
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot cascade synthesis of dibenzothiophene-based heterobiaryls from dibenzothiophene-5-oxide
Xiaofang
Hu†
ab,
Zeen
Qiao†
ac,
Li
Zhang
ac,
Jinzhong
Zhao
 b,
Ya-Zhou
Liu
a,
Jiangang
Zhang
*b and
Xiaofeng
Ma
*a
b,
Ya-Zhou
Liu
a,
Jiangang
Zhang
*b and
Xiaofeng
Ma
*a
aNatural Products Research Centre, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, People's Republic of China. E-mail: maxf@cib.ac.cn
bCollege of Foundation, Shanxi Agricultural University, 030800 Taigu, Shanxi, People's Republic of China. E-mail: zjg487992@163.com
cUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 31st October 2023
Abstract
A sulfoxide directed C–H metalation/boration/B2Pin2 mediated reduction/Suzuki coupling process to synthesize 4-substituted dibenzothiophene (DBT) in one-pot from dibenzothiophene-5-oxide (DBTO) was developed. A variety of DBT-based heterobiaryls were prepared in satisfactory to good yields. A mechanism was proposed. The application of this methodology was demonstrated by synthesizing a luminescent material.
Sulfur-containing heteroaromatic sub-units, such as dibenzothiophene (DBT) are widely found in unrefined fossil fuels and light-emitting materials.1–4 Moreover, due to the pioneering contribution by Yorimitsu and co-workers, these molecules have also been used extensively to the preparation of other aromatic and heterocyclic aromatic ring systems5–11 including spirocyclic diarylfluorenes,7 triphenylenes,8 carbazoles9,10 and dibenzophosphole oxides11via an aromatic metamorphosis (Scheme 1A).
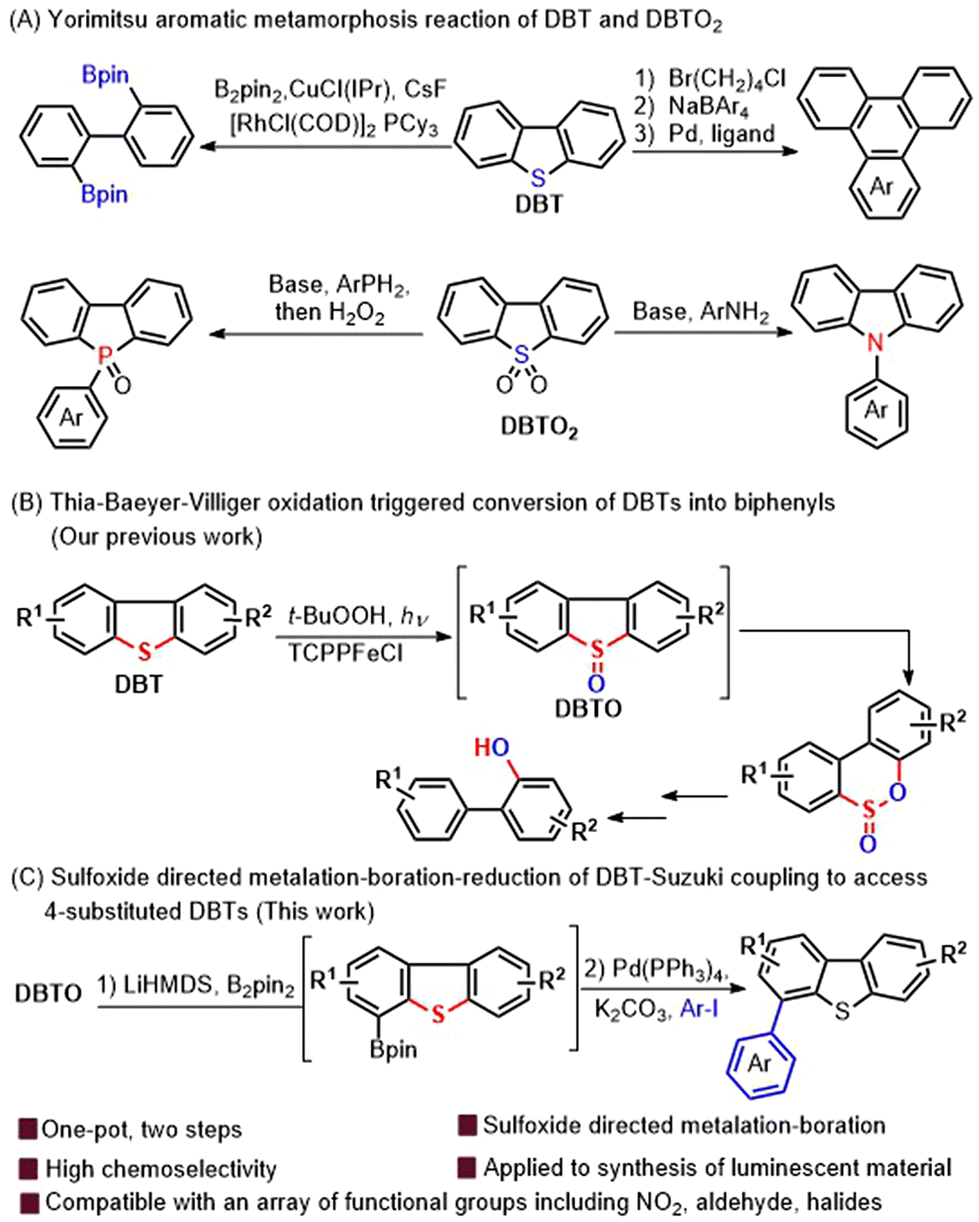 | ||
| Scheme 1 The transformation of DBTs to other aromatic molecules. | ||
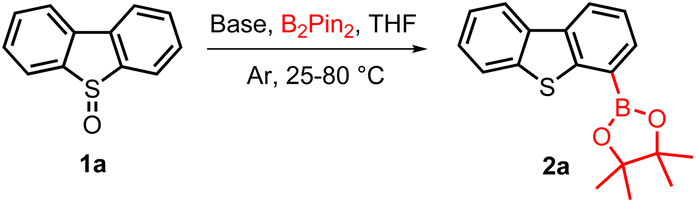
