
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tricyclic oxazinoindolones via Pd-catalyzed intramolecular addition of carboxylic acids to alkynes†
Subhamoy
Mukhopadhyay
,
Bhavya
Khaitan
 and
Shikha
Gandhi
*
and
Shikha
Gandhi
*
Department of Chemical Sciences, Indian Institute of Science Education and Research, Berhampur 760010, India. E-mail: sgandhi@iiserbpr.ac.in
First published on 14th November 2023
Abstract
A completely atom-economical synthesis of the oxazinoindolone core via the Pd-catalyzed intramolecular addition of carboxylic acids to alkynes has been developed. Oxazinoindolones have been known to have varied biological activities. The reaction proceeds via 6-exo-dig cyclization and affords the products in high yields (55–93%). The developed method demonstrates the applicability of a Pd(0) complex in combination with a substrate-tethered acid for the 1,2-addition of carboxylic acids to alkynes.
Fused polycyclic indoles constitute the structural core of a large number of bioactive natural products and pharmaceuticals.1 In particular, tricyclic oxazinoindolones, especially the 3,4-dihydro-1H-[1,4]oxazino[4,3-a]indol-1-ones, have attracted attention from the synthetic community owing to their varied biological activities, such as anticancer,2 antitubercular,3 and herbicidal activities.4 Among the different approaches adopted for the construction of this scaffold,5 an intramolecular cyclization of indole-2-carboxylic acid with an appropriate N-tethered alkyne, via a 6-exo-dig pathway, is arguably the most direct approach. In addition, it installs an exocyclic double bond in the oxazinone ring, making it suitable for further functionalization. Although iodolactonization utilizing this strategy with stoichiometric reagents has been reported (Scheme 1a),5d surprisingly, there have hardly been any reports on the synthesis of this core via a direct catalytic 1,2-addition of carboxylic acids to alkynes. This reaction is also completely atom-economical and thus offers an additional advantage. There has only been a single catalytic approach reported using AuCl3 as a catalyst which, however, has an extremely limited substrate scope (Scheme 1b).5e While this approach remains largely unexplored, a report published last year utilizes allenes generated from alkynes and an NHC-Au(I) complex as the catalyst for synthesizing the oxazinoindolone scaffolds (Scheme 1c).5b The procedure, however, requires an additional synthetic step and the use of relatively unstable allenes.
