
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective conjugate addition to nitroolefins catalysed by helical peptides with a single remote stereogenic centre†
David P.
Tilly
 *ab,
Catherine
McColl
ab,
Mingda
Hu
ab,
Iñigo J.
Vitórica-Yrezábal
a and
Simon J.
Webb
*a
*ab,
Catherine
McColl
ab,
Mingda
Hu
ab,
Iñigo J.
Vitórica-Yrezábal
a and
Simon J.
Webb
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: s.webb@manchester.ac.uk; david.tilly@manchester.ac.uk
bManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK
First published on 20th November 2023
Abstract
Two short pentapeptides rich in α-aminoisobutyric acid (Aib) residues have been shown to act as enantioselective organocatalysts for the conjugate addition of nucleophiles to nitroolefins. An L-alanine terminated peptide, (Aib)4(L-Ala)NHtBu, which has neither functionalised sidechains nor a highly designed reactive site, used an exposed N-terminal primary amine and the amide bonds of the backbone to mediate catalysis. Folding of this peptide into a 310 helical structure was observed by crystallography. Folding into a helix relays the conformational preference of the chiral alanine residue at the C-terminus to the primary amine at the N-terminus, 0.9 nm distant. The chiral environment and defined shape produced by the 310 helix brings the amine site into proximity to two exposed amide NHs. Reaction scope studies implied that the amine acts as a Brønsted base and the solvent-exposed NH groups of the helix, shown to weakly bind β-nitrostyrene, are needed to obtain an enantiomeric excess. Replacement of L-alanine with D-phenylalanine gave (Aib)4(D-Phe)NHtBu, a peptide that now catalysed the benchmark reaction with the opposite enantioselectivity. These studies show how achiral residues can play a key role in enantioselective catalysis by peptides through the promotion of folding.
Introduction
Catalysis by amino acids and short peptides has been proposed to be important for the prebiotic production of biomolecules.1 Catalytic peptides often use reactive sidechains to mediate reactivity, like those present in histidine and serine. Nonetheless, peptides composed of residues with simple alkyl sidechains, such as alanine, leucine, valine and α-amino-iso-butyric acid (Aib), can mediate catalysis through their amine and carboxylate termini, as well as the amide backbone.2Enantioselectivity during catalysis is another important outcome in a proposed prebiotic world. For chiral proteinogenic amino acids and their simple derivatives, the α-amine is available to stereoselectively catalyse reactions, with proline and the primary amino acids shown to act as stereoselective organocatalysts for aldol reactions.3–7 In peptides, any reactive sidechains of proteinogenic residues will also have adjacent stereogenic centres, which may make any catalysis enantioselective.2 Even in the absence of reactive sidechains, peptides with a chiral primary amino acid residue at the N-terminus can act as enantioselective primary amine catalysts, e.g. small di- to tetra-peptides are reported to catalyse asymmetric intermolecular aldol reactions in aqueous media with high asymmetric induction.2,7b,8,9 Some enzymes also use primary amines as catalytic groups, for example lysine residues in type I aldolases,10 with enantioselectivity arising from the shape of the active site.
The folding that forms the active site of enzymes can be replicated by peptides adopting different secondary structures. Turn motifs are one example, with tetrapeptides folded into β-turn motifs shown to catalyse aldol reactions,11–13 as have peptides containing a γ-turn.14 Similarly, α-helical structures, such as those formed by polyleucines,15 have been shown to act as enantioselective catalysts for aldol reactions16 and conjugate additions.17 Non-natural secondary structures are available to folded oligomers (foldamers). Foldamers, which replicate the folding of natural peptides and proteins, have been shown to act as organocatalysts.18–20 Formation of a chiral secondary structure by a foldamer can provide a source of enantioselectivity for an asymmetric transformation, in the place of a stereogenic centre close to the reactive site.19,21
In natural proteins and peptides, the 310 helix is a less common secondary structure than the α helix22 and there are few reports of 310 helical peptides acting as organocatalysts.23 However some non-proteinogenic quaternary residues like Aib can favour the adoption of this structure. Peptides with at least four Aib residues can fold into 310 helices that are stabilised by at least one intramolecular hydrogen bond.24 Since the Aib residue is achiral, a racemic mixture of right- (P) or left- (M) handed screw-senses is formed; these rapidly interconvert at room temperature in solution. However, attaching a chiral group either covalently25 or non-covalently26 to one terminus creates a relay of chiral conformations that propagate down the peptide body to the other terminus, leading to one screw-sense becoming favoured over the other.27 For example, Clayden and co-workers have shown that a single L-alanine residue at the C-terminus can produce a significant enhancement in the proportion of P helix (P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M of 88:12).28 Other L-amino acid residues at the C-terminus were found to produce a similar excess of P helix, including L-valine, L-tert-leucine and L-phenylalanine (P:M of 86:14, 83:17 and 85:15 respectively). Terminating with an N-(tert-butyl) amide gave strong helical induction across several chiral peptides, although only small differences were found between L-Ala containing peptides with Me and tBu secondary amide termini. The type of C-terminal capping group strongly affected the P:M ratio, with a L-alanine t-Bu ester C-terminus favouring the opposite, M, helical sense.28 In each case, the 310 helical structure of the peptide produces a chiral conformation at the N-terminus despite the stereogenic centre itself being remote from this site.
:M of 88:12).28 Other L-amino acid residues at the C-terminus were found to produce a similar excess of P helix, including L-valine, L-tert-leucine and L-phenylalanine (P:M of 86:14, 83:17 and 85:15 respectively). Terminating with an N-(tert-butyl) amide gave strong helical induction across several chiral peptides, although only small differences were found between L-Ala containing peptides with Me and tBu secondary amide termini. The type of C-terminal capping group strongly affected the P:M ratio, with a L-alanine t-Bu ester C-terminus favouring the opposite, M, helical sense.28 In each case, the 310 helical structure of the peptide produces a chiral conformation at the N-terminus despite the stereogenic centre itself being remote from this site.
Clayden and co-workers have used this C- to N-terminus chiral relay to show remote stereocontrol over an organocatalytic site. Combining an amine site and strong hydrogen bond donor (e.g. urea and thiourea) has provided enantioselective organocatalysts for Michael additions to nitroolefins,29–31 so they functionalised an (Aib)4(L-Ala)NHiPr pentapeptide at the N-terminus with thiourea and a secondary amine. The best compound enantioselectively catalysed the conjugate addition of dimethyl malonate to β-nitrostyrene, providing the R isomer as the major product in up to 64% ee.32
Both motifs thought to be needed to catalyse conjugate additions, amines and exposed hydrogen bond donors, should be present in even a simple 310 helical peptide. The peptide Aib4(L-Ala)NHtBu 1 would be one such potentially bifunctional organocatalyst (Fig. 1). This peptide is designed to have the primary amine of the N-terminal Aib as a sterically congested Brønsted base/H bond acceptor with chirality imparted by the 310 helix; this amine also has the potential to form imines or enamines. The solvent-exposed NH groups of the first two Aib residues could activate electrophiles, even in the absence of a highly designed hydrogen bond donor site. All sites might control the spatial positioning of reactants, with transfer of helix chirality to the conjugate addition transition state providing enantioselectivity. Simple peptides like 1, which are potentially accessible in a prebiotic world, could show how chirality can be relayed in a molecular environment dominated by achiral groups. Herein we assess the ability of unfunctionalised 310 helical peptides with a single stereogenic centre, which is remote from the reactive site, to act as enantioselective catalysts for conjugate additions.
