
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe perfluoroalkylthiolation/decarbonylation reaction of 1,3-diketones with perfluoroalkanesulfenic acids†
Jia-Hui
Li
ab,
Min
Jiang
b and
Jin-Tao
Liu
 *ab
*ab
aThe Education Ministry Key Lab of Resource Chemistry and Shanghai Key Laboratory of Rare Earth Functional Materials, Shanghai Normal University, Shanghai 200234, China. E-mail: jtliu@sioc.ac.cn; Fax: +86-21-64166128; Tel: +86 -21-54925188
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 15th November 2023
Abstract
The perfluoroalkylthiolation/decarbonylation reactions of 1,3-dicarbonyl compounds with in situ formed perfluoroalkanesulfenic acids were achieved. Using trifluoromethanesulfonic acid as an additive, a series of α-perfluoroalkylthiolated arylethanones were obtained in moderate to good yields. A possible mechanism was proposed based on the reaction results and control experiments.
Introduction
In recent decades, the introduction of fluorine-containing groups into organic compounds has received considerable attention and has become an important synthetic approach in labs and industries.1 Among various fluorine-containing groups, trifluoromethylthio (CF3S) plays an important role in organofluorine chemistry due to its high electronegativity, hydrophobicity, metabolic activity and bioavailability. Some compounds bearing fluoroalkylthio groups have been applied in the field of agrochemicals and pharmaceuticals.2On the other hand, carbonyl compounds constitute a large family of interesting building blocks for organic synthesis, and a wide range of bioactive and natural compounds possess a carbonyl moiety.3 For example, the structure of β-keto thioether is found in many bioactive molecules and some β-keto thioethers have been used as useful intermediates in organic synthesis,4 and various synthetic methods for β-keto arylthioethers have been developed.5 Consequently, introducing the fluorine-containing group into carbonyl compounds could be of great value for further applications, and the α-functionalization of the carbonyl group appears to be a privileged strategy.6 Since the trifluoromethylthiolation of ethyl formylacetate has been achieved in 1973,7 a large number of electrophilic trifluoromethylthiolation reagents have been developed and great success has been achieved in the trifluoromethylthiolation reactions of keto-esters, ketones and aldehydes by different research groups.8 However, the perfluoroalkylthiolation of carbonyl compounds has been less studied. In 2016, Shibata et al.9 reported the perfluoroalkylthiolation of α-methylene-β-keto esters with perfluoroalkyl-DAST giving α-perfluoroalkylthio-β-keto esters, but an electron-withdrawing group at the β-position of the substrate was necessary. Therefore, the development of practical methods for the perfluoroalkylthiolation of carbonyl compounds is of great importance.
Results and discussion
Recently, our group has been working on the chemistry of perfluoroalkylsulfenic acids (RfSOH). These solution-stable perfluoroalkanesulfenic acids are highly reactive and versatile fluorinated reagents due to the strong electron-withdrawing effect of the perfluoroalkyl group.10 Previous studies have shown that they could undergo addition reactions with some unsaturated compounds, such as alkenes, alkynes and allenes, to afford the corresponding perfluoroalkyl sulfoxides,11 and electrophilic reactions with carbon nucleophiles, such as indoles, activated benzenes and pyrroles to give perfluoroalkylthiolated compounds.12 To explore the reaction with more carbon nucleophiles and the synthetic applications of perfluoroalkanesulfenic acids, we further studied the reactions of perfluoroalkanesulfenic acids with carbonyl compounds. The results are reported in this paper.Initially, phenyl acetone was chosen as the substrate. Based on our previous work,12 a mixture of phenyl acetone, 4.0 equivalents of i-propyl perfluorobutyl sulfoxide (1a) and 2.0 equivalents of trifluoromethanesulfonic acid (TfOH) was directly stirred at 100 °C. However, 19F NMR monitoring showed that the reaction was complicated and no perfluoroalkylthiolated product was isolated (Scheme 1, eqn (1)). Considering the higher nucleophilicity of 1,3-diketone, we prepared 1,3-diphenylpropane-1,3-dione (2a) and studied its reaction with perfluorobutanesulfenic acid generated in situ from i-propyl perfluorobutyl sulfoxide (1a). Interestingly, the reaction occurred readily and α-perfluorobutylthio acetophenone (3a) was formed as the major product, whereas the expected product, 2-perfluorobutylthio-1,3-diphenyl-1,3-propanedione was not obtained (Scheme 1, eqn (2)). Furthermore, benzoic acid was also isolated from the reaction system. It is obvious that a perfluorobutylthiolation/decarbonylation process was involved in this reaction.
 | ||
| Scheme 1 The reaction of carbonyl compounds with in situ generated perfluorobutanesulfenic acid. | ||
Inspired by these results, we screened different conditions to find a suitable protocol for the selective formation of 3a. It was found that decreasing the temperature led to a lower yield of 3a and a longer reaction time (entries 2 and 3). Investigating the ratio of reactants indicated that increasing the amount of 1a or additive was beneficial for the perfluoroalkylthiolation/decarbonylation reaction (entries 4–7). With a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:6 (2a/1a/TfOH), the yield of 3a could be improved to 85% (entry 7). Control experiments showed that TfOH was important for the reaction (entry 8). Other organic acids such as trifluoroacetic acid and acetic acid failed to promote this reaction with full recovery of 2a (entries 9 and 10). We also studied the solvent effect. It was found that the reaction hardly occurred in N,N-dimethylformamide (DMF), acetonitrile or tetrahydrofuran (entries 11–13), and much lower yields were obtained in dichloromethane or n-hexane (entries 14 and 15). Thus, the optimal conditions were set to 4.0 equivalents of perfluoroalkyl sulfoxide and 6.0 equivalents of TfOH in toluene at 100 °C (Table 1).
:4:6 (2a/1a/TfOH), the yield of 3a could be improved to 85% (entry 7). Control experiments showed that TfOH was important for the reaction (entry 8). Other organic acids such as trifluoroacetic acid and acetic acid failed to promote this reaction with full recovery of 2a (entries 9 and 10). We also studied the solvent effect. It was found that the reaction hardly occurred in N,N-dimethylformamide (DMF), acetonitrile or tetrahydrofuran (entries 11–13), and much lower yields were obtained in dichloromethane or n-hexane (entries 14 and 15). Thus, the optimal conditions were set to 4.0 equivalents of perfluoroalkyl sulfoxide and 6.0 equivalents of TfOH in toluene at 100 °C (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | 2a/1a/TfOH | Temp. (°C) | Time (h) | Solvent | 3a (%) |
| a Reaction conditions: 2a (0.2 mmol), i-propyl perfluorobutyl sulfoxide 1a (1.0–4.0 equiv.), TfOH (0–6.0 equiv.), toluene. b Determined by 19F NMR spectroscopy using PhCF3 as the internal standard. c Trifluoroacetic acid was used instead of TfOH. d Acetic acid was used instead of TfOH. | |||||
| 1 | 1:4:2 |
100 | 1 | Toluene | 66 |
| 2 | 1:4:2 |
80 | 4 | Toluene | 60 |
| 3 | 1:4:2 |
60 | 6 | Toluene | 56 |
| 4 | 1:1:2 |
100 | 1 | Toluene | 38 |
| 5 | 1:3:2 |
100 | 1 | Toluene | 53 |
| 6 | 1:4:4 |
100 | 1 | Toluene | 77 |
| 7 | 1:4:6 |
100 | 1 | Toluene | 85 |
| 8 | 1:4:0 |
100 | 1 | Toluene | NR |
| 9c | 1:4:6 |
100 | 4 | Toluene | NR |
| 10d | 1:4:6 |
100 | 4 | Toluene | NR |
| 11 | 1:4:6 |
100 | 1 | DMF | Trace |
| 12 | 1:4:6 |
100 | 1 | CH3CN | Trace |
| 13 | 1:4:6 |
100 | 1 | THF | Trace |
| 14 | 1:4:6 |
100 | 1 | DCM | 28 |
| 15 | 1:4:6 |
100 | 1 | n-Hexane | 25 |

With the optimized reaction conditions in hand, we then examined the reactions of other perfluoroalkyl sulfoxides with 2a. As shown in Scheme 2, the reactions of 2a with in situ formed n-C6F13SOH, n-C8F17SOH, n-C3F7SOH or CF3SOH also took place smoothly to give the corresponding products 3b–3e in moderate to good yields. Furthermore, a range of 1,3-diarylpropane-1,3-diones 2 with different substituents in the phenyl group reacted readily with 1a, giving the expected products in moderate to good yields. 