The direct boration of C–H bonds plays an extremely important role in the synthesis of organoborons with high efficiency and selectivity.12–16 C–H boration assisted by directional groups is one of the most powerful methods to achieve high regioselectivity of C–H conversion. Due to continuous development in recent decades, this type of reaction can realize a variety of types of C–H bond functionalization. In order to achieve better functional group compatibility, various directing-groups including ketones, amides, sulfonates, pyridines, acetals, imidazoles, oxazolines, pyrazoles, and even sp3-nitrogen based groups such as NMe2 have been developed.12–16 Meanwhile, similar types of functionalization of C–H bonds by using sulfur containing directing groups especially sulfoxides have also been developed for the formation of C–C and C–O bonds.17–23 For example, sulfoxides have been used as directing groups for transition-metal-catalyzed C–H activation, or more traditionally, for stoichiometric metalation24,25 to make new C–C bonds. However, using sulfoxides as directing groups for metalation–boration, especially metalation–boration of sp2 C–H bonds is much more challenging due to the intrinsic sensitivity to boration conditions,25,26 even though the produced organoborons are widely used in organic functional group transformation. Thanks to our continual efforts towards realizing the high-value utilization of DBT derivatives and the synthesis of biaryls (Scheme 1B),27–30 we have recently achieved a diverse conversion of DBT to a range of biphenyls via a photoinduced Thia-Baeyer–Villiger oxidation of DBTOs.27 We reported herein a sulfoxide group directed C–H metalation–boration of DBT derivatives. In particular, we have developed a sulfoxide directed regioselective boration of DBTO, which was followed by reduction of the sulfoxide to sulfide and the Suzuki coupling reaction, yielding 4-aromatic substituted DBT derivatives (heterobiaryls) in one-pot (Scheme 1C).