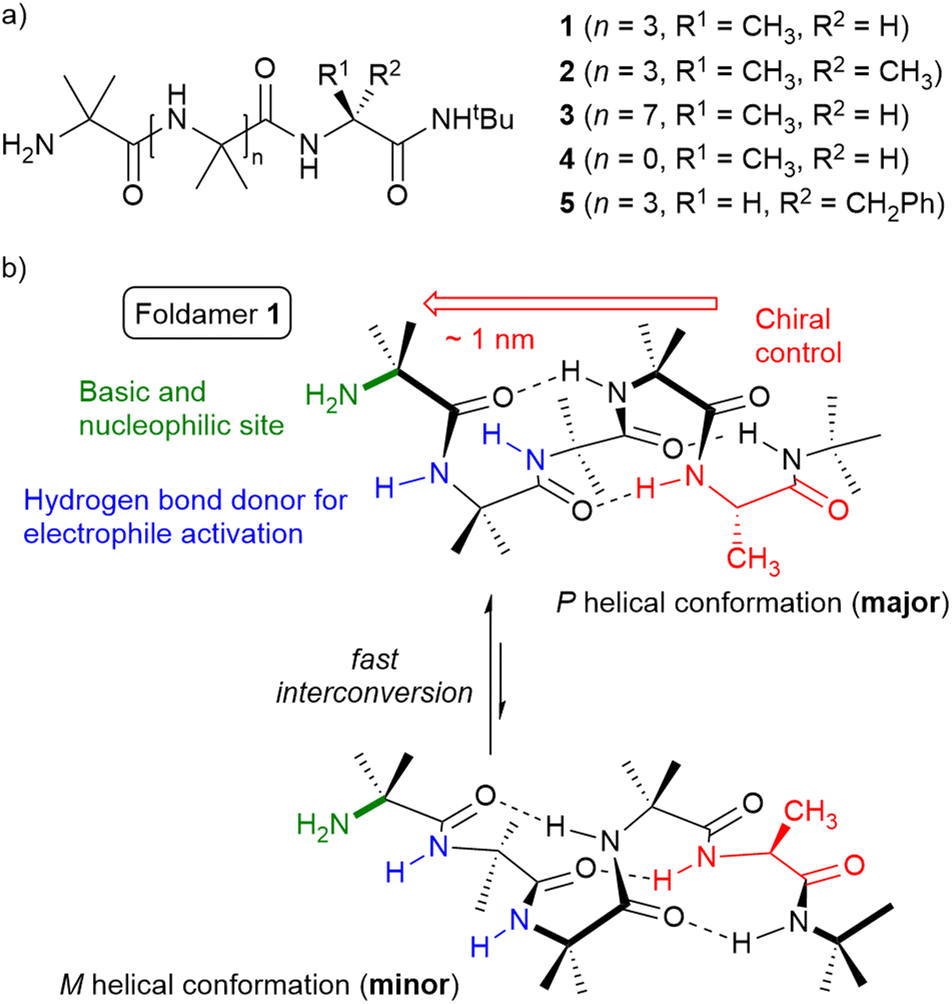 | ||
| Fig. 1 (a) Peptides 1 to 5 for application as organocatalysts. (b) Proposed bifunctional organocatalyst peptide 1 that incorporates a primary amine (green) and hydrogen bond donor sites (blue) on a helical scaffold, with overall chirality controlled by a C-terminal L-alanine residue (red). | ||
Results and discussion
Peptides 1 and 3 and control peptides 2 and 5 were synthesised through modifications of published conditions.24d,33,34 Tetrameric Aib foldamer N3Aib4OH was synthesised33 and ligated to either AibNHtBu, L-AlaNHtBu or D-PheNHtBu, using EDC·HCl as coupling agent to give the corresponding peptides with azide at the N-terminus (Scheme 1). The reduction of the azido groups to the corresponding primary amines 1, 2 and 5 was performed by hydrogenation using palladium on carbon. To investigate the effect of increasing the distance between the stereogenic centre and the reactive site, nonamer (Aib)8(L-Ala)NHtBu 3 was synthesised in 21% yield by first coupling the tetramer N3Aib4OH and pentamer 1 using EDC·HCl, followed by hydrogenation of the resulting peptide 7 in the presence of palladium on carbon (Scheme 1a). The analogue shorter than 1, dipeptide 4, was synthesised by coupling together the respective Aib and (L-Ala)tBu monomers followed by hydrogenation (Scheme 1b). | ||
| Scheme 1 (a) Synthesis of peptides 1, 2, 3 and 5. Conditions: a. EDC·HCl (1.5 equiv.), DIPEA (1.5 equiv.), CH2Cl2, 20 °C, 4 h, then AibNHtBu (1 equiv.), CH3CN, 82 °C, 3 days; b. Pd/C, EtOH, H2, 20 °C, 1 day; c. EDC·HCl (1.5 equiv.), DIPEA (1.5 equiv.), CH2Cl2, 20 °C, 4 h, then (L-Ala)NHtBu (1 equiv.), CH3CN, 82 °C, 3 days; d. EDC·HCl (1.5 equiv.), DIPEA (1.5 equiv.), CH2Cl2, 20 °C, 4 h, then peptide 1 (1 equiv.), CH3CN, 82 °C, 7 days; e. EDC·HCl (1.1 equiv.), HOBt (1.3 equiv.), DIPEA (2 equiv.), CH2Cl2, 20 °C, 0.5 h, then (D-Phe)NHtBu (1 equiv.), CH2Cl2, 20 °C, 2 days. (b) Synthesis of compound 4. Conditions: f. EDC·HCl (1.5 equiv.), DIPEA (1.5 equiv.), CH2Cl2, 20 °C, 0.5 h, then (L-Ala)NHtBu (1 equiv.), CH3CN, 20 °C, 3 days; b. Pd/C, EtOH, H2, 20 °C, 1 day. | ||
Both (Aib)4(L-Ala)NHtBu and (Aib)4(L-Phe)NHtBu cores have been reported to favour the P helical screw-sense. Clayden and co-workers measured the “helical excess” (he, defined as ([P] − [M])/([P] + [M]))34a,35 for the peptides CbzAib*Aib4(L-Xxx)NHtBu, where Aib* is a spectroscopic “reporter” of he33,36 For Xxx = Ala, he = +75% and the P:M helical ratio, hr, was 88:12. For Xxx = Phe, the respective values are he = +70% and hr = 85:15;28 the latter values will be inverted for D-Phe, the chiral residue in peptide 5.