1,3-Diketones containing both electron-rich and electron-deficient phenyl groups were compatible with the reaction conditions. For example, the reactions of diketones bearing methyl or methoxy groups gave the corresponding products 3f and 3g in good yields. Diketones containing a halide or trifluoromethyl substituent also reacted well to give products 3h–k in moderate yields. It was found that the yield of 3g with a methoxy substituent was higher than those of α-perfluoroalkylthio-β-ketones bearing electron-withdrawing groups, indicating that the electronic effect of the substituent had some influence on the reaction. Diketones bearing dichloro or dimethyl substituents were also used and the corresponding products 3l and 3m were obtained in reasonable yields. A similar result was obtained with 1,3-bis(2-naphthyl)-1,3-propanedione (2n). However, 1,3-diketone 2o containing a cyano substituent in both phenyl groups failed to give the desired product 3o due to the weak nucleophilicity of 1,3-diketone 2o. Furthermore, the perfluoroalkylthiolation/decarbonylation reaction didn't occur when ethyl benzoylacetate, pentane-2,4-dione or 1,3-cyclohexanedione was used as the substrate with full recovery of starting materials 2.
 | ||
| Scheme 2 Scope of the perfluoroalkylthiolation/decarbonylation reaction. | ||
We also studied the reaction of 1a with 1,3-diketones 2p–s containing different phenyls. As shown in Table 2, both products were obtained in the cases of 1,3-diketones containing methyl, methoxy or chlorine-substituted phenyls, but only product 3g was obtained when compound 2s bearing a methoxyl and a cyano substituent was used as the substrate.
|
|
||
|---|---|---|
| Entry | Substrate | Products |
| Reaction conditions: 2 (0.2 mmol), 1 (4.0 equiv.), TfOH (6.0 equiv.), toluene, 100 °C, isolated yield. | ||
| 1 |

|

|
| 2 |

|

|
| 3 |

|

|
| 4 |

|

|

In order to investigate the effect of substituents at the methylene position on the reaction, we synthesized 2-ethyl-1,3-diphenyl-1,3-propanedione (2t) and studied its reaction with 1a under standard conditions. 19F NMR monitoring showed that a small amount of perfluoroalkylthiolated product was formed. After separation by column chromatography, 3a and 5 were isolated in 2% and 8% yield, respectively (Scheme 3). It is interesting to find that heating the solution of 2t in the presence of trifluoromethanesulfonic acid at 100 °C for 1 hour gave the decarbonylation product 5 in 26% yield, while 2t remained unchanged in the absence of TfOH under similar conditions. According to the literature, the decarbonylation of β-substituted diketones occurs under basic conditions.13 We hypothesized that 2-perfluoroalkylthiodiketone might be formed as an intermediate under the reaction conditions, which then underwent decarbonylation to give compound 3. Therefore, we prepared 2-trifluoromethylthio-1,3-diphenylpropane-1,3-dione (6a) by the reaction of 2a with CF3SOCl and studied its reaction under standard conditions (Scheme 4). After being treated with TfOH in toluene at 100 °C, 6a was fully converted to 3e.
 | ||
| Scheme 3 Reactions of 2t. | ||
 | ||
| Scheme 4 Preparation and transformation of 6. | ||
Based on the experimental results and the related literature, a plausible mechanism was proposed for the above reactions as shown in Scheme 5. In the presence of trifluoromethanesulfonic acid, the sulfur atom in perfluoroalkanesulfenic acid becomes more electrophilic and is easily attacked by nucleophilic 1,3-diketone 2 to give intermediate 6. Subsequently, the decarbonylation of 6 takes place rapidly to give the final product 3 under the reaction conditions (Scheme 5).