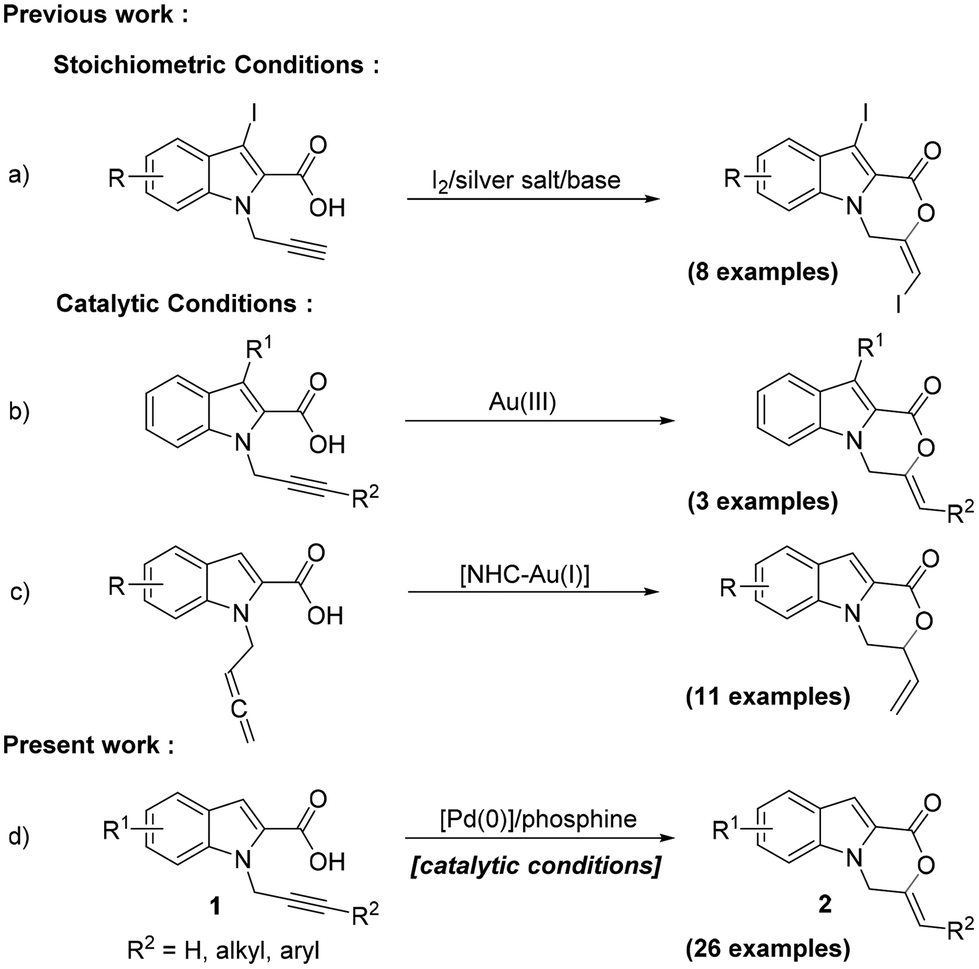 | ||
| Scheme 1 Oxazinoindolone synthesis via intramolecular addition of carboxylic acids to alkynes/allenes. | ||
Transition metal catalysis has been employed for the addition of carboxylic acids to alkynes, for both the inter- and intramolecular versions.6 The use of Pd has also been reported, mostly employing the Pd(II) catalysts.7 The application of a Pd(0) complex in combination with an acid for the intramolecular 1,2-addition of carboxylic acid to alkynes has however not been reported.8 We were intrigued to explore the application of such a species for the synthesis of the oxazino-indol-1-one core via a 6-exo-dig cyclization of 1. The presence of a carboxylic acid moiety in the substrate itself might make the addition of an external acid optional. We hereby report the results of our study on the cyclization of substrates 1 using Pd(PPh3)4 with tri-n-butylphosphine as the ligand and no additional acid (Scheme 1d).
We commenced our studies using 1a as the model substrate, 5 mol% of Pd(dba)2 as the catalyst and 10 mol% of PPh3 as the ligand (Table 1, entry 1). The reaction was conducted in toluene at 105 °C. After 24 h, 2a was isolated in 18% yield. We attempted to improve the yield by changing the phosphine ligand. An improvement was observed when tBuXPhos and JohnPhos were used as the ligands (Table 1, entries 2 and 3). However, the yield was still not optimal. We then changed the Pd source to Pd(PPh3)4. When no additional ligand was added, after 12 h, the product was isolated in 50% yield (Table 1, entry 4). With Pd(PPh3)4 as the Pd source, we then explored different phosphines as ligands (Table 1, entries 5–9). A jump in the yield was observed with PCy3 and JohnPhos as the ligands (Table 1, entries 5 and 6). With P(nBu)3 after 9 h, the product could be isolated in 85% yield (Table 1, entry 7).9 The reaction proved to be highly selective for 6-exo-dig cyclization and a (Z)-isomer of the product. Other phosphines did not offer any significant improvement (Table 1, entries 8 and 9). We also tested the reaction with Pd(OAc)2 and after 9 h at 105 °C, the product was obtained in only 34% yield (Table 1, entry 10). The yield increased to 63% when