The 1H NMR spectra of 1, 2, 3 and 5 confirm that pentamers 1, 5 and nonamer 3 have an he. The methyl group resonances of the Aib residues in 1, 3 and 5 are anisochronous whereas for the achiral foldamer 2 these methyl group resonances average on the 1H NMR timescale to appear isochronous; only an he = 0 can give isochronous resonances.
Circular dichroism (CD) measurements showed a negative band at the diagnostic wavelength of 205 nm for peptides Aib4(L-Ala)NHtBu 1 and Aib8(L-Ala)NHtBu 3, which is consistent with the reported excess of P 310 helicity (ESI Fig. S1†).28 This CD signal is stronger for the longer peptide 3, commensurate with the increased number of residues in this peptide. The achiral peptide Aib5NHtBu 2 gave no CD signal as expected and peptide 5 was not assayed as it has a chromophoric sidechain. The infrared spectra of 1 and 3 in the solid state show the respective amide I bands at 1644 cm−1 and 1657 cm−1. The latter is close to the range expected for a 310 helix (1662–1666 cm−1) whereas the former suggests a significant amount of other conformations are present.24c
Solid state structure of foldamer 1
Crystals of (Aib)4(L-Ala)NHtBu 1 suitable for crystallographic structure determination were obtained by slow evaporation from acetonitrile. The solid-state structure shows 1 is present in two different conformations in the unit cell (Fig. 2). Both conformations show distorted 310 helical structures, but one has an M helical screw-sense and the other has a P helical screw-sense. The observation of both screw-senses in the unit cell shows that favourable interactions between these pseudo-enantiomeric peptide conformations in the unit cell are greater than the energetic penalty for a mismatch between the conformational preference of the L-Ala residue and an M helical sense. Examples of crystal packing forces counteracting the helical preference of an N-terminal chiral group are reported. This can include the adoption of both helical screw-senses in the unit cell,36c as seen for 1, as well as the adoption of the reverse screw-sense in the solid state compared to in solution.37 | ||
| Fig. 2 (a)–(d) Crystallographic structures showing the two diastereomeric conformations adopted by 1 in the unit cell. P helical diastereomer in (a) longitudinal and (b) axial view. M helical diastereomer in (c) longitudinal and (d) axial view. Dashed lines show hydrogen bonds. Water molecules of solvation are shown in the longitudinal views. C atoms are shown in grey, N in light blue, O in red. The H atoms of the CH3 groups have been removed for clarity. | ||
Three intramolecular i + 3 hydrogen bonds are present in each conformer; between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the first Aib and the NH of the fourth Aib, the CO of the second Aib and the NH of the Ala, and the CO of the third Aib and the terminal NHtBu. This hydrogen bonding pattern leaves two amide NH groups available to external hydrogen bond acceptors. In both conformations, the N-terminal primary amine group is in a chiral environment created by the helical conformation.
O of the first Aib and the NH of the fourth Aib, the CO of the second Aib and the NH of the Ala, and the CO of the third Aib and the terminal NHtBu. This hydrogen bonding pattern leaves two amide NH groups available to external hydrogen bond acceptors. In both conformations, the N-terminal primary amine group is in a chiral environment created by the helical conformation.
Peptide 1 co-crystallised with two molecules of water, each bound to different conformer. Each water forms a hydrogen bond between oxygen and the NH of the third Aib. For the M helical conformer, this is part of a bifurcated hydrogen bond that includes an NH of the amine (N⋯O distances of 2.924 Å and 3.168 Å respectively, Fig. 2c and ESI Fig. S106†). This bifurcated geometry is not present for the P helical conformer, with the NH of the amine too far to effectively hydrogen bond to the water molecule (N⋯O distance of 3.597 Å, see ESI Fig. S107†). This water instead adopts an end-on orientation relative to the amide NH of the third Aib (N⋯O distance of 2.896 Å, Fig. 2a). The presence of this bound water in the unit cell illustrates the ability of these NH groups to form intermolecular hydrogen bonds to reagents with hydrogen bond acceptor properties. The proximity between these free amide NH groups and the amine also illustrates the potential of 1 to act as a bifunctional catalyst.
Catalysis of conjugate additions to β-nitrostyrene
β-Nitrostyrene ((E)-(2-nitrovinyl)benzene) 8 has been widely used as an electrophile for screening catalysts of enantioselective conjugate additions.38 Le Bailly et al. showed Aib4Ala peptides functionalised with Takemoto's designed aminothiourea catalysts38b could catalyse dimethyl malonate addition to β-nitrostyrene 8 in up to 64% ee.32 The diastereo- and enantioselective addition of 1-methyl-2-oxocyclopentanecarboxylate 9 to β-nitrostyrene 8 to give the stereoisomers of methyl 1-(2′-nitro-1′-phenylethyl)-2-oxocyclopentane-1-carboxylate 10 has been well characterised (Scheme 2).39 This reaction was therefore selected as a benchmark for evaluating the catalytic efficacy of 1. | ||
| Scheme 2 Stereoisomers of methyl 1-(2′-nitro-1′-phenylethyl)-2-oxocyclopentane-1-carboxylate 10a–10d produced by the addition of 1-methyl-2-oxocyclopentanecarboxylate 9 to β-nitrostyrene 8. | ||
Conversions and diastereomeric ratios were determined by 1H NMR spectroscopy of the crude reaction mixtures in CDCl3 (Fig. 3a and b). In the absence of catalyst, the reaction between 8 (154 mM) and 9 (3.5 eq., 539 mM) proceeded slowly, reaching 58% conversion after 6 days, with a diastereomeric ratio of 77:23. The presence of catalyst (Aib)4(L-Ala)NHtBu 1 (20 mol%, 31.2 mM) increased the reaction rate to give full conversion after 5 days, with the diastereomeric ratio increasing to a value of 96:4.