We started our investigation by using DBTO (1a) as the model substrate in the presence of LiHMDS and B2Pin2. Initial screening to determine the amounts of base and B2Pin2 showed that good results were only obtained when 6.0 equiv. of LiHMDS and 4.0 equiv. of B2Pin2 were employed. Under these conditions, we could detect the formation of desired product TD4B (2a) in 40% yield when the reaction was carried out at room temperature (Table 1, entry 1). Increasing the temperature to 45 °C and 55 °C improved the yield to 59% and 72%, respectively (Table 1, entries 2 and 3), while further heating the reaction mixture only led to a decrease in yield (Table 1, entries 4 and 5). The highest yield was obtained when 5.0 equiv. of B2Pin2 was employed, which delivered the product in 83% isolated yield (Table 1, entry 6). However, further increasing the amount of B2Pin2 dramatically reduced the yield to 61% due to the increase of the DBT by-product, which was generated from the reduction of DBTO in the presence B2Pin2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 (Table 1, entry 7). In contrast, using 1–3 equiv. of B2Pin2 led to the formation of the products in less than 20% yield (Table 1, entry 8). Other bases including KHMDS, NaHMDS, n-BuLi, t-BuOLi, MeOLi and Li2CO3 were also examined, and in all these cases, only the reduction of DBTO took place (Table 1, entries 9–14).
31 (Table 1, entry 7). In contrast, using 1–3 equiv. of B2Pin2 led to the formation of the products in less than 20% yield (Table 1, entry 8). Other bases including KHMDS, NaHMDS, n-BuLi, t-BuOLi, MeOLi and Li2CO3 were also examined, and in all these cases, only the reduction of DBTO took place (Table 1, entries 9–14).
|
|
||||
|---|---|---|---|---|
| Entry | Base | B2pin2 (eq.) | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), base (6.0 eq.), B2Pin2 (4.0–6.0 eq.), THF (1.2 mL), at 25–80 °C under Ar atmosphere for 13 h. b NMR yield using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. d 2.0 eq. of base was used. | ||||
| 1 | LiHMDS | 4.0 | 25 | 40 |
| 2 | LiHMDS | 4.0 | 45 | 59 |
| 3 | LiHMDS | 4.0 | 55 | 72 (70c) |
| 4 | LiHMDS | 4.0 | 65 | 62 |
| 5 | LiHMDS | 4.0 | 80 | 14 |
| 6 | LiHMDS | 5.0 | 55 | 88 (83 ) |
| 7 | LiHMDS | 6.0 | 55 | 61 |
| 8 | LiHMDS | 1.0–3.0 | 55 | <20 |
| 9 | NaHMDS | 5.0 | 55 | — |
| 10 | KHMDS | 5.0 | 55 | — |
| 11 | n-BuLi | 5.0 | 55 | — |
| 12 | t-BuOLi | 5.0 | 55 | — |
| 13 | MeOLi | 5.0 | 55 | — |
| 14d | Li2CO3 | 5.0 | 55 | — |
The development of contemporary organic chemistry requires the efficient and environmentally sustainable preparation of valuable target molecules. In this context, the one-pot synthesis of a target molecule in the same reaction vessel is considered as an effective approach.32–34 This strategy could save time, reduce chemical waste, and lower costs by avoiding the need for lengthy work-up and purification processes, and thus enhancing the efficiency of the synthesis of target molecules. We have also applied the one-pot strategy in our previous reports for the synthesis of complex molecules.28,35,36 Therefore, after determining the optimal reaction conditions (Table 1, entry 6), we wondered if we could use the formed arylboronates directly for the coupling reactions. Initially, the Suzuki coupling reaction carried out by adding Pd catalyst and aryl halides directly into the reaction mixture resulted in the formation of a messy reaction complex. Fortunately, when the same reaction was performed by removing the solvent after the boration reaction, followed by adding the Pd catalyst, iodobenzene and solvent, and then heating the reaction mixture overnight, the coupling product 3a was produced in 69% yield over the two steps.37 The procedure was then used to investigate the scope of this one-pot two-step method to synthesize 4-aryl substituted DBT from DBTO via the TD4B intermediate, and the results are summarized in Table 2. Aryl halides with electron withdrawing groups including NO2, aldehyde, and ketone at the ortho-, meta-, and para-positions afforded the corresponding Suzuki coupling products (3b–3d) in 35% to 76% yields. The influence of electron donating groups on aryl halides was then examined. When 4-iodotoluene was subjected to a mixture of boration reaction, the corresponding product 3e was isolated in 33% yield after two steps, while iodomesitylene offered the product 3f in 42% yield. 