
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphological evolution of poly(glycerol monomethacrylate)-stat-glycine–phenylalanine–phenylalanine–methacrylamide-b-poly(2-hydroxypropylmethacrylate) (P(GMA-stat-(MAm-GFF))-b-PHPMA) block copolymer nano-objects via polymerization-induced self-assembly†
T. P. Tuyen
Dao
abc,
Lubomir
Vezenkov
c,
Gilles
Subra
 c,
Muriel
Amblard
c,
Vincent
Ladmiral
b and
Mona
Semsarilar
*a
c,
Muriel
Amblard
c,
Vincent
Ladmiral
b and
Mona
Semsarilar
*a
aInstitut Européen des Membranes, IEM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: mona.semsarilar@umontpellier.fr
bInstitut Charles Gerhardt Montpellier, ICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France
cInstitut des Biomolécules Max Mousseron, IBMM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France
First published on 12th December 2022
Abstract
Here we report a study on the reversible addition–fragmentation chain transfer (RAFT) polymerization-induced self-assembly (PISA) of self-assembling peptide-containing diblock copolymers. The self-assembling tripeptide (SAP) monomer used (MAm-GFF) is a methacrylamide-functionalized Gly–Phe–Phe sequence. The incorporation of this self-assembling GFF tripeptide into the stabilizing PGMA segment of a well-known poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) block copolymer (PGMA-b-PHPMA) has been shown in a previous study to induce the formation of complex self-assembled structures. The present article explores the effect of varying the block length (both hydrophilic and hydrophobic blocks), block composition (different SAP units in the hydrophilic block), solvent composition and temperature on the morphologies of PISA synthesized P(GMA-stat-(MAm-GFF))-b-PHPMA. The polymerization of HPMA from the SAP-containing P(GMA-stat-(MAm-GFF)) stabilizing block led to well-defined diblock copolymers, which self-assembled into diverse morphologies ranging from vesicles, worms, or micelles to unreported highly anisotropic structures. Moreover, these morphologies were found to be thermoresponsive. At 70 °C, more classic morphologies were observed as hydrogen bonds are weakened at high temperatures. However, these structures transform into unprecedented morphologies (urchin-like) once cooled to 4 °C probably due to the reduced hydrophilicity of the PHPMA block.
Introduction
Self-assembly of amphiphilic molecules in solution is a very promising bottom-up approach in nanomaterials science. It has found many potential applications in the fields of photonics, electronics, sustainable energy, bioengineering or drug delivery for instance.1–8 Besides the well-known morphologies (micelles, worm-like micelles or vesicles), the discovery of more complex architectures (such as tubes, rods, platelets, sheets, wires, ribbons, interwound fibrils, bundles, etc.) has stimulated many studies aiming to understand and control their formation. Molecular self-assembly results from the combination of various weak forces such as hydrophobic interactions, electrostatic interactions, hydrogen bonding and π–π stacking.Block copolymer self-assembly has usually been performed using the post-polymerization approach whereby preformed block copolymers are self-assembled in a selected solvent. This approach is, however, limited to relatively low concentrations (typically less than 1%), and is both time- and labor-intensive as it involves multiple steps (polymerization, purification, and self-assembly), is not easily reproducible and often does not lead to a unique type of morphology. Polymerization-induced self-assembly (PISA), proceeding via the simultaneous formation and self-assembly of amphiphilic block copolymers, is a powerful method to prepare various morphologies at high concentrations (up to 50% solid content), fast (the polymerization time) and with high reproducibility.9–13 In addition, PISA can be used with different types of polymerization techniques and in various media.14–16
Few studies have reported the synthesis of self-assembled nanostructures based on peptides via PISA.17–19 Peptides are able to self-assemble via a range of non-covalent interactions. This ability is responsible for the secondary structure of proteins. We recently studied the effect of the insertion of self-assembling peptide (SAP) moieties into diblock copolymers on their PISA behaviour. Our group started this approach by inserting SAPs into very well-studied PGMA-b-PHPMA amphiphilic copolymers to investigate their influence on the final PISA-generated structures.1 This study showed that the insertion of only a few monomer units containing the SAP (tripeptide of Gly–Phe–Phe abbreviated as GFF) could result in the formation of long anisotropic fibrous morphologies, confirming the decisive influence of the GFF SAP on the self-assembly of P(GMA-stat-(MAm-GFF))-b-PHPMA.
In another report, we used SAP containing monomers as the core-forming blocks, either on their own (forming a pure polypeptide block) or as a statistical copolymer with HPMA and GMA.20 In all cases, placing the SAP-containing monomer in the core resulted in the formation of a range of interesting morphologies (fibre-, flake-, and leaf-like) never reported previously.