 | ||
| Fig. 3 Reaction of 9 with 8 in CDCl3 at ambient temperature catalysed by 1. (a) Conversion determined by integration of 1H NMR signals monitoring the disappearance of nitroalkene CHNO2 signal at 7.9 ppm and the appearance of CH2NO2 signals at 4 ppm and 5.0–5.2 ppm. (b) Diastereomeric ratio (major: blue circles, minor: red circles) determined by integration of 1H NMR signals of the CH2NO2 moiety. (c) Enantiomeric excess determined by integration of the HPLC chromatogram. HPLC chiralcel OD-H column, hexane/i-PrOH (80:20), flow rate 1.0 mL min−1, 220 nm; minor diastereomer: (red circles) 10b = 9.064 min, 10d = 12.85 min, ee = 58%; major diastereomer: (blue circles) 10a = 10.39 min, 10c = 15.35 min, ee = 54%. Diastereomeric ratio: 92:8. | ||
The relative configuration of the product mixture 10 was determined by chiral phase HPLC on adducts pre-purified by silica gel chromatography, followed by comparison with literature data.39 This analysis showed an enantiomeric excess in each diastereomer, with ee = +54% for the major diastereomer (major) and +58% for the minor diastereomer (minor); isomer 10a, methyl (S)-1-((R)-2′-nitro-1′-phenylethyl)-2-oxocyclopentane-1-carboxylate, was the major product. The observation of a 54% ee confirms involvement of 1 in this reaction and is remarkably close to the best ee (64%) reported by Clayden and co-workers for a highly designed peptide.32 The estimated +75% he for catalyst 1 (the value for CbzAib*Aib4(L-Ala)NHtBu)28 would be anticipated to be the maximum ee value available. The achiral analogue 2 was also tested under the same conditions. It also catalysed the reaction but did not produce an ee in the products (Table 1).
| Catalyst | Isolated yielda (%) | Time (days) | Conv.b (%) | drc | eed major % (minor %) |
|---|---|---|---|---|---|
| a Isolated yields after chromatography on silica. b Conversion determined by integration of the appropriate 1H NMR signals in the crude reaction mixture. c Ratio of diastereomers (dr) measured by integration of the appropriate 1H NMR signals in the crude reaction mixture. d Enantiomeric excesses (ee) were measured by integration of peaks from chiral stationary phase HPLC on the purified product. n.a. = not applicable. A positive ee value indicates an excess of 10a, values in parentheses are for the minor diastereomer. e Significantly greater by-product formation. | |||||
| None, 20 °C, CDCl3 | 30e | 6 | 58 | 77:23 |
n.a. |
| t BuNH2, 20 °C, CDCl3 | 88 | 1 | 100 | 68:32 |
n.a. |
| 1, 20 °C, CDCl3 | 98 | 5 | 100 | 96:4 |
+54 (+58) |
| 1, 40 °C, CDCl3 | 94 | 2.5 | 100 | 91:9 |
+23 (+9) |
| 2, 20 °C, CDCl3 | 96 | 6 | 100 | 95:5 |
0 (0) |
| 3, 20 °C, CDCl3 | 93 | 5 | 100 | 88:12 |
+5 (+19) |
| 5, 20 °C, CDCl3 | 95 | 8 | 100 | 90:10 |
−42 (−1) |
| 6, 20 °C, CDCl3 | 10 | 6 | 26 | 65:35 |
0 (0) |
Monitoring the evolution of the enantiomeric excess during the reaction showed little change from day one to day five (see the ESI†), indicating that the reaction mixture was not equilibrating over this time period in the presence of 1. Increasing the temperature to 40 °C accelerated the reaction, which reached completion after 2 days, with a small decrease in the diastereomeric ratio (91:9) but a larger decrease in ee in each diastereomer (+23% (major)/+9% (minor)).
The relay of chirality from the L-Ala residue to the environment around the N-terminal amine should be less efficient with a greater number of intervening Aib residues.35 To assess this effect, the benchmark Michael addition of 9 to 8 was performed in the presence of 20 mol% of the nonameric peptide (Aib)8(L-Ala)NHtBu 3. The reaction proceeded with good conversion and a high diastereomeric ratio of 88:12 over the same period as the reaction catalysed by 1. Significantly lower enantioselectivity was obtained (+5% (major)/+19% (minor)), which is lower than might be anticipated given the decay in relayed helicity from a C-terminus alanine measured by Clayden and co-workers (e.g. a decrease of 6.1% per Aib residue for peptides in methanol).35 A shorter peptide, dipeptide (Aib)(L-Ala)NHtBu 4, was also investigated although this analogue of 1 is too short to fold into a helical structure. However, and perhaps due to this lack of folding, dipeptide (Aib)(L-Ala)NHtBu was not soluble in chloroform at the required concentrations.
The replacement of the L-Ala residue in 1 with D-Phe in peptide 5 should give a peptide with an excess of M-helical conformation, with a ratio of approximately 15:85 P:M (based upon the he expected for CbzAib*Aib4(D-Phe)NHtBu).28 If the conformation around the N-terminal catalytic site controls enantioselectivity, organocatalysis by this control peptide should produce an ee opposite to that of 1. It would also show that other chiral residues at the C-terminus can produce an ee, with the most important factor the ability of a given residue to induce an he in the peptide. The benchmark Michael addition of 9 to 8 was performed in the presence of 20 mol% of peptide (Aib)4(D-Phe)NHtBu 5. The reaction proceeded with good conversion and gave a high diastereomeric ratio of 90:10 over 8 days. Chiral HPLC separation of the product mixture showed an enantiomeric excess in each diastereomer, with ee = −42% for the major diastereomer and −1% for the minor diastereomer; isomer 10c, methyl (R)-1-((S)-2′-nitro-1′-phenylethyl)-2-oxocyclopentane-1-carboxylate, was the major product. This inversion of enantioselectivity in the major diastereomer confirms that it is the conformational preference of the C-terminal chiral residue that determines enantioselectivity at the N-terminal reactive site.
To confirm that the primary amine group in each of 1, 2, 3 and 5 is playing an important role in catalysis, the reaction of 9 with 8 was carried out in the presence of N3Aib5NHtBu 6 (20 mol%), an analogue of 2 deprived of its amino group (Scheme 1). The reaction at ambient temperature in CDCl3 proceeded with a significantly lower reaction rate, reaching only 26% conversion after six days to give the product mixture with a lower diastereomeric ratio (65:35), confirming that the amine group has a role in catalysing the reaction. Similarly, tBuNH2, a primary amine with steric hindrance reminiscent of peptides 1–3 and 5 but without any amide hydrogen bond donors to activate an electrophile, catalysed the reaction. Full conversion was observed after 2.5 days with a diastereomeric ratio of 68:32 in the product, showing the primary amine is the functionality that contributes most to the rate acceleration. On the other hand, the significant dr decrease for the reactions catalysed by either 6 or tBuNH2 suggests that a combination of primary amine with adjacent hydrogen bond donors provides the best diastereoselectivity.
In general these reactions proceeded with good yields but were slow. To increase the reaction rate, catalysis by 1 was performed without solvent, and in an attempt to increase the ee the temperature was decreased to 0 °C under these conditions. Full conversion was obtained after 2.5 days to give the adduct with a diastereomeric ratio of 9:1 and improved enantiomeric excess values of 69% (major) and 14% (minor). Unlike in chloroform, dipeptide Aib(L-Ala)NHtBu 4 was soluble in the absence of solvent. The reaction reached full conversion after 4 days at 0 °C, providing the adducts with a diastereomeric ratio of 82:18 – lower than obtained using 1 as catalyst – and no enantiomeric excess was found in the products. Without solvent, nonameric foldamer 3 was not soluble in the reactants and this reaction was unsuccessful.