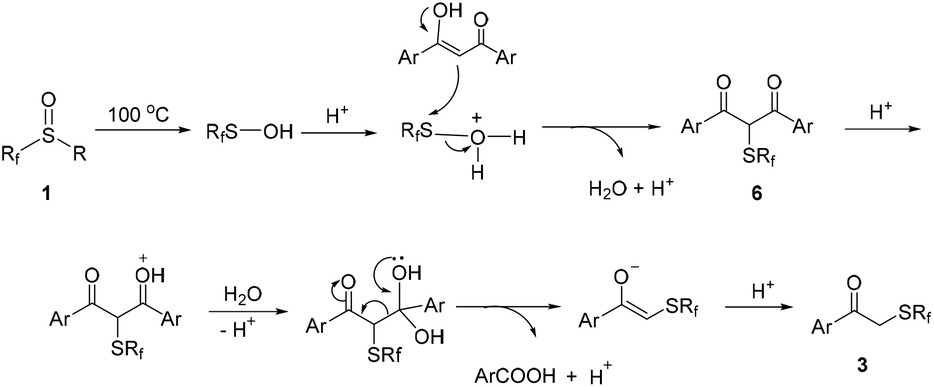 | ||
| Scheme 5 The proposed mechanism. | ||
Conclusions
In conclusion, we have demonstrated the reaction of perfluoroalkanesulfenic acids with 1,3-diketones. In the presence of trifluoromethanesulfonic acid, various 1,3-diarylpropane-1,3-diones reacted readily with perfluoroalkanesulfenic acids formed in situ from perfluoroalkyl sulfoxides to give the corresponding α-perfluoroalkylthio-β-ketone products in moderate to good yields. Mechanism studies indicated that 2-perfluoroalkylthiodiketone might be formed as the intermediate in the reaction system. Having advantages including cheap reagents, easy-to-handle and transition metal-free conditions, this protocol is expected to find applications in organic synthesis.Experimental
General information
Unless otherwise mentioned, solvents and reagents were purchased from commercial sources and used as received. Solvents were freshly distilled using a standard procedure prior to use. Melting points were measured on RY-I apparatus and uncorrected. 1H NMR spectra were recorded in CDCl3 on a Bruker AM 400 spectrometer (400 MHz) with TMS as the internal standard. 19F NMR spectra were recorded on a Bruker AM 400 (376 MHz) spectrometer with CFCl3 as the external standard. 13C NMR spectra were recorded in CDCl3 on a Varian AM-400 spectrometer (100 MHz) or an Agilent AM-400 (100 MHz) spectrometer with TMS as the internal standard. For compounds 3, the peaks for fluorine-containing carbon atoms in perfluoroalkyl groups are difficult to find in 13C NMR spectra because of their multiplicity. High Resolution Mass Spectra (HRMS) were obtained with a Waters Micromass GCT (EI) or an IonSpec FT-ICR mass spectrometer (FI).General procedure for the perfluoroalkylthiolation/decarbonylation reaction
To a sealed tube containing a solution of 1,3-diketones 2 (0.2 mmol) and sulfoxide 1 (0.8 mmol) in dry toluene (2.0 mL) was added TfOH (1.2 mmol). The mixture was stirred at 100 °C for 1 hour. After the completion of the reaction, solvent was removed under reduced pressure and the resulting crude product was purified by column chromatography on silica gel to give the corresponding product 3 (PE:EA = 30/1, v/v).
Compound 3a: brown liquid, 85% yield, 63 mg. 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0 Hz, 2H), 4.55 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −81.01 (t, J = 8.0 Hz, 3F), −87.41 to −81.49 (m, 2F), −121.54 to −120.63 (m, 2F), −125.54 to −125.62 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.86, 134.74, 134.29, 128.99, 128.39, 37.11 ppm; MS (EI) m/z (%): 370 (1) [M]+, 135 (3), 119 (3), 105 (100), 91 (6), 77 (60), 69 (5), 51 (8); HRMS (FI) calcd for C12H7F9OS [M]+ requires 370.0068, found 370.0063.