the reaction was conducted with Pd(OAc)2 and P(nBu)3, indicating Pd(0) to be the active species (Table 1, entry 11). Ag(OTf)2 and Cu(OTf)2 as the catalysts showed no conversion (Table 1, entries 12 and 13). When 2 eq. of TfOH were used, even after 48 h, no product could be isolated (Table 1, entry 14). We also tested base-promoted cyclization with K2CO3, which did not yield any product after 48 h (Table 1, entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), catalyst (5 mol%), ligand (10 mol%) and toluene (300 μL) were heated at 105 °C in a Schlenk tube under an Ar atmosphere. b Isolated yield. c 15 mol% ligand. d 10 mol% catalyst. e 2 eq. of TfOH were used. f 3 eq. of K2CO3 were used. | ||||
| 1 | Pd(dba)2 | PPh3 | 24 | 18 |
| 2 | Pd(dba)2 | t BuXphos | 24 | 23 |
| 3 | Pd(dba)2 | JohnPhos | 24 | 32 |
| 4 | Pd(PPh3)4 | — | 12 | 50 |
| 5 | Pd(PPh3)4 | PCy3 | 9 | 74 |
| 6 | Pd(PPh3)4 | JohnPhos | 9 | 73 |
| 7 | Pd(PPh3)4 | P(nBu)3 | 9 | 85 |
| 8 | Pd(PPh3)4 | ((p-CF3)C6H4)3P | 9 | 49 |
| 9 | Pd(PPh3)4 | dppf | 9 | 46 |
| 10 | Pd(OAc)2 | — | 9 | 34 |
| 11c | Pd(OAc)2 | P(nBu)3 | 9 | 63 |
| 12d | AgOTf | — | 9 | 0 |
| 13d | Cu(OTf)2 | — | 9 | 0 |
| 14e | TfOH | — | 48 | 0 |
| 15f | K2CO3 | — | 48 | 0 |
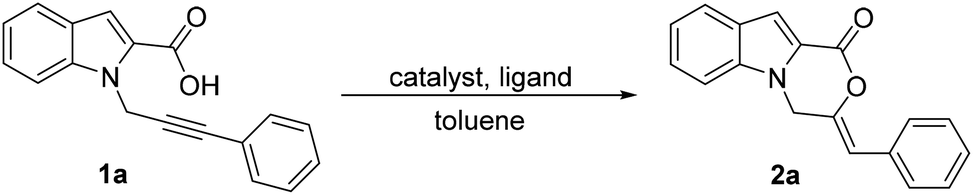
With the optimized reaction conditions in hand, the substrate scope of the reaction was then explored (Table 2). Substrates 1a–j with different R2 substituents were tested. Both the electron-donating and electron-withdrawing phenyl substituents were well tolerated. The substrate containing a para-fluoro phenyl ring attached to the alkyne moiety provided the product in 88% yield (Table 2, 2b). The substrates with para-chloro- and meta-bromo-substituted phenyl rings were also suitable and formed the cyclized products in high yields (Table 2, 2c and 2d). The introduction of electron-donating groups such as methyl and methoxy at different positions of the phenyl ring tethered to alkyne led to the formation of 6-exo-dig cyclized products selectively in good yields (Table 2, 2e–2g). A slight decrease in yields in such cases might be due to the reduced rate of carboxylate addition to the triple bond of the alkyne. A meta-chloro- and para-methyl-substituted phenyl ring was also well tolerated (Table 2, 2h). Notably, even an aliphatic methyl substituent on the alkyne was also found to be suitable (Table 2, 2i). An unsubstituted terminal alkyne moiety was also tested, and gratifyingly, the product was isolated in an excellent yield (Table 2, 2j). A biphenyl substituent could also be employed and the product was formed in 70% yield (Table 2, 2k). A heterocyclic group, 2-thiophene, was also tested and satisfyingly, the product was isolated in a high yield (Table 2, 2l). The structure and geometry of compound 2a were also confirmed by X-ray crystallography.10
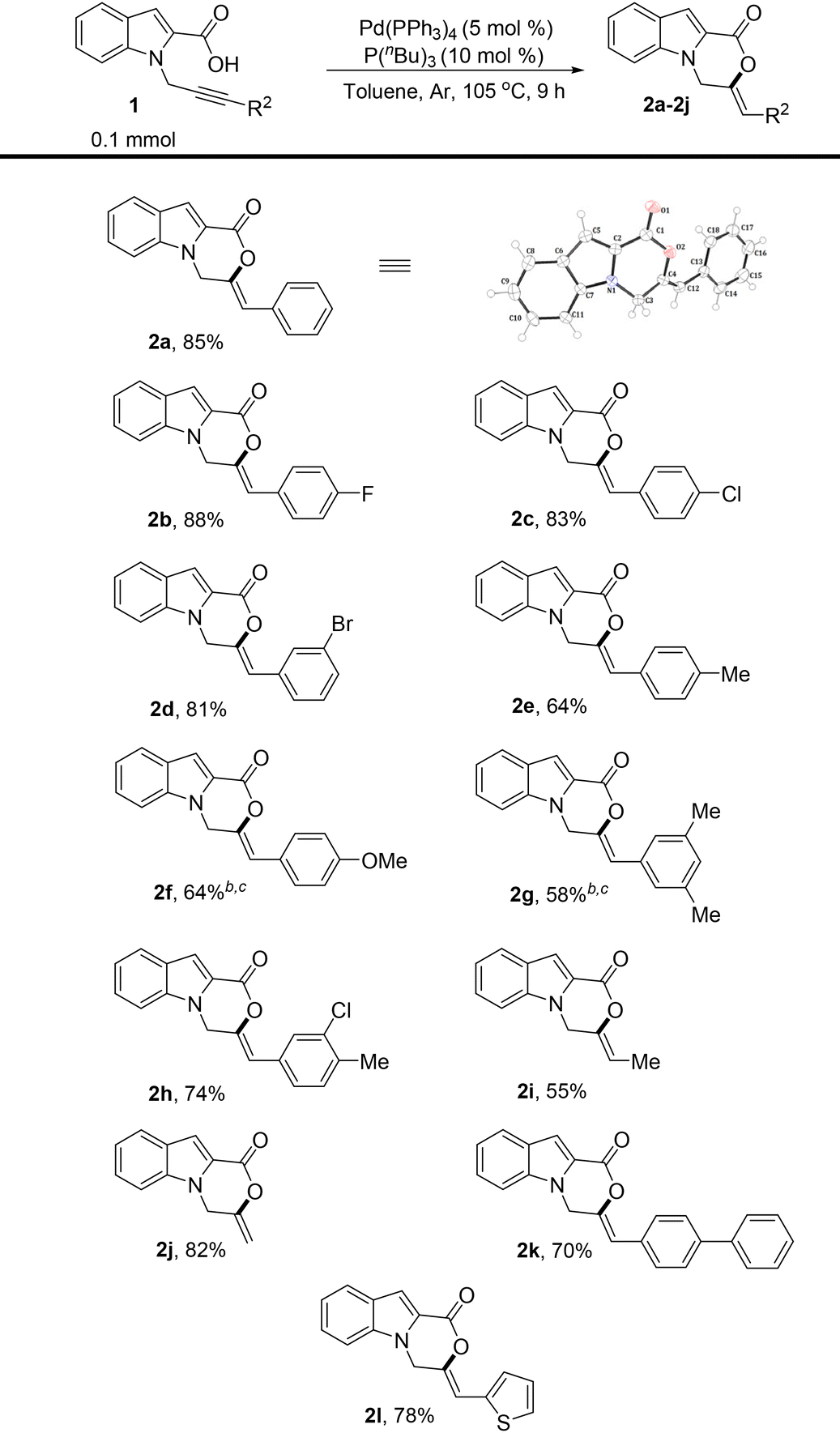
The substituent effects on the indole ring were also explored (Table 3). The introduction of electron-releasing substituents such as methoxy and methyl at different positions of the indole ring was tested, and the products were obtained in excellent yields when R2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ph was used (Table 3, 2m, 2n, and 2q). Notably, the terminal alkyne also cyclized well in all the cases tested (Table 3, 2o and 2p). The combination of electron-releasing substituents on the indole ring with different R2 substituents on alkyne was also explored, and remarkably, the reaction proved to be extremely tolerant of the different substitutions tested (Table 3, 2r–2v). The substrates containing electron-withdrawing groups on the indole ring worked well, albeit under slightly modified conditions, and afforded the products in good and high yields (Table 3, 2w–2z). A slight decrease in the yields in the case of 5-chloro-substituted indole ring might be attributed to the stronger electron-withdrawing effect of chlorine, thus reducing the rate of cyclization.
Ph was used (Table 3, 2m, 2n, and 2q). Notably, the terminal alkyne also cyclized well in all the cases tested (Table 3, 2o and 2p). The combination of electron-releasing substituents on the indole ring with different R2 substituents on alkyne was also explored, and remarkably, the reaction proved to be extremely tolerant of the different substitutions tested (Table 3, 2r–2v). The substrates containing electron-withdrawing groups on the indole ring worked well, albeit under slightly modified conditions, and afforded the products in good and high yields (Table 3, 2w–2z). A slight decrease in the yields in the case of 5-chloro-substituted indole ring might be attributed to the stronger electron-withdrawing effect of chlorine, thus reducing the rate of cyclization.
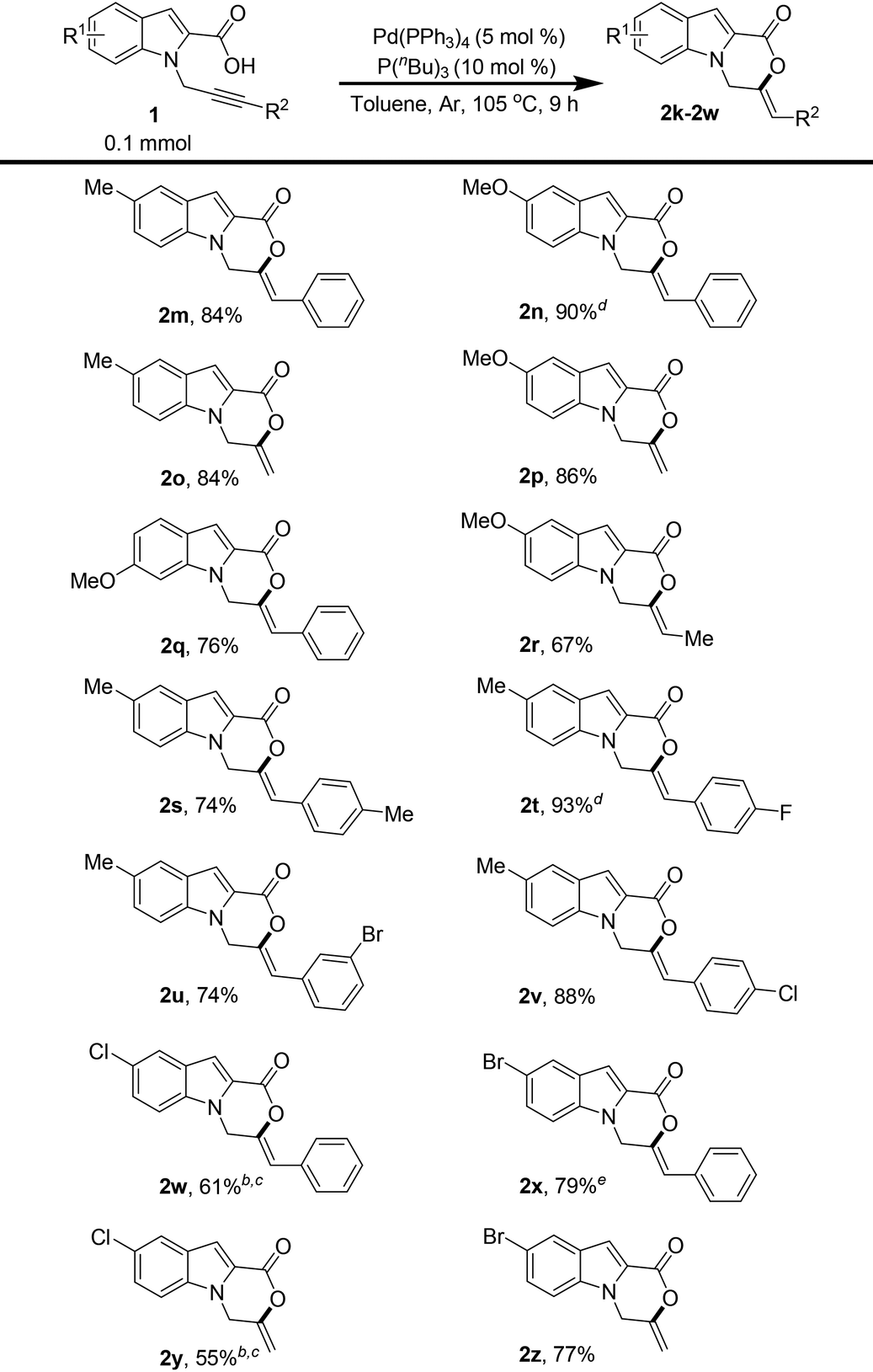
To further demonstrate a synthetic application of the product, compound 2p was reduced by hydrogenation on Pd/C. The reduction proceeded successfully and product 3 was isolated in 63% yield (Scheme 2).
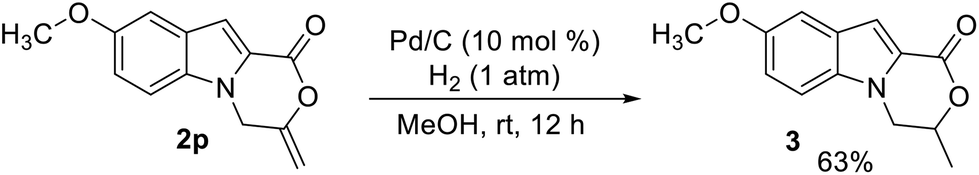 | ||
| Scheme 2 Synthetic transformation of 2p. | ||
Based on the mechanism proposed by Han11 and Hua12 and a mechanistic study by us,13 we hypothesize that the reaction proceeds via the hydropalladation of alkyne generating intermediate A (Scheme 3). A subsequent isomerization of A leads to intermediate B which undergoes reductive elimination to form the product and regenerate Pd(0) species. The mechanistic studies by Han et al.11 support the hydropalladation/isomerization sequence.
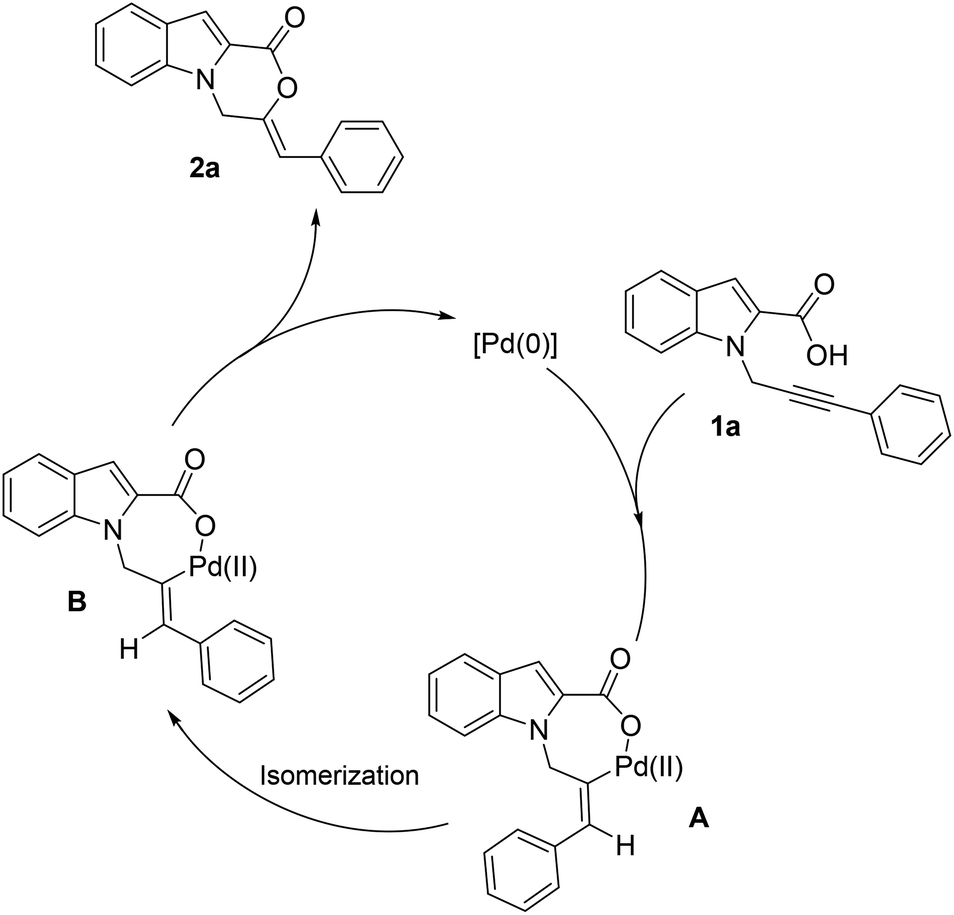 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed the Pd-catalyzed synthesis of the oxazinoindolone core via the intramolecular 1,2-addition of indole carboxylic acid to an N-tethered alkyne for the first time. The reaction selectively gave the 6-exo-dig cyclization products with the installation of an exocyclic double bond in the oxazinone ring. The reaction is completely atom economical, high yielding and widely tolerable to changing substitutions on the substrate. A Pd(0) complex in combination with an acid of the substrate is proposed to be the active catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB), Government of India (CRG/2018/000130) and IISER Berhampur is gratefully acknowledged. S. M. and B. K. thank IISER Berhampur for the fellowship. We thank Dr C. S. Purohit, School of Chemical Sciences, NISER Bhubaneshwar for help in recording the X-ray data.References
- (a) I. Ngantchou, B. Nyasse, C. Denier, C. Blonski, V. Hannaert and B. Schneider, Bioorg. Med. Chem. Lett., 2010, 20, 3495–3498 CrossRef CAS; (b) V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CrossRef CAS; (c) L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329–332 CrossRef CAS; (d) J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Org. Lett., 2007, 9, 4127–4129 CrossRef CAS.
- USPTO, US Patent, 20090192147:A1, 2009 Search PubMed.
- C. Maaliki, J. Fu, S. Villaume, A. Viljoen, C. Raynaud, S. Hammoud, J. Thibonnet, L. Kremer, S. P. Vincent and E. Thiery, Bioorg. Med. Chem., 2020, 28, 115579 CrossRef CAS.
- USPTO, US Patent, 5356862, 1994 Search PubMed.
- For reported methods, see: (a) F. Xiao, P. Liao, X. Lu, J. Wang, X.-Q. Dong and C.-J. Wang, Org. Lett., 2022, 24, 8592–8597; (b) R. Pedrazzani, E. Pinosa, G. Bertuzzi, M. Monari, S. Lauzon, T. Ollevier and M. Bandini, Chem. Commun., 2022, 58, 8698–8701 RSC; (c) C. Maaliki, J. Fu, S. Villaume, A. Viljoen, C. Raynaud, S. Hammoud, J. Thibonnet, L. Kremer, S. P. Vincent and E. Thiery, Bioorg. Med. Chem., 2020, 28, 115579 CrossRef CAS; (d) S. Hammoud, E. Anselmi, K. Cherry, J.-C. Kizirian and J. Thibonnet, Eur. J. Org. Chem., 2018, 2018, 6314–6327 CrossRef CAS; (e) S. Taskaya, N. Menges and M. Balci, Beilstein J. Org. Chem., 2015, 11, 897–905 CrossRef CAS; (f) J. K. Vandavasi, W.-P. Hu, G. C. Senadi, S. S. K. Boominathan, H.-Y. Chen and J.-J. Wang, Eur. J. Org. Chem., 2014, 2014, 6219–6226 CrossRef CAS; (g) V. A. Vaillard, R. A. Rossi and S. E. Martín, Org. Biomol. Chem., 2011, 9, 4927–4935 RSC.
- (a) V. Cadierno, Curr. Org. Chem., 2021, 25, 2260–2303 CrossRef CAS; (b) V. Cadierno, Catalysts, 2020, 10, 1206 CrossRef CAS; (c) N. T. Patil, R. D. Kavthe and V. S. Shinde, Tetrahedron, 2012, 68, 8079–8146 CrossRef CAS; (d) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3159 CrossRef CAS.
- (a) R. Mancuso, P. Russo, M. Lettieri, D. Santandrea, C. Cuocci and B. Gabriele, Adv. Synth. Catal., 2022, 364, 3917–3926 CrossRef CAS; (b) R. Mancuso, I. Ziccarelli, M. Novello, C. Cuocci, R. Centore, N. Della Ca’, D. Olivieri, C. Carfagna and B. Gabriele, J. Catal., 2022, 405, 164–182 CrossRef CAS; (c) G. Abbiati, E. M. Beccalli and E. Rossi, in Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 231–290 Search PubMed.
- For Pd(0)- and acid-catalysed allylation of acids with alkynes via allene intermediates, see: (a) Y. Ogiwara, K. Sato and N. Sakai, Org. Lett., 2017, 19, 5296–5299 CrossRef CAS; (b) Z. Huo, N. T. Patil, T. Jin, N. K. Pahadi and Y. Yamamoto, Adv. Synth. Catal., 2007, 349, 680–684 CrossRef CAS.
- Please refer to the ESI for the screening studies of solvent and catalyst loading.†.
- CCDC 2256568 (2a) contains the supplementary crystallographic data for this paper.†.
- R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto and L.-B. Han, J. Am. Chem. Soc., 2011, 133, 17037–17044 CrossRef CAS.
- J. Li, R. Hua and T. Liu, J. Org. Chem., 2010, 75, 2966–2970 CrossRef CAS.
- Please refer to the ESI for the mechanistic study.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2256568. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01672b |
| This journal is © The Royal Society of Chemistry 2023 |