Exploring reaction scope
To examine the scope of conjugate additions catalysed by 1, different electrophiles and nucleophiles were selected based upon literature precedent, especially methods for the determination of product ee (Fig. 4 and ESI, Table S2†). Changing the β-substituent in the nitroalkene electrophile (Fig. 4a) was explored first (Table 2). Reaction of 9 with (E)-3-methyl-1-nitrobut-1-ene 11 gave good conversion (80% after 4 days with catalysis by 1) with a diastereomeric ratio of 99:1, although the enantiomeric excess was low (ee = +10%). Changing the conditions to 0 °C without solvent, (E)-(1-nitroprop-1-ene) 12 reacted with 9 to give the adducts in a diastereomeric ratio of 89:11, with ee of +28% (major) and +18% (minor); a poorer ee than that obtained when 8 reacted with 9 under the same conditions. These observations with 11 and 12 suggest that steric bulk in the electrophile may be important for maintaining the ee of the catalysed reaction. Reactions with either 1-methyl pyrrole-2,5-dione40 or 1,1-bis(phenylsulfonyl)ethylene41 were too slow to be explored further.
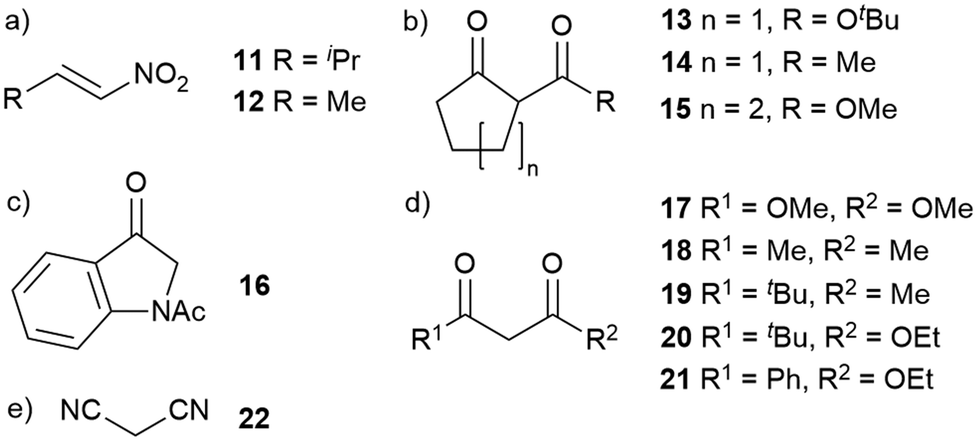 | ||
| Fig. 4 (a) Electrophiles and (b–e) nucleophiles for substrate screening studies. | ||
| Electrophile, conditions | Isol. yielda (%) | Time (days) | Conv.b (%) | drc | eed maj. % (min. %) |
|---|---|---|---|---|---|
| a Isolated yields after chromatography on silica. b Conversion determined by integration of the appropriate 1H NMR signals in the crude reaction mixture. c Ratio of diastereomers (dr) measured by integration of the appropriate 1H NMR signals in the crude reaction mixture. d Enantiomeric excesses (ee) were measured by integration of peaks from chiral stationary phase HPLC on the purified product. The values in parentheses are the ee values of the minor diastereomers. | |||||
| 11, 20 °C, CDCl3 | 80 | 4 | 89 | 99:1 |
+10 (0) |
| 12, 0 °C, no solvent | 80 | 5 | 88 | 89:11 |
+28 (+18) |
Nucleophile structure was then varied (Fig. 4b–e and Table 3) and these compounds reacted with β-nitrostyrene 8. In general, however, changes to the nucleophile structure did not improve the ee. Increasing the bulkiness of the ester group from methyl in 9 to tert-butyl ester in 13 decreased the reaction rate at ambient temperature in CDCl3 (8 days to reach full conversion) and the diastereomeric ratio (from 96:4 to 81:19). The ee also decreased to +17% (major) and +5% (minor). Performing the reaction without solvent at 0 °C only slightly increased the diastereomeric ratio and enantiomeric excess (+27% (major), +2% (minor)). Replacing the tert-butoxy ester in 13 with methyl to give diketone 14 restored the reaction rate, yet the diastereomeric ratio (86:14) and enantiomeric excess (+18%) of Michael adducts remained low. A larger analogue of 9, cyclohexanone 15, reacted slowly – 25 days to reach full conversion – and in the absence of catalyst the reaction did not proceed. The diastereomeric ratio (92:8) was similar to that obtained with 13 and the enantiomeric excesses were similar (+16% (major), +5% (minor)). Performing the reaction without solvent and at 0 °C significantly increased the reaction rate, with full conversion obtained after 5 days, and slightly increased diastereoselectivity (dr = 94:6) and enantiomeric excess (+17% (major), +16% (minor)).
| Nucleophile, conditions | Isol. yielda (%) | Time (days) | Conv.b (%) | drc | eed maj. % (min. %) |
|---|---|---|---|---|---|
| a Isolated yields after chromatography on silica. b Conversion determined by integration of the appropriate 1H NMR signals in the crude reaction mixture. c Ratio of diastereomers (dr) measured by integration of the appropriate 1H NMR signals in the crude reaction mixture. n.a. = not applicable. d Enantiomeric excesses (ee) were measured by integration of peaks from chiral stationary phase HPLC on the purified product. The values in parentheses are the ee values of the minor diastereomers. n.d. = not determined. | |||||
| 13, 20 °C, CDCl3 | 50 | 8 | 100 | 81:19 |
+17 (+5) |
| 13, 0 °C, no solvent | 51 | 10 | 100 | 85:15 |
+27 (+2) |
| 14, 20 °C, CDCl3 | 70 | 5 | 100 | 86:14 |
+18 (0) |
| 15, 20 °C, CDCl3 | 80 | 25 | 100 | 92:8 |
+16 (+5) |
| 15, 0 °C, no solvent | 84 | 5 | 100 | 94:6 |
+17 (+16) |
| 16, 20 °C, CDCl3 | 71 | 25 | 87 | 85:15 |
+18 (+2) |
| 17, 20 °C, CDCl3 | 91 | 23 | 100 | n.a. | 0 |
| 18, 20 °C, CDCl3 | 67 | 5 | 100 | n.a. | +23 |
| 19, 20 °C, CDCl3 | 79 | 5 | 100 | 79:21 |
+52 (+44) |
| 20, 20 °C, CDCl3 | 84 | 23 | 100 | 95:5 |
+46 (n.d.) |
| 21, 20 °C, CDCl3 | 94 | 15 | 100 | 55:45 |
+8 (+9) |
| 22, 20 °C, CDCl3 | 100 | 1.5 | 100 | n.a. | +2 |
Addition of nucleophile 16 to 8 has been reported by Liu et al.,42 so was explored here. At ambient temperature in CDCl3 the uncatalyzed reaction did not proceed, but catalysis by pentamer 1 provided the adducts albeit with a slow reaction rate; 87% conversion after 25 days. The products had a diastereomeric ratio of 85:15 (identical to the ratio obtained by using tBuNH2 as the catalyst) and the major diastereomer had an enantiomeric excess of +18%.