1-tert-Butyl-4-iodobenzene and 2-iodoanisole were also successfully transformed into the 4-aryl-substituted DBT product in 79% (3g) and 54% (3h) yields, respectively. Moreover, the aryl iodides substituted with OCF3 at the para- and ortho-positions were also suitable substrates for this protocol and were coupled with the in situ formed TD4B intermediate directly, the corresponding coupling products (3i, 3j) were produced in 65% and 72% yields, respectively. We then investigated the selective coupling reaction of the in situ TD4B formed between different aryl halides. Thus, aryl iodides with other halide substituents such as F, Cl, and Br on the phenyl ring were subjected to the standard reaction procedure, it was found that all the aryl dihalides were reacted selectively with the aryl iodide part and produced the corresponding adducts (3k–3n) in around 60% yield over two steps. Heterocyclic aryl iodides, such as 2-iodothiophene can also be converted smoothly to obtain the corresponding 4-aryl heterocyclic substituted DBT product (3o) in 50% yield. Similarly, heterocyclic aryl dihalides including 2-fluoro-5-iodopyridine and 2-bromo-4-iodopyridine also reacted smoothly with the TD4B intermediate, giving the corresponding products (3p and 3q) in 55% and 38% yields, respectively. Substituted DBTO such as 2,8-dibenzyl-DBTO was also used for this transformation, which after a one-pot two-step reaction was converted to multi-substituted DBT in 62% yield (3r).38

|
Moreover, we also successfully synthesized the luminescent material PhImPOTD39 in 62% yield through the reaction between DBTO and PhImPOI by our one-pot two-step method (Scheme 2), which greatly shortened the steps and improved the reaction efficiency.
 | ||
| Scheme 2 Synthesis of PhImPOTD via a one-pot process from DBTO. | ||
Control experiments showed that DBT does not undergo metalation–boration under otherwise identical conditions, which confirmed that the boration of DBTO took place prior to the sulfoxide reduction (Scheme 3A, eqn (1)). Replacing B2Pin2 with (PinB)2O only results in the recovery of the DBTO, no 2a was detected, which demonstrated that a Pummerer-type reaction40 did not take place in our case (Scheme 3A, eqn (2)). Subjecting DBTO to B2Pin2 (5.0 equiv.) in the absence of LiHMDS led to the reduction of DBTO to DBT in around 15% yield (Scheme 3A, eqn (3)). Similarly, when 4-Bpin-DBTO (4) was treated with B2Pin2 (5.0 equiv.), we isolated 2a in 30% yield. Based on these observations and prior works, a plausible reaction mechanism for the formation of 2a is proposed in Scheme 3. First, ortho-lithiation directed by sulfoxide forms organolithium intermediate I which in the first case (pathway-I)41 undergoes a nucleophilic substitution reaction with B2pin2 to generate an intermediate boronate complex II, which was followed by the reduction of a pendant S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group with a proximate B–B unit generated 2a, accompanied by the formation of lithium 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-olate (PinBOLi). The presence of PinBOLi was confirmed by GC-MS. Meanwhile, nucleophilic substitution of intermediate I with B2pin2 may generate 4-Bpin-DBTO (4) (pathway-II). Then, coordination of the oxygen atom in compound 4 with boron from B2pin2 forms intermediate III, which was followed by a 1,2-migration of the terminal boryl moiety to the oxygen, giving reduction product 2 accompanied by the formation of (PinB)2O. At the present stage, we prefer the first pathway (pathway-I), as we confirmed the presence of PinBOH, whereas (PinB)2O was not detected by GC-MS and HRMS; however, we cannot rule out the secondary pathway (pathway-II) as we did detect compound 4 in the reaction mixture by HRMS. With TD4B (2) in hand, under the classic Suzuki coupling reaction conditions, the 4-substituted DBT derivatives would be generated in the presence of aryl halides catalyzed by the Pd catalyst.