Compound 3b: yellow solid, m.p. 32–34 °C, 86% yield, 80 mg; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0 Hz, 2H), 4.55 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.90 (t, J = 10.2 Hz, 3F), −86.95 to −87.67 (m, 2F), −119.76 (s, 2F), −121.51 (s, 2F), −122.89 (s, 2F), −126.22 to −126.34 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.87, 134.74, 134.30, 129.00, 128.40, 37.17 ppm; MS (EI) m/z (%): 470 (3) [M]+, 365 (40), 135 (20), 105 (100), 91 (86), 77 (98), 65 (30), 51 (90); HRMS (EI) calcd for C14H7F13OS [M]+ requires 470.0005, found 470.0011.
Compound 3c: yellow solid, m.p. 63–65 °C, 86% yield, 98 mg; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0 Hz, 2H), 4.55 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.90 (t, J = 11.2 Hz, 3F), −87.21 to −87.29 (m, 2F), −119.67 (s, 2F), −121.26 (s, 2F), −121.82 to −122.34 (m, 4F), −122.87 (s, 2F), −126.19 to −126.28 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.87, 134.74, 134.28, 128.98, 128.39, 37.13 ppm; MS (EI) m/z (%): 570 (2) [M]+, 551 (8), 465 (10), 179 (15), 105 (100), 91 (56), 77 (85), 51 (50); HRMS (FI) calcd for C16H7F17OS [M]+ requires 569.9941, found 569.9947.
Compound 3d: yellow liquid, 84% yield, 54 mg; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0, 2H), 4.55 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.08 (t, J = 11.2 Hz, 3F), −88.08 to −88.13 (m, 2F), −124.02 (t, J = 3.7 Hz, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.86, 134.73, 134.32, 129.01, 128.41, 37.04 ppm; MS (EI) m/z (%): 320 (2) [M]+, 215 (3), 169 (5), 105 (100), 91 (10), 77 (58), 69 (8), 51 (18); HRMS (FI) calcd for C11H7F7OS [M]+ requires 320.0100, found 320.0104.
Compound 3e: brown liquid, 61% yield, 27 mg; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.63 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0, 2H), 4.51 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −41.46 (s, 3F) ppm; 13C NMR (101 MHz, CDCl3) δ 192.00, 134.73, 134.26, 131.14 (q, J = 197 Hz), 128.99, 128.42, 38.39 (d, J = 2.0 Hz) ppm; MS (EI) m/z (%): 220 (2) [M]+, 182 (5), 105 (100), 83 (3), 77 (45), 69 (3), 65 (2), 51 (5); HRMS (FI) calcd for C9H7F3OS [M]+ requires 220.0164, found 220.0159. Some unidentified impurities were hard to separate from the reaction mixture. So, the clean 1H NMR spectrum was not obtained.
Compound 3f: white solid, m.p. 89–91 °C, 74% yield, 56 mg; 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 4.52 (s, 2H), 2.42 (s, 3H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.98 (t, J = 11.2 Hz, 3F), −87.39 to −81.46 (m, 2F), −121.53 to −120.61 (m, 2F), −125.50 to −125.61 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.45, 145.45, 132.27, 129.67, 128.52, 37.07, 21.76 ppm; MS (EI) m/z (%): 384 (1) [M]+, 292 (22), 265 (5), 149 (3), 119 (100), 105 (7), 91 (60), 65 (20); HRMS (FI) calcd for C13H9F9OS [M]+ requires 384.0225, found 384.0226.
Compound 3g: white solid, m.p. 69–71 °C, 89% yield, 71 mg; 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 4.50 (s, 2H), 3.88 (s, 3H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.99 (t, J = 11.2 Hz, 3F), −87.41 to −81.48 (m, 2F), −121.54 to −120.61 (m, 2F), −125.54 to −125.61 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 190.27, 164.44, 130.81, 127.73, 114.17, 55.59, 36.88 ppm; MS (EI) m/z (%): 400 (2) [M]+, 265 (3), 135 (100), 121 (5), 107 (7), 92 (9), 77 (12), 69 (3); HRMS (FI) calcd for C13H9F9O2S [M]+ requires 400.0174, found 400.0168.
Compound 3h: brown liquid, 78% yield, 63 mg; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 4.50 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −81.03 (t, J = 11.2 Hz, 3F), −87.39 to −87.46 (m, 2F), −120.56 to −120.63 (m, 2F), −125.56 to −125.64 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 190.76, 140.97, 133.01, 129.78, 129.37, 36.89 ppm; MS (EI) m/z (%): 404 (2) [M]+, 265 (6), 139 (100), 125 (8), 111 (47), 89 (3), 75 (36), 50 (5); HRMS (FI) calcd for C12H6ClF9OS [M]+ requires 403.9676, found 403.9682.
Compound 3i: white solid, m.p. 52–54 °C, 58% yield, 51 mg; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 4.49 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.94 (t, J = 11.2 Hz, 3F), −87.33 to −87.41 (m, 2F), −121.50 to −120.58 (m, 2F), −125.49 to −125.57 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 190.95, 133.43, 132.40, 129.84, 129.75, 36.83 ppm; MS (EI) m/z (%): 448 (2) [M]+, 350 (6), 265 (10), 185 (98), 183 (100), 157 (79), 155 (80), 76 (50); HRMS (FI) calcd for C12H6BrF9OS [M]+ requires 447.9174, found 447.9172.
Compound 3j: yellow solid, m.p. 26–28 °C, 82% yield, 64 mg; 1H NMR (400 MHz, CDCl3) δ 8.19–7.79 (m, 2H), 7.19–7.14 (m, 2H), 4.51 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.00 to −81.14 (m, 3F), −87.44 to −87.52 (m, 2F), −100.04 to −107.02 (m, 1F), −120.58 to −120.66 (m, 2F), −125.59 to −125.67 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 190.34, 166.40 (d, J = 257.1 Hz), 131.12, 131.22, 116.24 (d, J = 22.2 Hz), 36.90 ppm; MS (EI) m/z (%): 388 (2) [M]+, 123 (100), 109 (10), 95 (41), 83 (3), 75 (15), 69 (12), 50 (2); HRMS (FI) calcd for C12H6F10OS [M]+ requires 387.9974, found 387.9969.
Compound 3k: yellow solid, m.p. 40–41 °C, 56% yield, 49 mg; 1H NMR (400 MHz, CDCl3) 8.06 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 4.53 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −63.39 (s, 3F), −80.98 (td, J = 9.8, 2.7 Hz, 3F), −87.33 to −87.40 (m, 2F), −121.53 to −120.60 (m, 2F), −125.54 to −125.59 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.08, 137.32, 135.55 (q, J = 33.3 Hz), 128.79, 126.11 (q, J = 3.1 Hz), 123.31 (d, J = 273.7 Hz), 36.92 ppm; MS (EI) m/z (%): 438 (2) [M]+, 419 (20), 265 (10), 173 (100), 159 (20), 145 (85), 125 (9), 95 (10); HRMS (FI) calcd for C13H6F12OS [M]+ requires 437.9942, found 437.9940.
Compound 3l: brown liquid,78% yield, 68 mg; 1H NMR (400 MHz, CDCl3) 8.02 (d, J = 4.0 Hz, 1H), 7.77 (dd, J = 8.3, 2.1 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 4.47 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.93 (t, J = 7.5 Hz, 3F), −87.30 to −87.38 (m, 2F), −121.52 to −120.60 (m, 2F), −125.53 to −125.61 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 189.86, 139.17, 134.15, 133.94, 131.17, 130.35, 127.33, 36.73 ppm; MS (EI) m/z (%): 438 (3) [M]+, 175 (98), 173 (100), 159 (59), 145 (95), 138 (20), 109 (85), 75 (70); HRMS (FI) calcd for C12H5Cl2F9OS [M]+ requires 437.9298, found 437.9286.
Compound 3m: brown liquid, 74% yield, 59 mg; 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 2H), 7.26 (s, 1H), 4.53 (s, 2H), 2.38 (s, 6H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.93 (t, J = 7.5 Hz, 3F), −87.30 to −87.38 (m, 2F), −121.53 to −120.60 (m, 2F), −125.50 to −125.62 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 192.16, 138.77, 136.00, 134.82, 126.15, 37.36, 21.22 ppm; MS (EI) m/z (%): 398 (2) [M]+, 265 (15), 133 (100), 119 (46), 105 (85), 91 (30), 77 (75), 69 (38); HRMS (FI) calcd for C14H11F9OS [M]+ requires 398.0381, found 398.0378.
Compound 3n: light yellow solid, m.p. 69–71 °C, 77% yield, 65 mg; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.03–7.96 (m, 2H), 7.9–7.82 (m, 2H), 7.66–7.57 (m, 2H), 4.69 (s, 2H) ppm; 19F NMR (376 MHz, CDCl3) δ −80.88 to −80.95 (m, 3F), −87.26 to −87.35 (m, 2F), −120.47 to −120.54 (m, 2F), −125.45 to −125.56 (m, 2F) ppm; 13C NMR (101 MHz, CDCl3) δ 191.78, 136.04, 132.37, 132.07, 130.48, 129.70, 129.25, 129.03, 127.93, 127.28, 123.56, 37.23 ppm; MS (EI) m/z (%): 420 (8) [M]+, 265 (3), 155 (100), 141 (10), 127 (50), 105 (5), 77 (8), 69 (7); HRMS (FI) calcd for C16H9F9OS [M]+ requires 420.0225, found 420.0223.
Compound 6a: yellow liquid, 8% yield, 5.2 mg; 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 4H), 7.59 (t, J = 8.0 Hz, 2H), 7.46 (t, J = 8.0 Hz, 4H), 6.40 (s, 1H) ppm; 19F NMR (376 MHz, CDCl3) δ −40.09 (s, 1.5F), −46.53 (s, 1.5F) ppm. MS (EI) m/z (%): 324 (28) [M]+, 303 (5), 255 (8), 237 (9), 221 (5), 105 (100), 82 (8), 77 (36); HRMS (EI) calcd for C16H11F3O2S [M]+ requires 324.0426, found 324.0425.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21572257) is gratefully acknowledged.References
- (a) T. Umemoto, Chem. Rev., 1996, 96, 1757–1778 CrossRef CAS; (b) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 CrossRef CAS; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS; (e) F. A. Carey and R. J. Sundberg, Nucleophilic Substitution, in Advanced Organic Chemistry: Part A: Structure and Mechanisms, ed. F. A. Carey and R. J. Sundberg, Springer US, Boston, MA, 2007 Search PubMed; (f) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048–5050 ( Angew. Chem. , 2012 , 124 , 5134–5136 ) CrossRef CAS; (g) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 ( Angew. Chem. , 2012 , 124 , 9082–9090 ) CrossRef CAS; (h) X. Wang, Y. Zhang and J. Wang, Sci. Sin.: Chim., 2012, 42, 1417–1427 CAS; (i) B. E. Smart, Chem. Rev., 1996, 96, 1555–1556 CrossRef CAS; (j) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC; (k) Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed; (l) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31–42 RSC; (m) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- (a) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS; (c) T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer, Berlin, 2000 CrossRef; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS; (e) M. Li, J. Guo, X. S. Xue and J. P. Cheng, Org. Lett., 2016, 18, 264–267 CrossRef CAS; (f) X. H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS.
- (a) T. Silverstone, J. Fincham and J. Plumley, Br. J. Clin. Pharmacol., 1979, 7, 353–356 CrossRef CAS; (b) R. B. Strelkov and L. F. Semenov, Radiobiologiya, 1964, 4, 756–759 CAS; (c) L. M. Yagupolskii, I. I. Maletina, K. I. Petko, D. V. Fedyuk, R. Handrock, S. S. Shavaran, B. M. Klebanov and S. Herzig, J. Fluorine Chem., 2001, 109, 87–94 CrossRef CAS.
- (a) R. J. Cremlyn, An Introduction to Organo-Sulfur Chemistry, Wiley & Sons, New York, 1996 Search PubMed; (b) H. Q. Zhang, J. M. Xi and W. W. Liao, J. Org. Chem., 2020, 85, 1168–1180 CrossRef CAS; (c) H. Y. Chen, S. Kim, J. Y. Wu, E. T. Birzin, W. Chan, Y. T. Yang, J. Dahllund, F. DiNinno, S. P. Rohrer, J. M. Schaeffer and M. L. Hammond, Bioorg. Med. Chem. Lett., 2004, 14, 2551–2554 CrossRef CAS.
- (a) M. Liao and B. Wang, Green Chem., 2007, 9, 184–188 RSC; (b) J. Li, K. Ji, R. Zheng, J. Nelson and L. Zhang, Chem. Commun., 2014, 50, 4130–4133 RSC; (c) R. M. P. Dias and A. C. B. Burtoloso, Org. Lett., 2016, 18, 3034–3037 CrossRef CAS; (d) Y. Xu, X. Huang, G. Lv, R. Lai, S. Lv, J. Li, L. Hai and Y. Wu, Eur. J. Org. Chem., 2020, 4635–4638 CrossRef CAS; (e) X. B. Xu, Z. H. Lin, Y. Liu, J. Guo and Y. He, Org. Biomol. Chem., 2017, 15, 2716–2720 RSC; (f) A. Jana, P. Bhaumick and L. H. Choudhury, New J. Chem., 2020, 44, 8442–8453 RSC; (g) A. Cadu, R. A. Watile, S. Biswas, A. Orthaber, P. J. R. Sjöberg and J. S. M. Samec, Org. Lett., 2014, 16, 5556–5559 CrossRef CAS.
- (a) S. G. Li and S. Z. Zard, Org. Lett., 2013, 15, 5898–5902 CrossRef CAS; (b) Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W. D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970–976 CrossRef CAS; (c) L. Xu, H. Wang, C. Zheng and G. Zhao, Adv. Synth. Catal., 2017, 359, 2942–2948 CrossRef CAS; (d) H. B. Yang, X. Fan, Y. Wei and M. Shi, Org. Chem. Front., 2015, 2, 1088–1093 RSC.
- H. Bayreuther and A. Haas, Chem. Ber., 1973, 106, 1418–1422 CrossRef CAS.
- (a) L. Hu, M. Wu, H. Wan, J. Wang, G. Wang, H. Guo and S. Sun, New J. Chem., 2016, 40, 6550–6553 RSC; (b) X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 ( Angew. Chem. , 2013 , 125 , 3541–3544 ) CrossRef CAS; (c) C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316–9320 ( Angew. Chem. , 2014 , 126 , 9470–9474 ) CrossRef CAS.
- I. Saidalimu, S. Suzuki, T. Yoshioka, E. Tokunaga and N. Shibata, Org. Lett., 2016, 18, 6404–6407 CrossRef CAS.
- (a) X. B. Li, Z. F. Xu, L. J. Liu and J. T. Liu, Eur. J. Org. Chem., 2014, 1182–1188 CrossRef CAS; (b) J. H. Xu, M. Jiang, L. P. Song and J. T. Liu, Eur. J. Org. Chem., 2021, 1919–1923 CrossRef CAS.
- (a) X. B. Li, J. Zhao, J. Jiang and J. T. Liu, J. Fluorine Chem., 2016, 185, 24–30 CrossRef CAS; (b) X. B. Li, J. Zhao, Q. Liu, M. Jiang and J. T. Liu, Chin. J. Org. Chem., 2019, 39, 183–191 CrossRef CAS.
- J. H. Li, J. H. Xu, M. Jiang, L. P. Song and J. T. Liu, Eur. J. Org. Chem., 2022, e202200885 CrossRef.
- T. Hirashita, S. Kuwahara, S. Okochi, M. Tsuji and S. Araki, Tetrahedron Lett., 2010, 51, 1847–1851 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d3ob01482g |
| This journal is © The Royal Society of Chemistry 2023 |