Acyclic 1,3-dicarbonyl compounds were also assessed. Reaction of dimethyl malonate 17 with 8 proceeded slowly with primary amine catalysis (tBuNH2 or 1), but after 23 days no ee was measured for the reaction catalysed by 1. Acetylacetone 18 reacted more quickly, with the catalyst 1 providing the adduct with 23% enantiomeric excess after 5 days. Adding steric bulk to the nucleophile by replacing a methyl with tert-butyl (diketone 19) further increased the ee to 52% (major) and 44% (minor) without significantly slowing the rate. Replacing the methyl groups of acetylacetone with tert-butyl and ethoxy (substrate 20) resulted in similarly slow reaction in the presence of 1 (no reaction without catalyst) but gave an ee of 46% in the product, when none had been observed with dimethyl malonate 17. A phenyl group in the place of tert-butyl (substrate 21) increased reaction rate but resulted in a lower ee (to 8%) as well as a decrease in dr (to approx. 1:1 from 95:5).
Proposed mechanism
The primary amine of peptide 1 (and of tBuNH2) might accelerate the reaction by acting as a base or by forming an enamine. It was noted that the reaction of 8 with either acetone, propanal or methylpyruvate in the presence of 1 did not proceed even after 21 days. These compounds are less acidic (e.g. in DMSO pKa ∼ 26.5, (acetone)43) than the 1,3-dicarbonyl compounds previously employed (e.g. in DMSO, pKa ∼ 13.3 (acetylacetone),44 pKa ∼ 15.9 (dimethyl malonate)45), suggesting the ease of proton transfer to the primary amine (pKa of a primary alkylammonium ∼ 11)46 is an important factor.To investigate whether the reactions proceed through intermediate N-terminal enamines with 1 or enolates with proton transfer to 1, a stoichiometric mixture of ketoester 9 and foldamer 1 in CDCl3 at ambient temperature was analysed by NMR spectroscopy. 1H and 13C NMR spectra showed negligible changes in the chemical shifts compared to the reagents analysed separately (see ESI, Fig. S39 and S40†), with only small shifts in the positions of the peptide NH resonances. Adding dry 4 Å molecular sieves to the mixture did not alter these observations. Although small amounts of reactive imine/enamine could be present (below the 1H NMR spectroscopy detection limit of ∼5 mol%), imine/enamine species are not significant products. Furthermore, adding malonitrile 22, which cannot form imines, to 8 in the presence of peptide 1 provided the Michael adducts, albeit with negligible enantioselectivity (Table 3). This reaction did not proceed in the absence of 1, suggesting that 1 is acting as a base with this nucleophile. This latter example shows the absence of significant enamine formation does not prevent 1 from catalysing conjugate addition, although enamine intermediates cannot be conclusively excluded in all cases.
To determine if the electrophiles interact with the amide NH groups on the peptides, a 1H NMR spectroscopic titration of β-nitrostyrene 8 (from 0 to 57 equivalents) into peptide 6 (29 mM) in CDCl3 at 25 °C was performed (see ESI, Fig. S41†). The addition of nitroalkene induced significant downfield shifts in two of the three furthest upfield foldamer NH signals (Δδ > 0.17 and >0.47 ppm, ESI Fig. S41 and S42†) indicating increased hydrogen bonding. The NH resonance that shifts the most is that which is the furthest upfield, at 6.12 ppm, consistent with a solvent-exposed NH. Fitting of these titration data gave an association constant of 0.84 M−1 (see section 7.2 in the ESI†), which would give 2.4 mol% of peptide 6 associated with 8 under the catalytic conditions (concentrations of 6 of 30 mM).
Based on these stereochemical observations and the solid state structure of 1, a possible intermediate structure is tentatively proposed (Fig. 5) that is inspired by those proposed by Le Bailly et al. and Rénio et al.32,47 Weak hydrogen bonds between the nitro group of the β-nitrostyrene and the exposed N-terminal amide NHs of the foldamer (NHa and NHc, which undergo the largest Δδ upon addition of 8, see Fig. S41†) position the electrophile adjacent to the amine. Steric effects are proposed to orient the phenyl group away from the helix. The primary amine hydrogen bonds with the acidic proton of the nucleophile, mediating its transfer as the reaction proceeds. This defined spatial arrangement of the reagents is proposed to favour approach of the nucleophile to the si face of the electrophilic carbon, producing a R configuration at the 1′ position. To obtain the S configuration at the 1 position (α to the ester), the ester should also be oriented away from the helix. The proposed transfer of the nucleophile proton to the primary amine would account for the sharp drop in reaction rates as the pKa of the nucleophile increases.
 | ||
| Fig. 5 Proposed ternary complex of the P-helical conformer of 1 with 8 and 9 that accounts for the stereochemistry in the major product 10a. X-ray crystallography shows peptide 1 should fold into a P 310 helix. The amine is proposed to act as a base and the exposed N-terminal NH groups are shown as binding to the nitroalkene, as suggested by 1H NMR spectroscopy titration data. | ||
Conclusions
Several 310 helical peptides bearing only the N-terminal primary amine and amide backbone functional groups common to simple unmodified peptides have been shown to catalyse conjugate addition to nitroalkenes. Two peptides, (Aib)4(L-Ala)NHtBu 1 and (Aib)4(D-Phe)NHtBu 5 produced significant but opposite enantiomeric excesses (ee) in a benchmark conjugate addition reaction. Folding into 310 helices is proposed to allow the chirality of each C-terminal residue to be relayed over 0.9 nm to the reactive amine site. Despite the absence of bespoke recognition groups in 1, the best ee for the 1-catalysed addition to β-nitrostyrene is similar to that reported for an Aib-rich peptide functionalised with Takemoto's designed secondary amine/urea catalyst.32,38b The inversion of ee when using (Aib)4(D-Phe)NHtBu 5 as the catalyst indicates that the conformational preference of the chiral C-terminal residue, which is relayed along the 310 helix to the N-terminal reactive site, determines enantioselectivity.Catalysis of conjugate additions was only successful for the reaction of nitroalkenes with compounds that could be relatively easily transformed into an enolate (or enolate analogue). In conjunction with the lack of spectroscopically identifiable imine/enamine intermediates, this latter observation is consistent with the primary amine acting as a Brønsted base. The observation of a good ee was limited to catalysis by 1 or 5, with reactant screening showing a 1,3-dicarbonyl substrate with steric bulk on one side was required. Replacing the phenyl substituent on nitroalkene 8 with an alkyl substituent still produced an ee in the adduct mixture, but to a lower extent. The best ee was obtained for the 1-catalysed reaction between 8 and 9, which was +54% in chloroform solution and +69% as a neat mixture. Decreasing the peptide length by removing Aib residues from 1 led to a loss of catalysis, which was ascribed to poorer solubility of peptide 4. We speculate that the lower than expected ee for catalysis by nonameric peptide 3 compared to pentapeptide 1 may be due to the shorter peptide adopting additional conformations, as implied by the amide I band in the FTIR spectrum. These may permit the adoption of alternative transition states that produce greater stereocontrol.