O group with a proximate B–B unit generated 2a, accompanied by the formation of lithium 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-olate (PinBOLi). The presence of PinBOLi was confirmed by GC-MS. Meanwhile, nucleophilic substitution of intermediate I with B2pin2 may generate 4-Bpin-DBTO (4) (pathway-II). Then, coordination of the oxygen atom in compound 4 with boron from B2pin2 forms intermediate III, which was followed by a 1,2-migration of the terminal boryl moiety to the oxygen, giving reduction product 2 accompanied by the formation of (PinB)2O. At the present stage, we prefer the first pathway (pathway-I), as we confirmed the presence of PinBOH, whereas (PinB)2O was not detected by GC-MS and HRMS; however, we cannot rule out the secondary pathway (pathway-II) as we did detect compound 4 in the reaction mixture by HRMS. With TD4B (2) in hand, under the classic Suzuki coupling reaction conditions, the 4-substituted DBT derivatives would be generated in the presence of aryl halides catalyzed by the Pd catalyst.
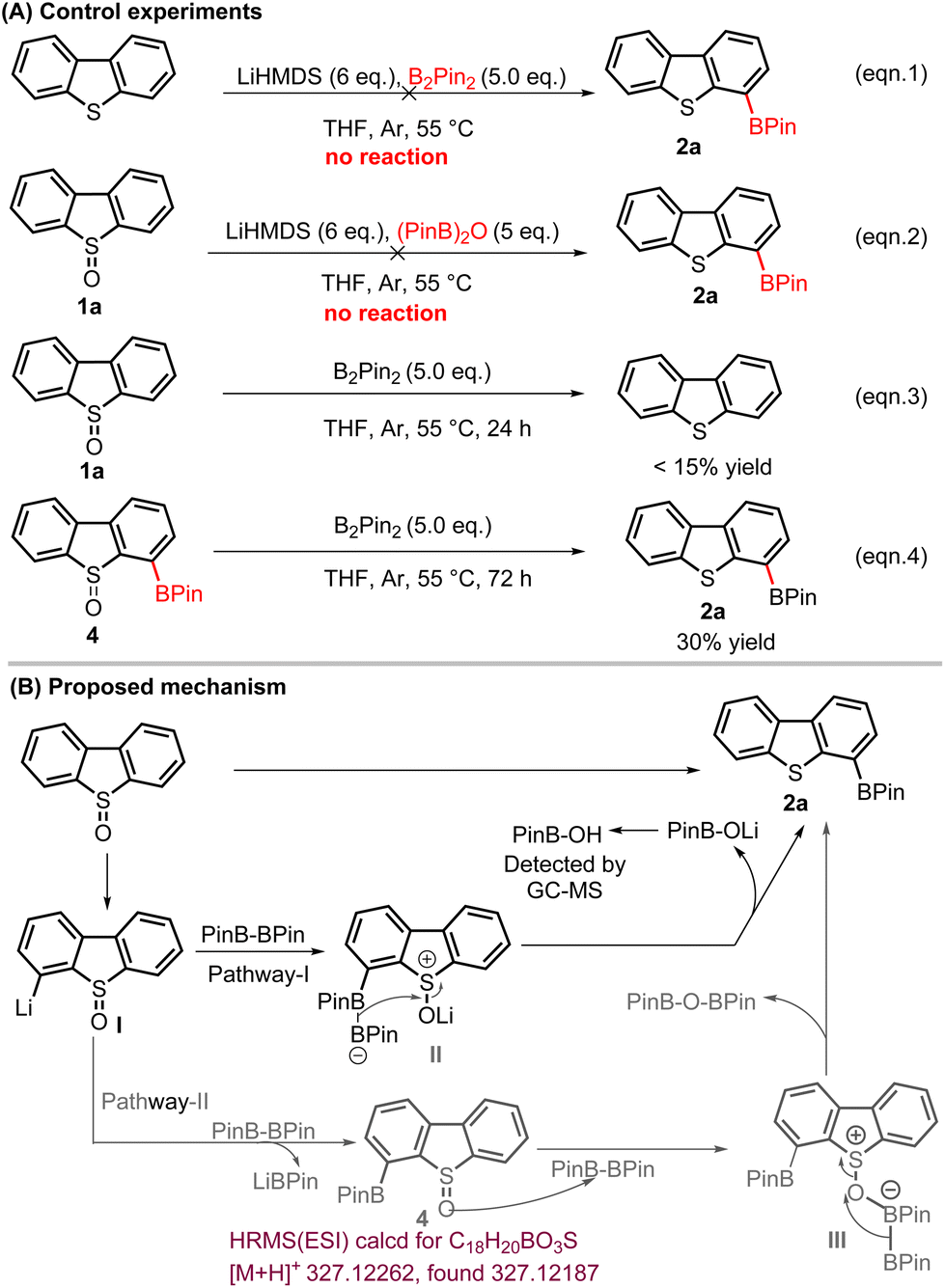 | ||
| Scheme 3 The control experiments and the proposed mechanism for the formation of TD4B from DBTO. | ||
Conclusions
In conclusion, a range of 4-substituted DBT derivatives were successfully synthesized via a sulfoxide directed C–H metalation–boration/B2Pin2 mediated reduction/Suzuki coupling tandem process. This methodology greatly reduced the number of steps for synthesizing such substances and made the reaction more efficient and controllable. The produced TD4B intermediate could be used for further derivatization.