Here, we report a more detailed study of the P(GMA-stat-GFF)-b-PHPMA formulation. Our previous report dealt with only one stabilizing block composition.1 Here, different parameters influencing the morphology and its evolution, namely, the number of SAP monomers in the CTA, the block length, solvent (pure water and a mixture of water/alcohol) and temperature were studied. The synthetic approach in this work is illustrated in Scheme 1.
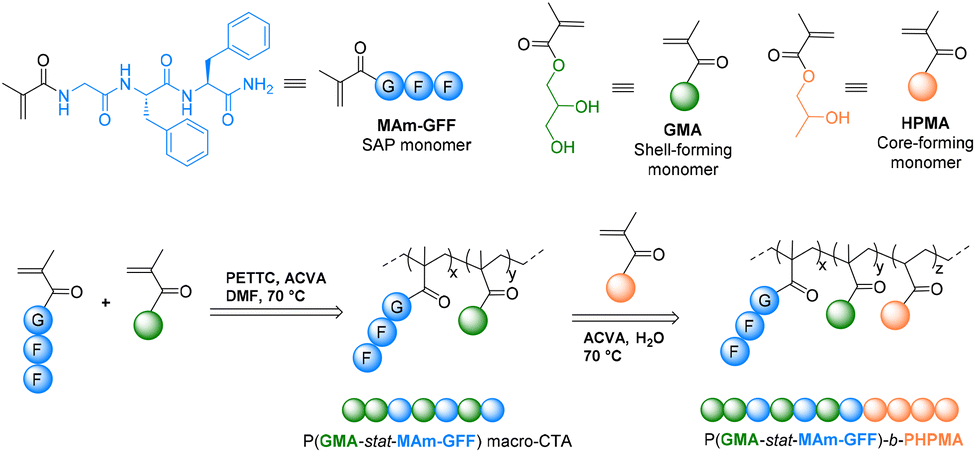 | ||
| Scheme 1 Schematic representation of the synthesis of the SAP-containing methacrylamide (MAm-GFF) and P(GMAx-stat-(MAm-GFF)y)-b-PHPMAz diblock copolymers. | ||
Experimental section
Materials
Glycerol monomethacrylate (GMA), 2-hydroxypropyl methacrylate (HPMA), 4,4′-azobis(4-cyanopentanoic acid) (ACVA) and all organic solvents were purchased from Sigma-Aldrich. DMSO-d6 was purchased from Eurisotop. MAm-GFF and 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)-sulfanyl pentanoic acid (PETTC) were prepared according to our previous reports.1,21Syntheses of P(GMA-stat-MAm-GFF) macromolecular chain transfer agents
P(GMAx-stat-(MAm-GFF)y) macromolecular chain transfer agents (mCTAs) were synthesized via RAFT solution copolymerization according to the following procedure. A round bottom Schlenk flask equipped with a stirrer bar was charged with GMA (x eq.), MAm-GFF (y eq.), PETTC RAFT agent (1 eq.), ACVA initiator (0.1 eq.) and DMF at the volume needed to solubilize all of the solids (the final concentration of solids was about 20% w/w). The flask was subsequently sealed with a rubber septum, placed in an ice bath and degassed by bubbling nitrogen for 30 min. The flask was then immersed in a preheated oil bath at 70 °C under stirring for 24 h. The copolymerization was quenched by opening the flask to air. GMA and MAm-GFF conversions were determined from the 1H NMR spectrum in DMSO-d6 (Fig. S1†). The reaction solution was diluted in DMF and precipitated in cold THF. The yellow precipitate was then redispersed in water and freeze-dried. The final product was a light yellow powder. The mean DP was determined by 1H NMR and the molar mass distribution was determined using SEC-HPLC in DMAc or DMF.Preparation of diblock P(GMA-stat-(MAm-GFF))-b-PHPMA via dispersion RAFT-PISA
A representative PISA synthesis via RAFT dispersion polymerization of HPMA with the P(GMA-stat-(MAm-GFF)) macro-CTA was carried out as described below. For a target composition of P(GMA24-stat-(MAm-GFF)3)-b-PHPMA60, the purified P(GMA24-stat-(MAm-GFF)3) macro-CTA (100 mg, 0.018 mmol), HPMA (155.5 mg, 1.08 mmol) and ACVA initiator (1.0 mg, 0.0036 mmol) were weighed into a 10 mL glass vial containing a stirrer bar. 2.3 mL Milli-Q water was then added to obtain a reaction solution at 10% w/w. The vial was sealed, degassed for 30 minutes and placed in an oil bath previously set at 70 °C using a stirring hotplate. After 24 h, the reaction was opened to air, and cooled down to ambient temperature. The HPMA conversion was always 100% with no remaining HPMA-vinyl signals in 1H NMR spectra. Other aliquots were taken to perform SEC, DLS and TEM.In the following text, GxMy denotes the P(GMAx-stat-(MAm-GFF)y) macro CTAs and GxMyHz denotes the P(GMAx-stat-(MAm-GFF)y)-b-PHPMAz formulation.
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 568000 g mol−1 (EasiVial-Agilent).
568000 g mol−1 (EasiVial-Agilent).
TEM grids for imaging at 70 °C or 4 °C were prepared as follows: a 10% w/w PISA suspension was incubated at the desired temperature for 1 h, diluted to 0.1% w/w at the same temperature and this solution was deposited on a TEM grid and stained as described above. Adequately thermostated glassware was used for sample preparation.
Results and discussion
P(GMA-stat-MAm-GFF) macromolecular chain transfer agents (mCTAs)
Copolymers of the SAP-containing monomers MAm-GFF and GMA of various compositions were synthesized by RAFT in DMF using PETTC as the RAFT agent and ACVA as the initiator. The solid content in the reaction was about 20 wt% due to the low solubility of MAm-GFF in DMF. The copolymerization was carried out for 24 h. This duration led to the full conversion of GMA while 80% of MAm-GFF was copolymerized. According to the kinetics study performed using in situ1H NMR and reported in our previous work,1 MAm-GFF was indeed consumed slower than GMA. However, a statistical P(GMA-stat-(MAm-GFF)) copolymer was formed.The resulting mCTAs (mCTA 1, mCTA 2 and mCTA 3) were characterized by 1H NMR and SEC (Fig. S1, S2† and Table 1). The copolymerization proceeded as expected with a satisfactory level of control and the resulting mCTA had relatively low dispersity (<1.4). These 3 mCTAs afforded a clear solution in Milli-Q water at 10 wt%, enabling the aqueous dispersion PISA protocols.
TEM and DLS analyses were also performed to investigate the mCTA aqueous solutions used for the PISA experiments. 0.1 wt% DLS and TEM samples were prepared from 10 wt% stock solutions in Milli-Q water. As shown in our previous work, mCTA 2 formed large bundles of fibers/ribbons with diameters of 60–500 nm and an average length of 10 μm (Fig. 1). mCTA 1 also formed large bundles of fibers/ribbons. However, as shown in Fig. 1, the structures are not exactly identical. These structures likely still exist at 70 °C based on our previous study1 and the GMA/MAm-GFF ratios of mCTA 1 and mCTA 2 are similar. In contrast, no clear structure was found for the mCTA 3 aqueous solution. This suggests that this mCTA was molecularly dissolved at 0.1 wt% as expected given the GMA/MAm-GFF ratio (200/9) of this copolymer. Light scattering data confirmed the presence of objects with very high dispersity in both mCTA 1 and 2 solutions (Fig. S3†) in agreement with the anisotropic morphologies observed by TEM. No morphology was observed for the longer macro-CTA with fewer MAm-GFF units (mCTA 3), indicating full solubility under the tested conditions.
 | ||
| Fig. 1 Representative TEM images of mCTA 1 (G24M3) and mCTA 2 (G65M7) in Milli-Q water at 0.1 wt% and prepared at ambient temperature (about 25 °C). Note: no morphology was detected for mCTA 3. | ||
RAFT aqueous dispersion polymerisation of HPMA using the MAm-GFF-containing mCTAs
Amphiphilic P(GMAx-stat-(MAm-GFF)y)-b-PHPMAz (GxMyHz) block copolymers (BCPs) featuring different PHPMA block lengths were synthesized from the three MAm-GFF-containing mCTAs under aqueous RAFT PISA conditions. The polymerization proceeded without any issues, as expected, reaching full conversion (>99%) within 24 h and producing relatively well-defined block copolymers (Đ < 1.35, Fig. 2). The Mn and Đ values of all diblock copolymers are given in Table S1.†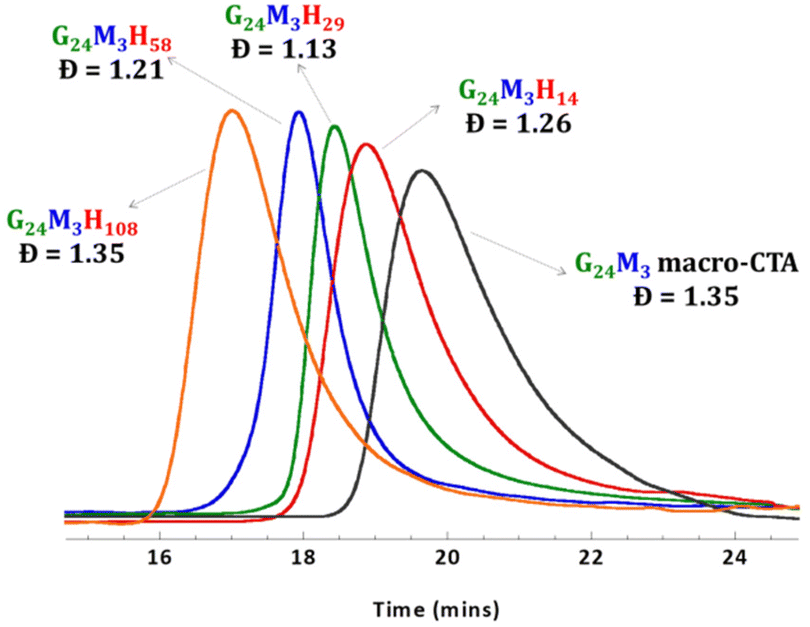 | ||
| Fig. 2 DMF SEC data (refractive index detector) for the P(GMA24-stat-(MAm-GFF)3) mCTA (mCTA 1) and the diblock copolymers G24M3H14–108. | ||
In all cases, the polymerization resulted in stable suspensions when low to moderate DPPHPMAs were targeted. These BCP suspensions were stable for months (>12). However, the block copolymers with a higher DPPHPMA phase separated over time into an excluded liquid phase and a viscous phase that could not be re-dispersed even after continuous vigorous stirring for up to 7 days (Fig. 3). Among the BCPs prepared from mCTA 1, only G24M3H14 formed a stable suspension. G24M3H29, G24M3H58 and G24M3H108 led to phase separation within two hours after polymerization. Similarly, the suspensions of G65M7H28 and G65M7H42 were stable while those of G65M7H54 and G65M7H63 phase-separated one hour after the polymerization, and G65M7H200 formed a gel during the PISA process (at 70 °C). In contrast, the diblock copolymers prepared using mCTA 3 formed stable suspensions. The cause of the phase separation is certainly due to the presence of SAP moieties, since such a phase separation has never been reported for any of the PGMA-b-PHPMA diblock copolymers synthesized under the same conditions (aqueous PISA at 10 wt%) even when very long PHPMA blocks (PGMA47-b-PHPMA310, PGMA78-b-PHPMA500, and PGMA112-b-PHPMA800)15,22 were targeted. As reported previously the presence of only a few units (3–7) of MAm-GFF is enough to profoundly change the morphologies of the self-assemblies of PGMA-b-PHPMA BCPs and the stabilities of their suspensions. Without doubt, the colloidal instability of these nanoparticle solutions is due to the reduced solubility of the SAP containing mCTAs. When the length of the mCTA is increased and the ratio of MAm-GFF/GMA is reduced as in the case of mCTA 3, stable colloidal solutions could be prepared even at very high DPs of PHPMA (≥500).
 | ||
| Fig. 3 Photographs of the G24M3H14 (left) and G24M3H58 (right) BCP reaction media after 24 h (aqueous RAFT-PISA carried out at 10 wt% in water at 70 °C). | ||
TEM analyses were performed for all diblock copolymers by simply diluting the stable suspension 100-fold or by re-dispersing the phase-separated solids (viscous phase that sedimented at the bottom of the vials) in Milli-Q water at 1 mg mL−1. Our initial study of this system had shown that G65M7H28 BCP PISA resulted in anisotropic rigid fibres that formed a fibrous network upon drying.1 Here, as shown in Fig. 4, the self-assembled morphologies observed post-mortem (at 25 °C) for the different GxMyHz series are diverse. In the G24M3Hz series, only G24M3H14 formed colloidally stable solutions. This series generally self-assembled into fibers of about 5 μm length, independently of their final colloidal stability. Increasing the length of the PHPMA block to 29 resulted in the formation of clustered fibers that formed separated bundles when z was increased to 58 and 108 with significantly more branching. This change in the morphology is no surprise as increasing the hydrophobicity of the BCP often results in the formation of higher order morphologies. However, maintaining the bundled fibrous networks (established as the specific feature of PISA formulations containing the MAm-GFF units) up to very high DPs of hydrophobic PHPMA (only 3 units of MAm-GFF with 108 units of HPMA) is remarkable. As reported in our previous work,1 in the absence of MAm-GFF, an increase in the length of PHPMA resulted in morphological changes towards worms and vesicles.
 | ||
| Fig. 4 Representative TEM images of G24M3H14–108 (top row), G65M7H42–200 (middle row) and G200M9H51–510 (bottom row) diblock copolymers prepared by RAFT-PISA in water at 10 wt% solids. Samples were diluted 100-fold at 30 °C. Red contour indicates the colloidally stable samples. | ||
For the G65M7Hz series (z ranging from 42 to 63), once again, bundles of fibers were observed (Fig. 4). The individual fibers seem identical in all the samples but they tended to arrange into different spherulitic structures (network-like for z = 42, bundles with a darker centre for z = 54 and sea urchin-like for z = 63). Finally, 55 nm diameter (measured by DLS) spheres were obtained for G65M7H200, suggesting almost complete suppression of the effect of the SAP since in the absence of the peptide sequence (G65H200), spheres of 87 nm were observed.23 It is important to note that some of these G65M7Hx formulations did not form stable suspensions. The TEM images of these samples were obtained by re-dispersing the colloidally unstable solution in water at a lower concentration (0.1 wt%) when possible, or by sampling and diluting the viscous/solid phase that sedimented at the bottom of the vials in Milli-Q water (at 1 mg mL−1).
The G200M9Hz series prepared from water-soluble mCTA 3 behaved quite differently. Only G200M9H51 led to spherulitic structures (Fig. 4). The other BCPs, G200M9H102, G200M9H250, and G200M9H510, self-assembled into more classical morphologies: fused spheres (dimers and trimers), spheres and vesicles, respectively (Fig. 4). These are the typical morphologies observed for the PGMA-b-PHPMA BCP suggesting that the self-assembly of G200M9Hx with x = 102–510 was not directed by the interactions of the GFF moieties but by the hydrophobicity/hydrophilicity balance as in the case of the PGMA-b-PHPMA system.
These results underline once again that the presence of very few units of MAm-GFF can have a significant impact on the morphology of PGMA-b-PHPMA polymerization-induced self-assembled objects. This impact of the SAP is all the more pronounced when the PGMA block is shorter. However, when the length of the stabilising block is relatively long and the ratio of GMA/MAm-GFF is low the effect of the peptide sequence is significantly reduced or completely suppressed particularly at high DPs of the core-forming block (PHPMA).
Temperature-induced morphological changes
Besides the composition of the block copolymers, experimental conditions such as ionic strength, pH and temperature are key parameters to access different self-assembled morphologies.24–27 The self-assembly of GxMyHz BCPs is driven not only by the hydrophilic–hydrophobic balance of the block copolymers but also by the non-covalent interactions (pi–pi and H-bonding) between the GFF sequences, more specifically due to the FF moieties. As the self-assembly based on FF is stabilized mostly via H bonding (FF–FF and FF–solvent) which is weakened or even completely suppressed at high temperature,28,29 the morphologies observed at ambient temperature (about 25–30 °C) might not be identical with the morphologies formed during the polymerization at 70 °C. In our previous work, the G65M7H28 suspension was shown to be composed of worm-like structures at 70 °C, and of flower-like fiber bundles at 20 °C.1 Therefore, the effect of temperature on the self-assembly of the G65M7Hz series was investigated. As expected, at 70 °C, no fibers or fiber bundles were observed for any G65M7Hz BCP. Instead, pure worms, a mixture of spheres and worms, beaded worms (pearl necklace-like morphology) and a mixture of worms and spheres were observed for G65M7H28, G65M7H42, G65M7H54, and G65M7H63, respectively (Fig. 5). In contrast, at both 70 °C and 30 °C, G65M7H200 formed spheres (Fig. 5). After a critical block length, high temperature has no effect on the self-assembly since the impact of the SAP becomes negligible. | ||
| Fig. 5 Representative TEM images of G65M7H28–200 diblock copolymers at 70 °C (top row), 30 °C (middle row) and 4 °C (bottom row) prepared by RAFT-PISA in water at 10 wt% solids. The samples were prepared at the corresponding temperatures (100-fold dilution and deposition on the TEM grid). | ||
This temperature-induced morphological change was also detected by DLS (Fig. 6). The suspension of G65M7H42 at 20 °C presented a broad average hydrodynamic diameter distribution, in agreement with the highly anisotropic structure observed in TEM. Upon heating from 20 °C to 70 °C, the average hydrodynamic particle diameter distributions gradually shifted towards smaller dimensions, and above 60 °C, only submicron particles were detected. This change was completely reversible as shown by the results of the DLS measurements carried out at 20 °C before and after heating to 70 °C, which were essentially identical. In contrast, the DLS data of G65M7H200 were not affected by heating up to 70 °C (Fig. 6). The particle hydrodynamic diameter distributions remained centred around 100 nm and became slightly narrower upon heating from 20 to 70 °C (Fig. 5), in agreement with the TEM images.
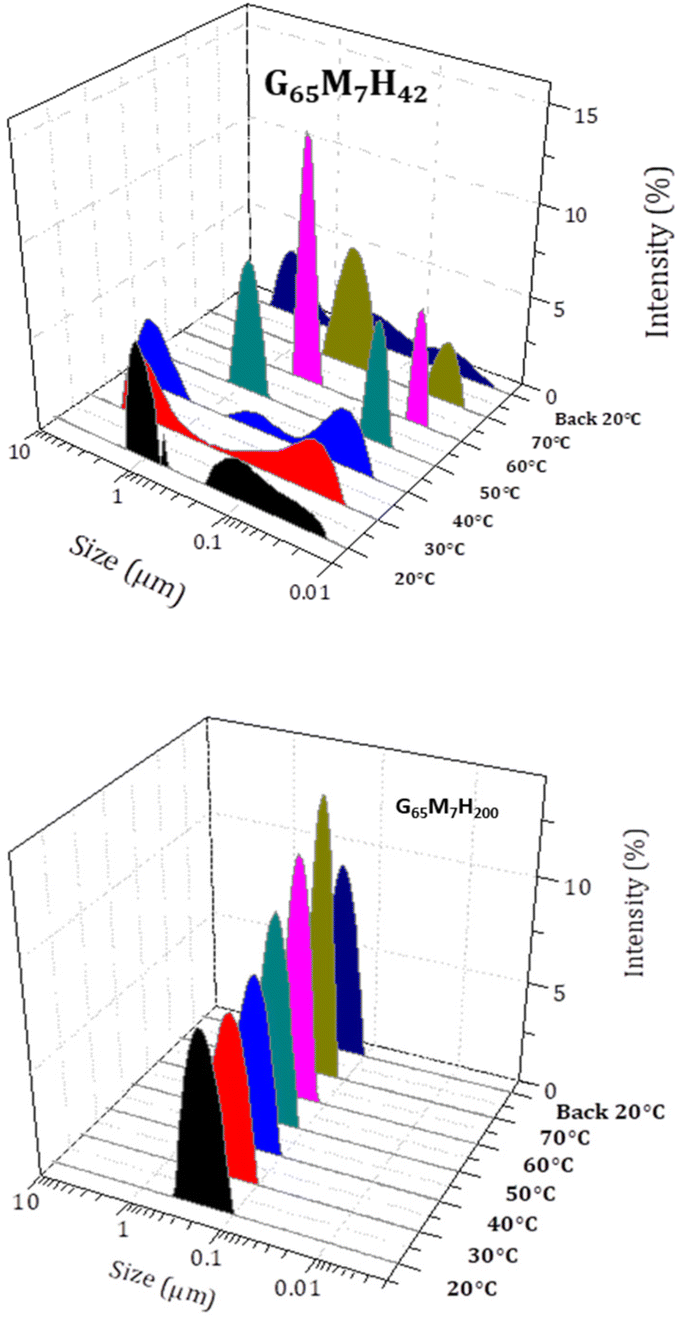 | ||
| Fig. 6 Evolution with temperature of the hydrodynamic diameter of G65M7H42 (top) and G65M7H200 (bottom) BCP objects at 0.1 wt% in Milli-Q water. | ||
Although heating disrupts the associative interactions between the GFF moieties, low temperatures have been reported to increase the hydrophilicity of PHPMA.30 Previous studies demonstrated that PGMA-b-PHPMA BCP morphologies undergo a worm-to-sphere transition upon cooling from 20 °C to 5 °C.31,32 Here, cooling also affected the P GxMyHz BCP morphologies. Upon cooling from 30 °C to 4 °C the self-assembled structures of the G65M7Hz series changed markedly from defined bundles to spheres. In the case of G65M7H28, cooling transformed the fiber bundles into not very defined fluffy bundles/balls (G65M7H28). In the case of G65M7H42, a temperature decrease led to the formation of individual spheres and small bundles of fibers, while for the G65M7H54 case the temperature change resulted in the formation of spheres coated with short fibers (sea urchins). For the longest hydrophobic block (G65M7H200) there was no transformation as the observed morphology at 30 °C was spheres. These unique structures have never been reported before once again highlighting the powerful structuring ability of the SAP moieties.
P(GMA-stat-(MAm-GFF))-b-PHPMA via RAFT dispersion PISA in water/ethanol mixtures
As mentioned above, some of the GxMyHz suspensions were not colloidally stable, likely due to the interactions between the GFF moieties located in the periphery of the self-assembled structures. To deal with this instability, RAFT polymerization was conducted in a water/ethanol binary mixture instead of pure water (MAm-GFF does not dissolve in water while it dissolves in ethanol up to 5% w/w at 30 °C). The RAFT dispersion polymerisation of HPMA using mCTA 2 (G65M7) was carried out in 50/50, 60/40 and 80/20 water/ethanol mixtures. These polymerization reactions reached 100% conversion of HPMA (G65M7H66), forming stable colloidal suspensions over the course of 12 months. Although the three reaction mixtures were slightly opaque, the samples in ethanol-rich binary mixtures (50/50 and 60/40) did not form defined structures as ethanol is a good solvent for PHPMA. In contrast, in an 80/20 water/ethanol mixture short rigid fibres of about 500 nm length were observed (Fig. 7). Unlike the fibrils formed in pure water (Fig. 4), these short fibers did not bundle. | ||
| Fig. 7 Representative TEM images of the G65M7H66 BCP prepared by RAFT-PISA in water/ethanol, 80/20 v/v. | ||
Conclusions
This work focuses on the morphology of P(GMA-stat-(MAm-GFF))-b-PHPMA (GMH) diblock copolymers synthesized via a one-step RAFT-mediated PISA approach. The effects of the composition of the GFF-containing corona-forming block and of the length of the core-forming block on the colloidal stability and the morphologies of the self-assembled structures were examined. Some of the PISA morphologies were colloidally stable for months while others sedimented and phase separated within a few hours after or even during the polymerization. Block extension of the shortest mCTA with the highest ratio of MAm-GFF/GMA units (mCTA 1) resulted in the formation of bundled fibres, similar to the morphology of mCTA1 alone. When a longer mCTA with a lower percentage of MAm-GFF (mCTA 2) was used, bundled fibers were again observed for low DPs of PHPMA. However, for a DP of PHPMA of above 54, first urchin-like structures and ultimately spheres were formed. This transformation suggests that above a threshold DP of the core-forming PHPMA block, the effect of the SAP becomes negligible and the self-assembly follows the “classical” regime dictated by the hydrophilic/hydrophobic balance under PISA conditions. Further reducing the MAm-GFF content in the mCTA (mCTA 3) resulted in the formation of urchin-like morphologies and finally spheres when the DP of PHPMA reached 510. All the examined formulations resulted in the formation of defined morphologies at 70 °C, 30 °C and 4 °C. At 70 °C, the impact of the SAP was weakened as only worms and spheres were observed (H-bonds are disrupted at high T). Upon cooling to ambient temperature, bundles of fibers were found as the main structures. The formation of such bundled fibres is the fingerprint of the GFF-sequence. Upon further cooling down to 4 °C, the morphology transformed from fiber bundles to spheres coated with short fibers that resemble sea urchins. Finally, the PISA of G65M7H66 in an 80/20 water/ethanol binary mixture resulted in the formation of colloidally stable non-bundled short fibers. This study confirms that including SAP moieties within the structure of block copolymers is an elegant approach to prepare self-assembled structures with rare and complex morphologies. Such a strategy provides a powerful tool for structural derivatization that can be used in combination with different SAP sequences and polymerization techniques. The reported novel morphologies could find use in the ever-changing fields of dynamic materials and drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The PDRA fellowship of T. P. Tuyen Dao was managed by the French National Research Agency (ANR): “investissement d'Avenir” LabEX CheMISyst, grant number ANR-10-LABX-05-01. The authors also thank CNRS for funding this work via the “Osez l'interdisciplinarité-2017” programme awarded to M. Semsarilar.Notes and references
- T. P. T. Dao, L. Vezenkov, G. Subra, M. Amblard, M. In, J.-F. Le Meins, F. Aubrit, M.-A. Moradi, V. Ladmiral and M. Semsarilar, Self-Assembling Peptide—Polymer Nano-Objects via Polymerization-Induced Self-Assembly, Macromolecules, 2020, 53, 7034–7043 CrossRef CAS.
- B. A. Parviz, D. Ryan and G. M. Whitesides, Using self-assembly for the fabrication of nano-scale electronic and photonic devices, IEEE Trans. Adv. Packag., 2003, 26(3), 233–241 CrossRef CAS.
- J. H. Fendler, Chemical Self-assembly for Electronic Applications, Chem. Mater., 2001, 13(10), 3196–32 CrossRef CAS.
- J. Zhang, Z. Sun and B. Yang, Self-assembly of photonic crystals from polymer colloids, Curr. Opin. Colloid Interface Sci., 2009, 14(2), 103–114 CrossRef CAS.
- C.-j. Liu, U. Burghaus, F. Besenbacher and Z. L. Wang, Preparation and Characterization of Nanomaterials for Sustainable Energy Production, ACS Nano, 2010, 4(10), 5517–5526 CrossRef CAS.
- S. Yadav, A. K. Sharma and P. Kumar, Nanoscale Self-Assembly for Therapeutic Delivery, Front. Bioeng. Biotechnol., 2020, 8, 127 CrossRef.
- S. I. Stupp, R. H. Zha, L. C. Palmer, H. Cui and R. Bitton, Self-assembly of biomolecular soft matter, Faraday Discuss., 2013, 166, 9–30 RSC.
- T. Shimizu, H. Minamikawa, M. Kogiso, M. Aoyagi, N. Kameta, W. Ding and M. Masuda, Self-organized nanotube materials and their application in bioengineering, Polym. J., 2014, 46(12), 831–858 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, Emerging Trends in Polymerization-Induced Self-Assembly, ACS Macro Lett., 2019, 8(8), 1029–1054 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polymerization techniques in polymerization-induced self-assembly (PISA), Polym. Chem., 2020, 11(22), 3673–3689 RSC.
- M. Semsarilar and V. Abetz, Polymerizations by RAFT: Developments of the Technique and Its Application in the Synthesis of Tailored (Co)polymers, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, RAFT-Mediated Polymerization-Induced Self-Assembly, Angew. Chem., Int. Ed., 2020, 59, 8368 CrossRef.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- N. J. Warren and S. P. Armes, Polymerization-Induced Self-Assembly of Block Copolymer Nano-objects via RAFT Aqueous Dispersion Polymerization, J. Am. Chem. Soc., 2014, 136(29), 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Industrially-relevant polymerization-induced self-assembly formulations in non-polar solvents: RAFT dispersion polymerization of benzyl methacrylate, Polym. Chem., 2015, 6(16), 3054–3062 RSC.
- V. Ladmiral, A. Charlot, M. Semsarilar and S. P. Armes, Synthesis and characterization of poly(amino acid methacrylate)-stabilized diblock copolymer nano-objects, Polym. Chem., 2015, 6, 1805–1816 RSC.
- L. Luppi, T. Babut, E. Petit, M. Rolland, D. Quemener, L. Soussan, M. A. Moradi and M. Semsarilar, Antimicrobial polylysine decorated nano-structures prepared through polymerization induced self-assembly (PISA), Polym. Chem., 2019, 10, 336–344 RSC.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Proapoptotic Peptide Brush Polymer Nanoparticles via Photoinitiated Polymerization-Induced Self-Assembly, Angew. Chem., Int. Ed., 2020, 59, 19136–19142 CrossRef CAS PubMed.
- T. P. T. Dao, L. Vezenkov, G. Subra, V. Ladmiral and M. Semsarilar, Nano-assemblies with core-forming hydrophobic polypeptide via polymerization-induced self-assembly (PISA), Polym. Chem., 2021, 12(1), 113–121 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Anionic Polyelectrolyte-Stabilized Nanoparticles via RAFT Aqueous Dispersion Polymerization, Langmuir, 2012, 28(1), 914–922 CrossRef CAS PubMed.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Critical Dependence of Molecular Weight on Thermoresponsive Behavior of Diblock Copolymer Worm Gels in Aqueous Solution, Macromolecules, 2018, 51(21), 8357–8371 CrossRef CAS.
- Y. Li and S. P. Armes, RAFT Synthesis of Sterically Stabilized Methacrylic Nanolatexes and Vesicles by Aqueous Dispersion Polymerization, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- J. Zhang, S. You, S. Yan, K. Müllen, W. Yang and M. Yin, pH-responsive self-assembly of fluorophore-ended homopolymers, Chem. Commun., 2014, 50, 7511–7513 RSC.
- R. Wakabayashi, A. Higuchi, H. Obayashi, M. Goto and N. Kamiya, pH-Responsive Self-Assembly of Designer Aromatic Peptide Amphiphiles and Enzymatic Post-Modification of Assembled Structures, Int. J. Mol. Sci., 2021, 22(7), 3459 CrossRef CAS.
- V. Bennevault, C. Huin, P. Guégan, K. Evgeniya, X. P. Qiu and F. M. Winnik, Temperature sensitive supramolecular self assembly of per-6-PEO-β-cyclodextrin and α,ω-di-(adamantylethyl)poly(N-isopropylacrylamide) in water, Soft Matter, 2015, 11(32), 6432–6443 RSC.
- D. Zhou, S. Dong, R. P. Kuchel, S. Perrier and P. B. Zetterlund, Polymerization induced self-assembly: tuning of morphology using ionic strength and pH, Polym. Chem., 2017, 8(20), 3082–3089 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Peptide self-assembly: thermodynamics and kinetics, Chem. Soc. Rev., 2016, 45(20), 5589–5604 RSC.
- R. Huang, Y. Wang, W. Qi, R. Su and Z. He, Temperature-induced reversible self-assembly of diphenylalanine peptide and the structural transition from organogel to crystalline nanowires, Nanoscale Res. Lett., 2014, 9(1), 653 CrossRef.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Order–Order Morphological Transitions for Dual Stimulus Responsive Diblock Copolymer Vesicles, Macromolecules, 2016, 49(3), 1016–1025 CrossRef CAS PubMed.
- R. Verber, A. Blanazs and S. P. Armes, Rheological studies of thermo-responsive diblock copolymer worm gels, Soft Matter, 2012, 8, 9915–9922 RSC.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, Sterilizable Gels from Thermoresponsive Block Copolymer Worms, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01203k |
| This journal is © The Royal Society of Chemistry 2023 |