Given that both Aib and alanine have been produced under proposed prebiotic conditions48 and given that enantioselective catalysis is a key biological function, the observation of a +69% ee for catalysis by 1 under solvent-free conditions shows how even achiral residues like Aib might play a role in enantioselective catalysis by simple prebiotic peptides. Enantioselective catalysis by 1, 3 and 5 are also examples of remote chiral control over catalysis by folded peptides49,50 and foldamers.51,52 This work contributes to our efforts to develop molecules able to transfer chiral information along nanometre distances; the effective relay of chiral information from either L-Ala or D-Phe along the 1 nm length of peptides 1 or 5 to the catalytic site is an important step towards this goal. Expanding the range of catalysed reactions and improving helical control will be key objectives in future work.
Author contributions
DT: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing-original draft preparation, writing-reviewing and editing, funding acquisition. CMcC and MH: investigation, data curation. IJV-Y: methodology, resources. SJW: conceptualization, methodology, validation, formal analysis, writing-original draft preparation, writing-reviewing and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPRSC (DT and SJW, grants EP/P027067/1 and EP/K039547).References
- R. Wieczorek, K. Adamala, T. Gasperi, F. Polticelli and P. Stano, Life, 2017, 7, 19 CrossRef PubMed.
- A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Chem. Rev., 2020, 120, 11479–11615 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- M. Agirre, A. Arrieta, I. Arrastia and F. P. Cossío, Chem. – Asian J., 2019, 14, 44–66 CrossRef CAS PubMed.
- (a) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 14, 1807–1821 RSC; (b) F. Peng and Z. H. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1–13 CrossRef CAS; (c) L. W. Xu and Y. Lu, Org. Biomol. Chem., 2008, 6, 2047–2053 RSC; (d) Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC; (e) U. V. S. Reddy, B. Anusha, Z. Begum, C. Seki, Y. Okuyama, M. Tokiwa, S. Tokiwa, M. Takeshita and H. Nakano, Catalysts, 2022, 12, 1674 CrossRef CAS.
- G. Bartoli and P. Melchiorre, Synlett, 2008, 1759–1772 CAS.
- (a) A. Cordova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes and Y. Xu, Chem. – Eur. J., 2006, 12, 5383–5397 CrossRef CAS PubMed; (b) S. S. V. Ramasastry, H. Zhang, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2007, 129, 288–289 CrossRef CAS PubMed.
- P. Dziedzic, W.-B. Zou, J. Hafren and A. Cordova, Org. Biol. Chem., 2006, 4, 38–40 RSC.
- W. Huang, H. Tian, H. Xu, L. Zheng, Q. Liu and S. Zhang, Catal. Lett., 2011, 141, 872–876 CrossRef CAS.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1375 CrossRef CAS PubMed.
- A. Y. Liu, X. A. Calicdan, G. N. Glover, X. Luo, G. T. Barroso, B. K. Hoppe, K. M. Boyle and L. S. Witus, ACS Omega, 2022, 7, 45336–45340 CrossRef CAS PubMed.
- Z.-H. Du, B.-X. Tao, M. Yuan, W.-J. Qin, Y.-L. Xu, P. Wang and C.-S. Da, Org. Lett., 2020, 22, 4444–4450 CrossRef CAS PubMed.
- F.-C. Wu, C.-S. Da, Z.-X. Du, Q.-P. Guo, W.-P. Li, L. Yi, Y.-N. Jia and X. Ma, J. Org. Chem., 2009, 74, 4812–4818 CrossRef CAS PubMed.
- R. Thiyagarajan, Z. Begum, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, S. Tokiwa, M. Takeshita and H. Nakano, RSC Adv., 2021, 11, 38925–38932 RSC.
- D. R. Kelly and S. M. Roberts, Chem. Commun., 2004, 18, 2018–2020 RSC.
- G. Carrea, G. Ottolina, A. Lazcano, V. Pironti and S. Colonna, Tetrahedron: Asymmetry, 2007, 18, 1265–1268 CrossRef CAS.
- K. Akagawa, R. Suzuki and K. Kudo, Asian J. Org. Chem., 2014, 3, 514–522 CrossRef CAS.
- Q. Lin, H. Lan, C. Ma, R. T. Stendall, K. Shankland, R. A. Musgrave, P. N. Horton, C. Baldauf, H.-J. Hofmann, C. P. Butts, M. M. Mueller and A. J. A. Cobb, Angew. Chem., Int. Ed., 2023, 62, e202305326 CrossRef CAS PubMed.
- (a) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC; (b) D. Becart, V. Diemer, A. Salaun, M. Oiarbide, Y. R. Nelli, B. Kauffmann, L. Fischer, C. Palomo and G. Guichard, J. Am. Chem. Soc., 2017, 139, 12524–12532 CrossRef CAS PubMed.
- M. K. Andrews, X. Liu and S. H. Gellman, J. Am. Chem. Soc., 2022, 144, 2225–2232 CrossRef CAS PubMed.
- (a) Y. Li, L. Bouteiller and M. Raynal, ChemCatChem, 2019, 11, 5212–5226 CrossRef CAS; (b) L. Zhou, K. He, N. Liu and Z.-Q. Wu, Polym. Chem., 2022, 13, 3967–3974 RSC.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1988, 201, 601–619 CrossRef CAS PubMed.
- A. Weyer, D. Díaz, A. Nierth, N. E. Schlörer and A. Berkessel, ChemCatChem, 2012, 4, 337–340 CrossRef CAS.
- (a) C. Toniolo and E. Benedetti, Macromolecules, 1991, 24, 4004–4009 CrossRef CAS; (b) S. J. Pike, T. Boddaert, J. Raftery, S. J. Webb and J. Clayden, New J. Chem., 2015, 39, 3288–3294 RSC; (c) M. G. Lizio, V. Andrushchenko, S. J. Pike, A. D. Peters, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, S. T. Mutter, J. Clayden, P. Bouř, E. W. Blanch and S. J. Webb, Chem. – Eur. J., 2018, 24, 9399–9408 CrossRef CAS PubMed; (d) A. D. Peters, S. Borsley, F. della Sala, D. F. Cairns-Gibson, M. Leonidou, J. Clayden, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, E. Takano, J. Burthem, S. L. Cockroft and S. J. Webb, Chem. Sci., 2020, 11, 7023–7030 RSC.
- (a) R. A. Brown, T. Marcelli, M. De Poli, J. Solà and J. Clayden, Angew. Chem., Int. Ed., 2012, 51, 1395–1399 CrossRef CAS PubMed; (b) M. De Poli, M. De Zotti, J. Raftery, J. A. Aguilar, G. A. Morris and J. Clayden, J. Org. Chem., 2013, 78, 2248–2255 CrossRef CAS PubMed; (c) S. J. Pike, J. E. Jones, J. Raftery, J. Clayden and S. J. Webb, Org. Biomol. Chem., 2015, 13, 9580–9584 RSC; (d) M. De Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef CAS PubMed.
- (a) F. G. A. Lister, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420–425 CrossRef CAS; (b) J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680–6691 CrossRef CAS PubMed; (c) N. Eccles, B. A. F. Le Bailly, F. della Sala, I. J. Vitórica-Yrezábal, J. Clayden and S. J. Webb, Chem. Commun., 2019, 55, 9331–9334 RSC; (d) K. Gratzer, V. Diemer and J. Clayden, Org. Biomol. Chem., 2017, 15, 3585–3589 RSC; (e) N. Eccles, F. della Sala, B. A. F. Le Bailly, G. F. S. Whitehead, J. Clayden and S. J. Webb, ChemistryOpen, 2020, 9, 338–345 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852–4863 RSC.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2014, 50, 7949–7952 RSC.
- (a) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051–7071 RSC; (b) X. Liu, L. Lin and X. Feng, Chem. Commun., 2009, 41, 6145–6158 RSC; (c) X. Fang and C.-J. Wang, Chem. Commun., 2015, 51, 1185–1197 RSC.
- (a) H. Huang and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 7170–7171 CrossRef CAS PubMed; (b) M. P. Lalonde, Y. G. Chen and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 6366–6370 CrossRef CAS PubMed.
- (a) Y.-D. Ju, L.-W. Xu, L. Li, G.-Q. Lai, H.-Y. Qiu, J.-X. Jiang and Y. Lu, Tetrahedron Lett., 2008, 49, 6773–6777 CrossRef CAS; (b) X.-J. Zhang, S.-P. Liu, X.-M. Li, M. Yan and A. S. C. Chan, Chem. Commun., 2009, 7, 833–835 RSC; (c) S. B. Tsogoeva and S. Wei, Chem. Commun., 2006, 13, 1451–1453 RSC.
- B. A. F. Le Bailly, L. Byrne and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 2132–2136 CrossRef CAS PubMed.
- J. Clayden, A. Castellanos, J. Solà and G. A. Morris, Angew. Chem., Int. Ed., 2009, 48, 5962–5965 CrossRef CAS PubMed.
- (a) L. Byrne, J. Solà, T. Boddaert, T. Marcelli, R. W. Adams, G. A. Morris and J. Clayden, Angew. Chem., Int. Ed., 2014, 53, 151–155 CrossRef CAS PubMed; (b) V. Diemer, J. Maury, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Chem. Commun., 2017, 53, 10768–10771 RSC.
- B. A. F. Le Bailly, L. Byrne, V. Diemer, M. Foroozandeh, G. A. Morris and J. Clayden, Chem. Sci., 2015, 6, 2313–2322 RSC.
- (a) D. Mazzier, M. Crisma, M. De Poli, G. Marafon, C. Peggion, J. Clayden and A. Moretto, J. Am. Chem. Soc., 2016, 138, 8007–8018 CrossRef CAS PubMed; (b) S. J. Pike, M. De Poli, W. Zawodny, J. Raftery, S. J. Webb and J. Clayden, Org. Biomol. Chem., 2013, 11, 3168–3176 RSC; (c) F. G. A. Lister, N. Eccles, S. J. Pike, R. A. Brown, G. F. S. Whitehead, J. Raftery, S. J. Webb and J. Clayden, Chem. Sci., 2018, 9, 6860–6870 RSC; (d) R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS PubMed.
- (a) S. Wang, F. Della Sala, M. J. Cliff, G. F. S. Whitehead, I. J. Vitórica-Yrezábal and S. J. Webb, J. Am. Chem. Soc., 2022, 144, 21648–21657 CrossRef CAS PubMed; (b) M. De Poli, L. Byrne, R. A. Brown, J. Solà, A. Castellanos, T. Boddaert, R. Wechsel, J. D. Beadle and J. Clayden, J. Org. Chem., 2014, 79, 4659–4675 CrossRef CAS PubMed.
- (a) D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299–365 CrossRef; (b) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- (a) J. Deutsch, H. J. Niclas and M. J. Ramm, J. Prakt. Chem./Chem.-Ztg., 1995, 337, 23–28 CrossRef CAS; (b) A. M. Flock, A. Krebs and C. Bolm, Synlett, 2010, 1219–1222 CAS.
- P. Chauhan, J. Kaur and S. S. Chimni, Chem. – Asian J., 2013, 8, 328–346 CrossRef CAS PubMed.
- S. Mossé and A. Alexakis, Org. Lett., 2005, 7, 4361–4364 CrossRef PubMed.
- Y.-Z. Liu, R.-L. Cheng and P.-F. Xu, J. Org. Chem., 2011, 76, 2884–2887 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- W. N. Olmstead, Z. Margolin and F. G. Bordwell, J. Org. Chem., 1980, 45, 3295–3299 CrossRef CAS.
- E. M. Arnett, S. G. Maroldo, S. L. Schilling and J. A. Harrelson, J. Am. Chem. Soc., 1984, 106, 6759–6767 CrossRef CAS.
- M. R. Crampton and I. A. Robotham, J. Chem. Res., Synop., 1997, 1, 22–23 RSC.
- M. Rénio, D. Murtinho and M. R. Ventura, Chirality, 2022, 34, 782–795 CrossRef PubMed.
- D. Ring, Y. Wolman, N. Friedmann and S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 765–768 CrossRef CAS PubMed.
- K. Ożga and Ł. Berlicki, ACS Catal., 2022, 12, 15424–15430 CrossRef.
- J. T. Blank and S. J. Miller, Biopolymers, 2006, 84, 38–47 CrossRef CAS PubMed.
- Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142, 17211–17223 CrossRef CAS PubMed.
- B. Legrand, J. Aguesseau-Kondrotas, M. Simon and L. Maillard, Catalysts, 2020, 10, 700 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectra of novel compounds, chromatograms, FTIR and CD spectra, crystallographic data. CCDC 2281755. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01594g |
| This journal is © The Royal Society of Chemistry 2023 |