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National Natural Science Foundation of China (22001246 and 22377123), the Sichuan Science and Technology Program (2022ZYD0047 and 2022JDRC0132), the Biological Resources Program (KFJ-BRP-008) from Chinese Academy of Sciences (CAS), the CAS Pioneer Hundred Talents Program and the Thousand Talents Program of Sichuan Province, for the financial support.References
- E. Block, Advances in Sulfur Chemistry, JAI Press, Greenwich, 1994 Search PubMed.
- R. J. Cremlyn, An Introduction of Organosulfur Chemistry, Wiley, Chichester, 1996 Search PubMed.
- J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879–890 CrossRef CAS.
- R. Javadli and A. De Klerk, Appl. Petrochem. Res., 2012, 1, 3–19 CrossRef CAS.
- H. Yorimitsu, D. Vasu, M. Bhanuchandra, K. Murakami and A. Osuka, Synlett, 2016, 1765–1774 CrossRef CAS.
- K. Nogi and H. Yorimitsu, Chem. Commun., 2017, 53, 4055–4065 RSC.
- M. Bhanuchandra, H. Yorimitsu and A. Osuka, Org. Lett., 2016, 18, 384–387 CrossRef CAS PubMed.
- D. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 7162–7166 CrossRef CAS PubMed.
- M. Bhanuchandra, K. Murakami, D. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10234–10238 CrossRef CAS PubMed.
- A. Kaga, K. Nogi and H. Yorimitsu, Chem. Eur. J., 2019, 25, 14780–14784 CrossRef CAS PubMed.
- M. Onoda, Y. Koyanagi, H. Saito, M. Bhanuchandra, Y. Matano and H. Yorimitsu, Asian J. Org. Chem., 2017, 6, 257–261 CrossRef CAS.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- L. Xu, G.-H. Wang, S. Zhang, H. Wang, L.-H. Wang, L. Liu, J. Jiao and P.-F. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS.
