038 CrossRef CAS.
- S. R. Kosuru, T.-H. Sun, L.-F. Wang, J. K. Vandavasi, W.-Y. Lu, Y.-C. Lai, S. C. N. Hsu, M. Y. Chiang and H.-Y. Chen, Inorg. Chem., 2017, 56, 7998–8006 CrossRef CAS PubMed.
- T. Xing, C. Jiang, M. R. J. Elsegood and C. Redshaw, Inorg. Chem., 2021, 60, 15543–15556 CrossRef CAS PubMed.
- N. Yuntawattana, T. M. McGuire, C. B. Durr, A. Buchard and C. K. Williams, Catal. Sci. Technol., 2020, 10, 7226–7239 RSC.
- M. Strianese, D. Pappalardo, M. Mazzeo, M. Lamberti and C. Pellecchia, Dalton Trans., 2020, 49, 16533–16550 RSC.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS PubMed.
- M. S. Zaky, A.-L. Wirotius, O. Coulembier, G. Guichard and D. Taton, ACS Macro Lett., 2022, 11, 1148–1155 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- R. Todd, S. Tempelaar, G. L. Re, S. Spinella, S. A. McCallum, R. A. Gross, J.-M. Raquez and P. Dubois, ACS Macro Lett., 2015, 4, 408–411 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- N. Zhao, C. Ren, Y. Shen, S. Liu and Z. Li, Macromolecules, 2019, 52, 1083–1091 CrossRef.
- X. Li, C. Chen and J. Wu, Molecules, 2018, 23, 189 CrossRef.
- B. Wang, Y. Wei, Z. J. Li, L. Pan and Y. S. Li, ChemCatChem, 2018, 10, 5287–5296 CrossRef CAS.
- E. Piedra-Arroni, C. Ladaviere, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13306–13309 CrossRef CAS.
- J. B. Zhu and E. Y. Chen, J. Am. Chem. Soc., 2015, 137, 12506–12509 CrossRef CAS.
- X.-Q. Li, B. Wang, H.-Y. Ji and Y.-S. Li, Catal. Sci. Technol., 2016, 6, 7763–7772 RSC.
- M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS.
- E. Piedra-Arroni, A. Amgoune and D. Bourissou, Dalton Trans., 2013, 42, 9024–9029 RSC.
- Q. Wang, W. Zhao, J. He, Y. Zhang and E. Y. X. Chen, Macromolecules, 2017, 50, 123–136 CrossRef CAS.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- B. Li, Y. Yang, H. Zhu and H. W. Roesky, Coord. Chem. Rev., 2021, 429, 213625 CrossRef CAS.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS.
- B. Gao, D. Li, Y. Li, Q. Duan, R. Duan and X. Pang, New J. Chem., 2015, 39, 4670–4675 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- M. J. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- Y. Wan, Y. Bai, H. Xu, J. He and Y. Zhang, Macromol. Rapid Commun., 2021, 42, 2000491 CrossRef CAS.
- M. C. D'Alterio, C. De Rosa and G. Talarico, ACS Catal., 2020, 10, 2221–2225 CrossRef.
- H.-Z. Fan, X. Yang, J.-H. Chen, Y.-M. Tu, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202117639 CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- X. Yang, W. Zhang, H.-Y. Huang, J. Dai, M.-Y. Wang, H.-Z. Fan, Z. Cai, Q. Zhang and J.-B. Zhu, Macromolecules, 2022, 55, 2777–2786 CrossRef CAS.
- M. J. L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- M. H. Chisholm and Z. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS PubMed.
- C. Kan, J. Ge and H. Ma, Dalton Trans., 2016, 45, 6682–6695 RSC.
- E. E. Marlier, J. A. Macaranas, D. J. Marell, C. R. Dunbar, M. A. Johnson, Y. DePorre, M. O. Miranda, B. D. Neisen, C. J. Cramer, M. A. Hillmyer and W. B. Tolman, ACS Catal., 2016, 6, 1215–1224 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- Y. Wei, L. Song, L. Jiang, Z. Huang, S. Wang, Q. Yuan, X. Mu, X. Zhu and S. Zhou, Dalton Trans., 2019, 48, 15290–15299 RSC.
- Y. Zhao, Y. Wang, X. Zhou, Z. Xue, X. Wang, X. Xie and R. Poli, Angew. Chem., Int. Ed., 2019, 58, 14311–14318 CrossRef CAS.
- P. Pander, R. Bulmer, R. Martinscroft, S. Thompson, F. W. Lewis, T. J. Penfold, F. B. Dias and V. N. Kozhevnikov, Inorg. Chem., 2018, 57, 3825–3832 CrossRef CAS.
- C. A. Baker, C. Romain and N. J. Long, Chem. Commun., 2021, 57, 12524–12527 RSC.
- D. J. Darensbourg, P. Ganguly and D. Billodeaux, Macromolecules, 2005, 38, 5406–5410 CrossRef CAS.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- M. Wisniewski, A. L. Borgne and N. Spassky, Macromol. Chem. Phys., 1997, 198, 1227–1238 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed.
- Y.-F. Lin and N.-Y. Jheng, Polymers, 2019, 11, 1530 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS PubMed.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed.
- D. J. Darensbourg and O. Karroonnirun, Organometallics, 2010, 29, 5627–5634 CrossRef CAS.
- F. Isnard, M. Lamberti, L. Lettieri, I. D'Auria, K. Press, R. Troiano and M. Mazzeo, Dalton Trans., 2016, 45, 16001–16010 RSC.
- Y. Lu, J. H. Swisher, T. Y. Meyer and G. W. Coates, J. Am. Chem. Soc., 2021, 143, 4119–4124 CrossRef CAS.
- J. P. MacDonald and M. P. Shaver, Polym. Chem., 2016, 7, 553–559 RSC.
- X. Pang, R. Duan, X. Li, C. Hu, X. Wang and X. Chen, Macromolecules, 2018, 51, 906–913 CrossRef CAS.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858–14861 CrossRef CAS PubMed.
- I. D. S. Vieira, E. L. Whitelaw, M. D. Jones and S. Herres-Pawlis, Chem. – Eur. J., 2013, 19, 4712–4716 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS.
- M. H. Chisholm, D. Navarro-Llobet and W. J. Simonsick, Macromolecules, 2001, 34, 8851–8857 CrossRef CAS.
- S. Doherty, R. J. Errington, N. Housley and W. Clegg, Organometallics, 2004, 23, 2382–2388 CrossRef CAS.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 50, 9188–9195 CrossRef CAS.
- C. Robert, T. E. Schmid, V. Richard, P. Haquette, S. K. Raman, M.-N. Rager, R. M. Gauvin, Y. Morin, X. Trivelli, V. Guérineau, I. del Rosal, L. Maron and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 6217–6225 CrossRef CAS PubMed.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 3502–3512 CrossRef CAS.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS.
- Y.-B. Wang, M.-Q. Wang, Y.-B. Shi, X.-L. Chen, D.-P. Song, Y.-S. Li and B. Wang, Macromol. Chem. Phys., 2022, 223, 2200079 CrossRef CAS.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- B. N. Mongal, A. Tiwari, C. Malapaka and U. Pal, Dalton Trans., 2019, 48, 10070–10077 RSC.
- Y. Wang, M. Li, S. Wang, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 10798–10805 CrossRef CAS.
- J. E. Mark, Polymer data handbook, Oxford University Press, New York, 1999 Search PubMed.
- M. Mandal, A. M. Luke, B. Dereli, C. E. Elwell, T. M. Reineke, W. B. Tolman and C. J. Cramer, ACS Catal., 2019, 9, 885–889 CrossRef CAS.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01273a |
| This journal is © The Royal Society of Chemistry 2023 |