
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelated translational motions in pseudo-rotaxane complexes controlled by a single chemical stimulus†
Naoko
Kotera
*a,
Pilar
Montellano
a,
Aldo C.
Catalán
a,
Anayeli
Carrasco-Ruiz
b,
Ruy
Cervantes
 a and
Jorge
Tiburcio
*a
a and
Jorge
Tiburcio
*a
aDepartment of Chemistry, Center for Research and Advanced Studies (Cinvestav), Avenida IPN 2508, Mexico City, 07360, Mexico. E-mail: jtiburcio@cinvestav.mx
bFacultad de Ciencias Básicas, Ingeniería y Tecnología, Universidad Autónoma de Tlaxcala, Apizaco 90341, Tlaxcala, Mexico
First published on 24th November 2023
Abstract
Coordinated motions are essential in the operation of molecular machines. This feature can be achieved by landscaping the energy surface along the movement coordinates. Herein, we present an approach of using a single stimulus to modify the free energy curve describing the threading and shuttling of a ring along a linear molecule. This approach has been realized by locating two identical ring-binding sites near the axle termini.
Molecular entangled chemical species, such as catenanes and rotaxanes, are attractive entities for the development of artificial molecular machines and motors.1 Their appeal relies on the ability to use an external stimulus to control the relative motions of their molecular components.2 A common precursor for rotaxanes and catenanes are pseudo-rotaxane complexes; the latter species are composed of a linear molecular axle threaded into a macrocyclic ring.3 As opposed to rotaxanes, in a pseudo-rotaxane, the ring can freely slip on and off, yielding a chemical equilibrium in solution between complex and free species. Additionally, when the axle features more than one recognition motif, the ring can undergo a translational motion between sites, generating a dynamic complex known as a molecular shuttle.4
It was demonstrated that axle end group bulkiness dictates ring threading rate;5 this relationship was rationalized by the tight fit between terminal groups on the guest and ring cavity size, in addition to conformational changes required during the slipping process.6 Conversely, it was proven that electrostatic interactions during the sliding process play a determinant role on the threading rate. Overall, these coulombic effects were applied to speed up,7 slow down,8 or even fully stop ring sliding motions.9
Similarly, for bistable molecular shuttles, the influence of the environment,10 macrocycle size,11 end groups,12 and length between stations13 on the ring translational motion was elucidated.
Typically, the ring-binding motif of the axle is located near the center of the axle, and electronically disconnected from the terminal groups. We hypothesized that by locating chemically sensitive binding sites at both axle termini, we could simultaneously affect ring sliding and shuttling motions with a single stimulus (Fig. 1).
 | ||
| Fig. 1 Energy curve (gray) representing translation of a ring along a linear molecule. The expected variation in the energy profile, induced by a stimulus affecting terminal groups and recognition sites, is shown in red. | ||
We previously investigated a similar design using bulky phosphonic groups symmetrically attached to a viologen unit in the axle and an anionic crown ether ring. The negatively charged phosphonate groups, i.e., in their basic form, become electrostatic stoppers, impeding the threading/dethreading motion and yielding the conversion of pseudo-rotaxane species into a rotaxane complex.14
Now, we proposed replacing the bulky phosphonic acid groups with smaller carboxylic acid units to allow unrestricted chemical exchange between bound and unbound species. This modification was designed to yield a correlated ring shuttling motion in the complex species. Moreover, upon the addition of a base, formation of negatively charged carboxylate groups was expected to yield changes in complex stability, sliding, and shuttling rates.
Herein, we report a viologen-based axle, featuring chemically sensitive propionic acid groups at both ends. These groups play a dual role: degenerating binding sites for a single crown ether ring and acting as handles to simultaneously control the sliding and shuttling ring motions.
We started by setting out to prove that a propionic acid group attached to a viologen core performs as a recognition motif for a crown ether. Consequently, we designed and prepared a nonsymmetric viologen guest, namely [1]2+, with a single binding site, and investigated its interaction with macrocyclic host [DSDB24C8]2− (Scheme 1).
 | ||
| Scheme 1 Chemical structures of host and guest compounds. | ||
Guest [1]2+ was synthesized in two steps, specifically through successive quaternization of 4,4′-bipyridine with benzyl bromide and 3-bromopropionic acid. This compound was fully characterized using proton and carbon-13 nuclear magnetic resonance (1H and 13C NMR) spectroscopies as well as mass spectrometry (see ESI, pages S2–S7†). The macrocycle [DSDB24C8]2− host was prepared as a tetramethylammonium salt and isolated as its anti-isomer according to our published method.15
Addition of [NMe4]2[DSDB24C8] to a colorless methanol solution of viologen [1][Br]2 produced a bright yellow color, suggesting the formation of a charge-transfer complex between the electron-rich crown ether and electron-poor viologen. This effect was accompanied by the appearance of a new set of resonances in the 1H NMR spectrum, recorded in CD3OD (Fig. 2a). In the presence of 0.5 mole equivalent of [DSDB24C8]2−, two sets of signals were observed, corresponding to bound and unbound species. However, in an equimolar solution, only resonances for the bound species were detected, supporting the formation of a host–guest complex with an association constant above 104 M−1, under the experimental conditions.
 | ||
Fig. 2 (a) 1H NMR spectra (500 MHz, CD3OD, 293 K, 5.0 × 10−3 M) of guest [1][Br]2 (blue trace); [1][Br]2 and [NMe4]2[DSDB24C8], 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 mole equivalent (black trace), [1][Br]2 and [NMe4]2[DSDB24C8], 1.0:1.0 mole equivalent (gray trace); and host [NMe4]2[DSDB24C8] (red trace); (b) partial 2D ROESY NMR spectrum of a solution formed from [1][Br]2 (5 × 10−3 M) and [NMe4]2[DSDB24C8] (2.5 × 10−3 M) with tm = 1.5 s (500 MHz, CD3OD, 293 K). EXSY-related peaks are shown in red; NOESY-related peaks, in blue. Proton assignments: blue, unbound [1][Br]2; black, complexed guest [1]2+; (c) ball-and-stick (C black, N blue, O red, H white, and S yellow) and space-filling (guest blue; host orange) representations of complex [1⊂DSDB24C8] determined using single-crystal X-ray diffraction. :0.5 mole equivalent (black trace), [1][Br]2 and [NMe4]2[DSDB24C8], 1.0:1.0 mole equivalent (gray trace); and host [NMe4]2[DSDB24C8] (red trace); (b) partial 2D ROESY NMR spectrum of a solution formed from [1][Br]2 (5 × 10−3 M) and [NMe4]2[DSDB24C8] (2.5 × 10−3 M) with tm = 1.5 s (500 MHz, CD3OD, 293 K). EXSY-related peaks are shown in red; NOESY-related peaks, in blue. Proton assignments: blue, unbound [1][Br]2; black, complexed guest [1]2+; (c) ball-and-stick (C black, N blue, O red, H white, and S yellow) and space-filling (guest blue; host orange) representations of complex [1⊂DSDB24C8] determined using single-crystal X-ray diffraction. | ||
To obtain a more accurate value of the association constant, we performed a titration of guest [1]2+ in methanol through successive additions of a methanol solution containing crown ether [DSDB24C8]2− and followed the response in the electronic spectrum at a wavelength of 380 nm (see ESI, page S26†). Data fitting using a 1:1 stoichiometry model provided a Ka value of 8.9(0.9) × 105 M−1; (ΔG° = −33.4(0.3) kJ mol−1), a value two orders of magnitude higher than that we found for paraquat2+/[DSDB24C8]2− system using a related method.16
The chemical shift resonances for the bound species were consistent with the formation of an interpenetrated complex. Upon binding, macrocycle aromatic (Hx, Hy, and Hz) as well as viologen (Hd, He, and Hf) signals shifted to a lower frequency (Hx: Δδ = −0.40; Hy: Δδ = −0.47; Hz: Δδ = −0.38; He,d: Δδ = −0.96; and Hf: Δδ = −0.40 ppm), indicating aromatic stacking between pyridinium and catechol rings. In contrast, propionic methylene resonances (Ha and Hb) shifted to a higher frequency (Ha: Δδ = +0.40; Hb: Δδ = +0.27 ppm), possibly due to hydrogen bonding with crown ether oxygen atoms in the cavity. Moreover, guest phenyl and benzyl resonances were practically unperturbed, indicating that the crown ether ring only resided on the propionic site. Furthermore, a 1H NMR rotating-frame Overhauser effect (ROESY) experiment demonstrated a through-space correlation between ethylene glycol macrocycle protons and ortho-N+ viologen protons at the propionic site (Fig. 2b), implying their locations near each other. 1H NMR experiments at temperatures between 313 and 183 K did not provide evidence of ring shuttling, assuring that the macrocycle stayed exclusively over the propionic station.
Integral values derived from the 1H NMR spectrum indicated the presence of a complex with one macrocycle per viologen, and no evidence of higher stoichiometries were obtained. An additional confirmation was provided using mass spectrometry. In the mass spectrum, a single peak was observed, corresponding to the ion pair complex [1 + DSDB24C8 + H]+ (exp: m/z 927.26758; calc.: m/z 927.26745; error: 0.1 ppm, ESI page S23†).
All these results agreed with the formation of a stable [2]pseudo-rotaxane complex [1⊂DSDB24C8] in solution, with the macrocycle residing on the propionic binding station.
The entangled structure of complex [1⊂DSDB24C8] was confirmed from an X-ray analysis (Fig. 2c).17 In the solid state, the host–guest complex was observed to adopt a threaded geometry with propionic methylene units standing at the center of the host cavity, forming weak hydrogen-bond interactions with the crown ether oxygen atoms (average C⋯O distances 3.43 Å, average C–H⋯O angles 146.29°). The macrocycle was observed to adopt a C-type conformation, favoring an aromatic π-stacking interaction between the two electron-rich sulfonated catechol rings and the electron-deficient pyridinium rings (with an average distance of 3.63 Å between host and guest aromatic rings and an average dihedral angle of 10.6°). Ion–dipole interactions were also indicated to play a major role in maintaining the interlaced structure, with the shortest N+⋯O distance being 3.09 Å, established between the nitrogen at the pyridinium and one of the oxygen atoms in the crown ether cavity.
Having established a positive role of the N+-propionic group as a binding motif for the anionic dibenzo 24-crown-8 ether macrocycle, we studied symmetrical guest [2]2+, a structure featuring two identical propionic binding sites and capable of forming a bistable [2]pseudo-rotaxane complex.
Guest [2]2+ was synthesized as a bromide salt by performing an alkylation of 4,4′-bipyridine with 3-bromopropionic acid18 and was characterized using NMR spectroscopy and mass spectrometry (see ESI, pages S8–S12 and S17†).
Compound [2][Br]2 formed a colorless solution in methanol; addition of one equivalent of [NMe4]2[DSDB24C8] produced a bright yellow solution suggesting the formation of a charge-transfer complex. As previously observed for complex [1⊂DSDB24C8], emergence of a new set of resonances in the 1H NMR spectrum recorded in CD3OD was observed, confirming the formation of a new host–guest complex, namely [2⊂DSDB24C8]. Mass spectrometry of this material demonstrated a peak related to the ion [2 + DSDB24C8 + H]+, proving a 1:1 stoichiometry (exp: m/z 909.24109; calc.: m/z 909.24163; error: 0.6 ppm, ESI page S24†). Due to the resonances for the complex being more intense than the sets observed for unbound host and guest, we were able to obtain a Ka value by conducting an electronic spectroscopy titration (see ESI, page S26†). Data fitting afforded a Ka of 7.2(0.7) × 105 M−1; (ΔG° = −32.9(0.3) kJ mol−1); similar to that found for complex [1⊂DSDB24C8].
Upon the binding to form complex [2⊂DSDB24C8], macrocycle aromatic protons (Hx, Hy, and Hz) and viologen ortho-N+ and meta-N+ protons (Hc and Hd) shifted to a lower frequency (Hx: Δδ = −0.38; Hy: Δδ = −0.47; Hz: Δδ = −0.35; Hd: Δδ = −0.95; and Hc: Δδ = −0.18 ppm), indicating aromatic π-stacking interactions between the electron-rich catechol and electron-poor viologen. Conversely, propionic methylene protons (Ha and Hb) shifted to a higher frequency (Ha: Δδ = +0.18 and Hb: Δδ = +0.08), implying hydrogen-bond interactions with the macrocycle cavity oxygen atoms. Interestingly, the observed shifts for protons Ha, Hb, and Hc were approximately half of those observed for analogous protons on the single-station complex [1⊂DSDB24C8], suggesting the formation of a mobile complex, with the ring dividing its residence time between two identical stations, at a rate faster than the NMR timescale. This feature was also implied by the broad resonances recorded in the 1H NMR spectrum (see ESI, page S19†).
To further understand the dynamic behavior of the [2⊂DSDB24C8] complex, we acquired its 1H NMR spectra at various temperatures, specifically from 183 to 313 K (Fig. 3). Broad single resonances corresponding to viologen aromatic protons (Hc and Hd) were observed above 293 K, but each peak transformed into two similar signals at low temperature (with coalescence temperatures of 263 K and 243 K, respectively). This result confirmed that the macrocycle underwent a shuttling motion between two degenerate binding stations on the axle. According to our interpretation of the results, above room temperature, the macrocycle translocated rapidly on the NMR time scale, averaging the resonances of both recognition sites, whereas, as the temperature decreased, the shuttling became slower, and the occupied and unoccupied stations were differentiated. The energy barrier for shuttling (ΔG‡shuttling) was calculated by applying the Eyring equation to the case of two equally populated sites19 and was found to be 51(1) kJ mol−1; from this quantity, a shuttling rate of 4.6 × 103 s−1 was estimated at 293 K (Table 1).
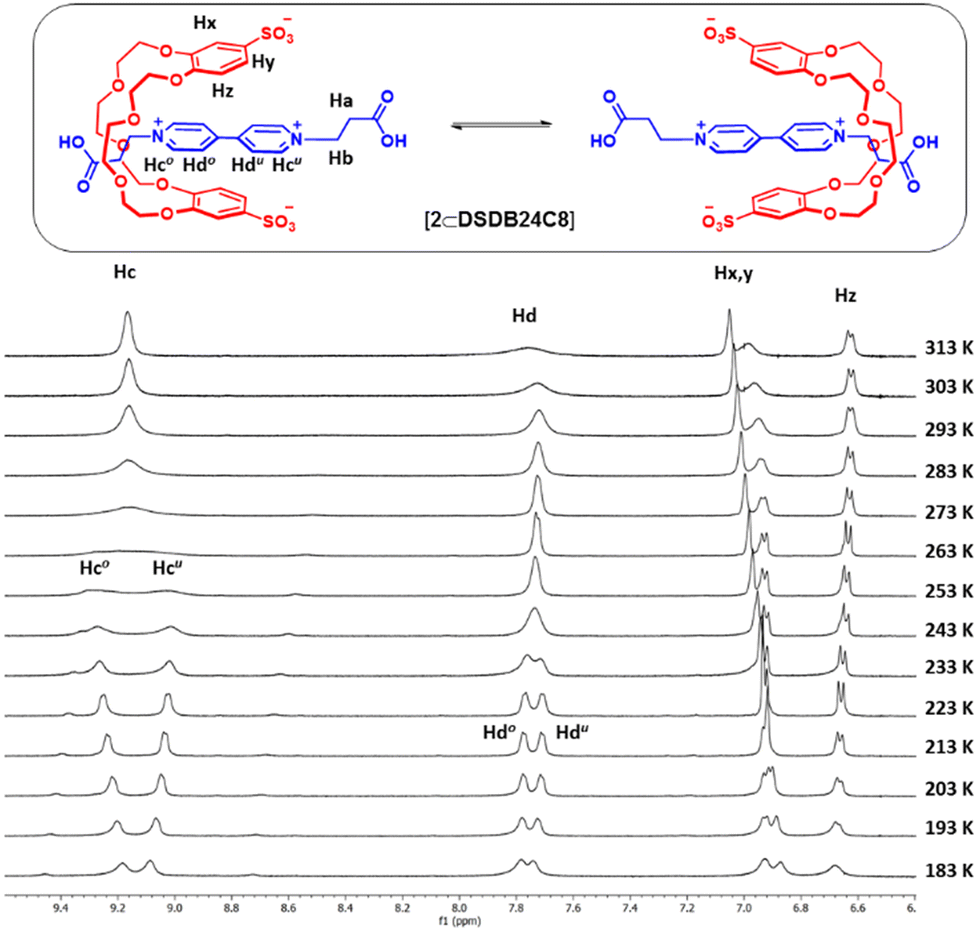 | ||
| Fig. 3 (Lower panel) Partial VT 1H NMR spectra (500 MHz, 5 × 10−3 M, CD3OD) for a 1:1 mixture of [NMe4]2[DSDB24C8] and [2][Br]2 (o = occupied site; u = unoccupied site). (Upper panel) Depiction of the shuttling motion of the macrocycle in the bistable complex. | ||
| [2⊂DSDB24C8] | [3⊂DSDB24C8]2− | |
|---|---|---|
| a Estimated errors are shown in parentheses. | ||
| ΔG° (kJ mol−1) | −32.9(0.3) | −10.7(0.1) |
| K a (M−1) | 7.2(0.7) × 105 | 8.1(0.8) × 101 |
| ΔG‡on (kJ mol−1) | 36.4(0.5) | 62.2(0.1) |
| t 1/2 (s) | 0.2 | 1.1 |
| ΔG‡shuttling (kJ mol−1) | 51(1) | 46(1) |
| SF (s−1) | 4.6 × 103 | 3.9 × 104 |
| SN | 9.8 × 102 | 4.3 × 104 |
To fully characterize the ring translational motion, the energy barrier for the slipping-on process (ΔG‡on) was calculated. Rate constants at each temperature were obtained by changing mixing times in the NMR ROESY spectra and measuring integral values of the cross peaks, specifically those originated from bound and unbound guest meta-N+ protons (see ESI, page S28–S31†).20 Furthermore, applying the Eyring equation provided a ΔG‡on of 36.4(0.5) kJ mol−1. From this value and association constant data, the first-order rate constant for the slipping-off route (koff) was estimated to be 3.5 s−1, corresponding to a half-life (t1/2) of approximately 0.2 s for the complex (Table 1).
We proposed that ring sliding and shuttling are correlated motions, due to both motions being connected to the free energy curve along the ring position coordinate. The ring would move from the solution into the axle in the first case and back-and-forth between stations along the axle in the second case. To characterize these motions, we introduced two new parameters:21 the shuttling frequency (SF), representing the average number of the ring traveling cycles between stations per second and numerically equal to the shuttling rate, and the dimensionless shuttling number (SN), indicating the number of ring traveling cycles between stations before complex dissociation and derived from the product of SF and complex t1/2 (Table 1).
Finally, we set out to achieve chemical control over the threading and shuttling motions. In situ addition of four equivalents of base [NEt(i-Pr)2] to a solution containing a 1:1 mixture of [2]2+ and [DSDB24C8]2− immediately changed the solution color, from bright yellow to pale yellow. In the 1H NMR spectrum, two sets of resonances were clearly observed: one set corresponding to unbound crown ether and zwitterionic [3], and the other to a new species in a minor proportion (see ESI, page S19†). As mentioned above, the integrals of the peaks indicated a 1:1 macrocycle-to-viologen stoichiometry. All resonances were sharp at 293 K, suggesting a slow exchange between the complex and free species. Although the chemical shifts differed from those of the protonated complex, their trends were similar. Upon the binding to form the unprotonated complex, aromatic macrocycle and meta-N+ protons shifted to a lower frequency (Hx: Δδ = −0.37; Hy: Δδ = −0.42; Hz: Δδ = −0.26; and Hd: Δδ = −0.97 ppm), indicating that aromatic stacking persisted in this complex, whereas methylene protons attached to the viologen shifted to a higher frequency (Ha: Δδ = +0.50 and Hb: Δδ = +0.12), suggesting the occurrence of hydrogen bonding. These observations were consistent with the formation of complex [3⊂DSDB24C8]2−.
The change in color and increase of the proportion of unbound species according to the 1H NMR spectrum were indicative of a less stable complex. From the 1H NMR data, the association constant for complex [3⊂DSDB24C8]2− was determined to be 8.1(0.8) × 101 M−1 at 293 K, a value at least four orders of magnitude lower than that of [2⊂DSDB24C8]. Removing the propionic acid proton yielded a drastic destabilization of the complex, probably due to the weakening of the hydrogen bonding, but also due to electrostatic repulsion between negatively charged sulfonate moieties on the ring and propionate groups on the axle.
Despite the remarkable reduction in stability, an interwoven complex was still formed, and its dynamic behavior was studied as mentioned above (see ESI, page S22†). Signal splitting at low temperature for ortho-N+, meta-N+ and methylene protons attached to the viologen proved the existence of shuttling. The free energy for shuttling (ΔG‡shuttling) was estimated to be 46(1) kJ mol−1. A comparison of the [2⊂DSDB24C8] and [3⊂DSDB24C8]2− complexes indicated that shuttling would occur one order of magnitude faster when the propionic end groups at the viologen are negatively charged.
Moreover, the energy barrier for slipping on (ΔG‡on) was also determined, using the same method as described above, yielding a value of 62.2(0.1) kJ mol−1 for complex [3⊂DSDB24C8]2−, with this value almost 26 kJ mol−1 higher than that of the neutral complex [2⊂DSDB24C8]. The observed difference can be explained by electrostatic repulsion between the electron-rich macrocycle cavity and the axle propionate groups during ring entry. Despite significant destabilization of the complex, the energy barrier for dethreading was elevated, yielding a longer half-life for complex [3⊂DSDB24C8]2−. Therefore, deprotonation of the carboxylic groups on the axle would accelerate ring SF and hinder complex dissociation, yielding a significantly higher SN in the anionic complex.
In summary, we presented a chemically sensitive viologen guest that pierces the cavity of a negatively charged crown ether to generate a pseudo-rotaxane complex. The presence of two identical recognition motifs on the viologen induces a ring shuttling motion. We demonstrated that by locating binding stations near axle termini, slipping and shuttling motions are simultaneously affected by a single chemical stimulus and are therefore correlated. We also introduced the use of SF and SN parameters to characterize these molecular motions. Removing protons at the propionic groups yield a less stable complex, with a longer half-life and higher SF.
Author contributions
N.K. Investigation, methodology, data curation, formal analysis, writing – original draft. P.M. investigation, methodology. A.C.C. investigation. A.C.R. formal analysis. R.C. formal analysis, data curation, writing – original draft. J.T. conceptualization, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from Conacyt project 255979 and SEP-Cinvestav Fund project 260. We thank Marco A. Leyva from Cinvestav for collecting the X-ray data. We also acknowledge LANCIC-IQ-UNAM (232619) for providing high-resolution mass spectra data. We thank both the anonymous reviewers for their insightful comments.References
- (a) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080 CrossRef CAS; (b) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094 CrossRef CAS.
- (a) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592 RSC; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed; (c) M. Baroncini, S. Silvi and A. Credi, Chem. Soc. Rev., 2020, 120, 200 CrossRef CAS.
- P. R. Ashton, D. Philp, N. Spencer and J. F. Stoddart, J. Chem. Soc., Chem. Commun., 1991, 1677 RSC.
- P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131 CrossRef CAS PubMed.
- F. M. Raymo;, K. N. Houk and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 9318 CrossRef.
- (a) M. A. Bolla, J. Tiburcio and S. J. Loeb, Tetrahedron, 2008, 64, 8423 CrossRef CAS; (b) J. Groppi, L. Casimiro, M. Canton, S. Corra, M. Jafari-Nasab, G. Tabacchi, L. Cavallo, M. Baroncini, S. Silvi, E. Fois and A. Credi, Angew. Chem., Int. Ed., 2020, 59, 14825 CrossRef CAS; (c) S. Chao, C. Romuald, K. Fournel-Marotte, C. Clavel and F. Coutrot, Angew. Chem., Int. Ed., 2014, 53, 6914 CrossRef CAS PubMed.
- (a) A. C. Catalán and J. Tiburcio, Chem. Commun., 2016, 52, 9526 RSC; (b) A. C. Catalán, A. A. Loredo, R. Cervantes and J. Tiburcio, ChemistryOpen, 2022, 11, e20220011 CrossRef PubMed.
- (a) A. E. Kaifer, W. Li, S. Silvi and V. Sindelar, Chem. Commun., 2012, 48, 6693 RSC; (b) A. Carrasco-Ruiz and J. Tiburcio, Org. Lett., 2015, 17, 1858 CrossRef CAS PubMed; (c) Y. Aeschi, L. Jucker, D. Häussinger and M. Mayor, Eur. J. Org. Chem., 2019, 21, 3384 CrossRef; (d) T. Ogoshi, D. Yamafuji, T. Aoki and T. Yamagishi, Isr. J. Chem., 2018, 58, 1246 CrossRef.
- Y. Kawaguchi and A. Harada, J. Am. Chem. Soc., 2000, 15, 3797 CrossRef.
- (a) S. Corra, C. de Vet, M. Baroncini, A. Credi and S. Silvi, Chem, 2021, 7, 2137 CrossRef CAS; (b) B. Wilson, L. Abdulla, R. Schurko and S. Loeb, Chem. Sci., 2021, 12, 3944 RSC; (c) S. Chen, D. Su, C. Jia, Y. Li, X. Li, X. Guo, D. A. Leigh and L. Zhang, Chem, 2022, 8, 243 CrossRef CAS.
- K. Hirose, Y. Shiba, K. Ishibashi, Y. Doi and Y. A. Tobe, Chem. – Eur. J., 2008, 14, 3427 CrossRef CAS.
- (a) T. Kumpulainen, M. R. Panman, B. H. Bakker, M. Hilbers, S. Woutersen and A. M. Brouwer, J. Am. Chem. Soc., 2019, 141, 19118 CrossRef CAS; (b) V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 20, 2185 RSC.
- (a) P. G. Young, K. Hirose and Y. Tobe, J. Am. Chem. Soc., 2014, 136, 7899 CrossRef CAS PubMed; (b) M. Douarre, V. Martí-Centelles, C. Rossy, I. Pianet and N. D. McClenaghan, Eur. J. Org. Chem., 2020, 5820 CrossRef CAS.
- R. A. Luna-Ixmatlahua, A. Carrasco-Ruiz, R. Cervantes, A. Vela and J. Tiburcio, Chem. – Eur. J., 2019, 25, 14042 CrossRef CAS PubMed.
- D. J. Hoffart, J. Tiburcio, A. de la Torre, L. K. Knight and S. J. Loeb, Angew. Chem., Int. Ed., 2008, 47, 97 CrossRef CAS.
- R. Cervantes, R. I. Sánchez and J. Tiburcio, Chem. – Eur. J., 2013, 19, 4051 CrossRef CAS PubMed.
- CCDC 2278846.†.
- H. Kamogawa and T. Suzuki, Bull. Chem. Soc. Jpn., 1987, 60, 794 CrossRef CAS.
- J. Sandström, Dynamic NMR Spectroscopy, Academic Press, 1982, ch. 6 and 7 Search PubMed.
- (a) T. Ogoshi, D. Yamafuji, T. Aoki and T. Yamagishi, J. Org. Chem., 2011, 76, 9497 CrossRef CAS PubMed; (b) G. Gholami, K. Zhu, G. Baggi, E. Schott, X. Zarate and S. J. Loeb, Chem. Sci., 2017, 8, 7718 RSC.
- These two parameters are reminiscent of TOF and TOF terms used in the field of catalysis. See for example: S. Kozuch and M. L. Martin, ACS Catal., 2012, 2, 2787 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2278846. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01741a |
| This journal is © The Royal Society of Chemistry 2024 |