- Z.-T. Jiang, B.-Q. Wang and Z.-J. Shi, Chin. J. Chem., 2018, 36, 950–954 CrossRef CAS.
- Y. Li and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 1770–1774 CrossRef CAS PubMed.
- X. Tang, C. M. Wang, T. H. Gao, L. Fan, L. Chen and L. P. Sun, Adv. Synth. Catal., 2018, 361, 26–38 CrossRef.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef CAS PubMed.
- Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129–5133 CrossRef CAS PubMed.
- K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef CAS PubMed.
- M. V. Barysevich, M. V. Laktsevich-Iskryk, A. V. Krech, V. N. Zhabinskii, V. A. Khripach and A. L. Hurski, Eur. J. Org. Chem., 2020, 937–943 CrossRef CAS.
- T. Sato, K. Nogi and H. Yorimitsu, ChemCatChem, 2020, 12, 3467–3471 CrossRef CAS.
- Gilman and Esmay found that DBTO could undergo metalation-reduction to produce 4-dibenzothiophenecarboxylic acid in 35.7% yield. For detail, see: H. Gilman and D. L. Esmay, J. Am. Chem. Soc., 1952, 74, 266–268 CrossRef CAS.
- C. Quesnelle, T. Iihama, T. Aubert, H. Perrier and V. Snieckus, Tetrahedron Lett., 1992, 33, 2625–2628 CrossRef CAS.
- G. Casoni, M. Kucukdisli, J. M. Fordham, M. Burns, E. L. Myers and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 11877–11886 CrossRef CAS PubMed.
- X. Ma, Y. Liu, L. Du, J. Zhou and I. E. Markó, Nat. Commun., 2020, 11, 914 CrossRef CAS PubMed.
- A. Wang, Y.-Z. Liu, Z. Shen, Z. Qiao and X. Ma, Org. Lett., 2022, 24, 1454–1459 CrossRef CAS PubMed.
- X. Zhang, Y.-Z. Liu, H. Shao and X. Ma, Molecules, 2022, 27, 8517 CrossRef CAS PubMed.
- H. Shi, J. Yang, Z. Qiao, L. Li, G. Liu, Q. Dai, L. Xu, W. Jiao, G. Zhang, F. Wang, X. Lu and X. Ma, Org. Biomol. Chem., 2023, 21, 7005–7017 RSC.
- F. Takahashi, K. Nogi and H. Yorimitsu, Eur. J. Org. Chem., 2020, 3009–3012 CrossRef CAS.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC and cited therein.
- Y. Hayashi, J. Org. Chem., 2021, 86, 1–23 CrossRef CAS PubMed.
- N. Umekubo, Y. Suga and Y. Hayashi, Chem. Sci., 2020, 11, 1205–1209 RSC.
- Z. Shen, Q. Tang, W. Jiao, H. Shao and X. Ma, J. Org. Chem., 2022, 87, 16736–16742 CrossRef CAS PubMed.
- P. Liang, H. Zhao, T. Zhou, K. Zeng, W. Jiao, Y. Pan, Y. Liu, D. Fang, X. Ma and H. Shao, Adv. Synth. Catal., 2021, 363, 3532–3538 CrossRef CAS.
- T. Ohe, N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef.
- When diphenyl sulfoxide, methyl phenyl sulfoxide, benzothiophene sulfoxide were subjected to the standard reaction in Table 2, we only detected the reduction of sulfoxides to sulfides.
- S. Chen, Y. Wu, S. Hu, Y. Zhao and D. Fang, J. Fluoresc., 2017, 27, 451–461 CrossRef CAS PubMed.
- H. Kawashima, T. Yanagi, C. C. Wu, K. Nogi and H. Yorimitsu, Org. Lett., 2017, 19, 4552–4555 CrossRef CAS PubMed.
- We are grateful to one of the reviewers who recommended this reaction route.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |