
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorphyrin- and Bodipy-helicene conjugates: syntheses, separation of enantiomers and chiroptical properties†
Vincent
Silber
 a,
Marion
Jean
b,
Nicolas
Vanthuyne
b,
Natalia
Del Rio
c,
Paola
Matozzo
c,
Jeanne
Crassous
*c and
Romain
Ruppert
*a
a,
Marion
Jean
b,
Nicolas
Vanthuyne
b,
Natalia
Del Rio
c,
Paola
Matozzo
c,
Jeanne
Crassous
*c and
Romain
Ruppert
*a
aInstitut de Chimie, UMR CNRS 7177, Université de Strasbourg, 4 rue Blaise Pascal, 67000 Strasbourg, France. E-mail: rruppert@unistra.fr
bAix-Marseille Université, UMR CNRS 7313, Centrale Marseille, iSm2, 13397 Marseille cedex 20, France
cISCR, UMR CNRS 6226, campus de Beaulieu, Université de Rennes, 35042 Rennes cedex, France. E-mail: jeanne.crassous@univ-rennes1.fr
First published on 25th October 2023
Abstract
The synthesis of several new compounds containing a chromophore and a helicenic moiety is reported. The preparation, characterisation and some physico–chemical studies are detailed. In particular, the two enantiomers of several chiral molecules of this type were separated by chiral HPLC (both analytically and in a preparative way) and their racemisation rates were determined for short-lived species. Electronic circular dichroism (ECD) and circular polarised luminescence (CPL) measurements were performed for the compounds with a very long racemisation half-life. Chiral porphyrins and Bodipys both gave ECD and CPL responses over a large area of the visible spectrum.
Introduction
Helicenes are inherently chiral, π-conjugated molecules that have been the subject of numerous studies during the last twenty years after improvements of synthetic procedures for their preparation and progress in chromatographic separation of their enantiomers.1–7 This interest was and is still due to their exceptional chiroptical properties. Future applications are expected in optoelectronics, non-linear optics, asymmetric synthesis or catalysis, chiroptical switches, and even magnetochirality.8–13 The two enantiomers of these helicenes are stable as soon as the number of ortho-fused rings is equal to or higher than six.14 However, one main drawback for optical applications is their poor absorbance in the visible area of the electronic spectrum. One way which might lead to an improvement of these optical properties, is to connect these helical aromatics to known chromophores. Porphyrins and Bodipys are poly-pyrrolic aromatics that absorb strongly in the visible area and both compounds are also good photosensitisers with emission in the visible region. To obtain compounds presenting CPL, covalent linkages of these molecules to chiral helicenes might be of interest (see Fig. 1). Several examples of helical porphyrinoids15–27 and Bodipy-helicenes conjugates were described earlier in the literature.28–38 Some of us have shown recently that a fused porphyrin–helicene conjugate presented interesting absorbance properties covering the full visible spectrum.27 | ||
| Fig. 1 Chemical structures of a helicene, a porphyrin, and a Bodipy. | ||
During this study, the presence of electron donating groups and the positioning of these substituents on the aromatic helicene attached to the porphyrin proved to be crucial in order to fuse together the two aromatic moieties. In this report, the preparation of new porphyrin conjugates with [4] or [6]helicenic groups bearing methyl donating groups will be detailed, and the Scholl reaction attempts to fuse the aromatics with the porphyrin core is described. Bodipy-[6]helicene conjugates were also prepared, and together with several other porphyrins bearing helicenic substituents were submitted to chiral HPLC to separate the enantiomers. For the stable enantiomers, CD spectra were obtained and for the compounds presenting emission, CPL spectra were also recorded.
Results and discussion
Synthesis and characterisation
All helicenes were prepared following described procedures, but with (or without) different donating substituents at one terminal aromatic.39,40 The aromatic aldehyde 3 was prepared according to the described procedure by substituting the starting aromatic phosphonate with two additional methyl groups. Even if the steric hindrance should decrease the cyclisation yield, the favourable inductive effect of the methyl groups and the limitation to only one possible cyclisation led to a rather good yield of 61% in the photocyclisation reaction compared to unsubstituted stilbene derivatives (see Scheme 1).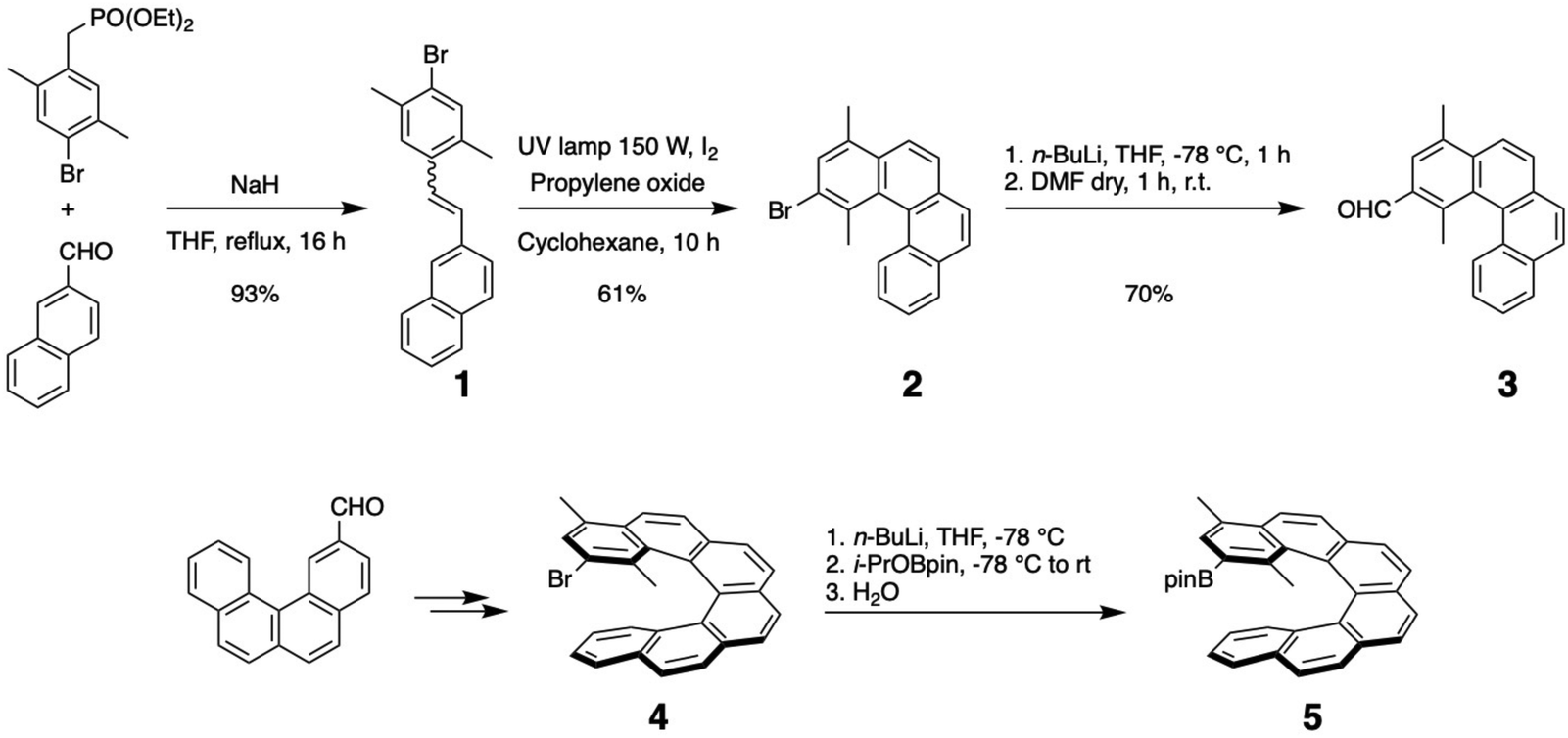 | ||
| Scheme 1 Preparation of dimethylated [4] and [6]helicenes 3 and 5. | ||
Following the same synthetic route, the bromo[6]helicene 4 bearing two methyl groups at the brominated terminal aromatic was prepared starting from the known benzo[c]phenanthrene-2-carbaldehyde (see Scheme 1, bottom).41 Classical chemistry afforded the [6]helicene 5 bearing a pinacolborate substituent, ready for coupling reactions.
A mixture of pyrrole, aldehyde 3 and 2,6-dimethyl-4-(tert-butyl)-benzaldehyde was then reacted in presence of trifluoroborate etherate under classical Lindsey conditions to obtain, after chromatographic separation, porphyrin 6 in 9% yield. After quantitative metalation with nickel(II), the nickel(II) porphyrin 7 was treated with iron(III) chloride to obtain the fused compound 8 with a respectable 43% yield (see Scheme 2). The nickel(II) ion was chosen, because nickel(II) porphyrins are very stable and diamagnetic, thus avoiding complexation with iron(III) during the course of the oxidation reaction. The cyclodehydrogenative oxidation (Scholl reaction) is known to afford fused nickel(II) porphyrins with many different aryl groups bearing donating substituents.42–49
 | ||
| Scheme 2 Preparation of fused nickel(II) porphyrin 8. | ||
To obtain an analogous nickel(II) porphyrin, now bearing one [6]helicenic meso-group, the synthetic route was modified, because the yield of the statistical Lindsey porphyrin synthesis used previously was rather low (9%). Starting from the known triarylporphyrin base 9, iodination and metalation with nickel(II) afforded nickel(II) porphyrin 10 in very good yield (see Scheme 3). A Suzuki–Miyaura coupling between this porphyrin and helicene 5 afforded then in 25% yield the nickel(II) porphyrin 11. Despite many attempts to run the cyclodehydrogenative oxidation with 11, the fused compound was never observed, in strong contrast with the previous Scholl reaction performed on porphyrin 7 to obtain fused nickel(II) porphyrin 8. We had encountered similar failures running Scholl reactions earlier and this was attributed to the steric hindrance of the terminal aromatic phenyl group which prevented the cyclisation.27
 | ||
| Scheme 3 Preparation of nickel(II) porphyrin 11 bearing one [6]helicenic meso group. | ||
Other chromophores of importance for potential CPL applications are the sensitisers derived from the Bodipy scaffold. As mentioned earlier, a few helicenic Bodipy derivatives have been previously described, but surprisingly not the simplest one (compound 12 shown in Scheme 4). Compound 12 was obtained by a very simple one-pot procedure following a described protocol.50
 | ||
| Scheme 4 Preparation of Bodipy 12 bearing one [6]helicene. | ||
The racemisation rate of the enantiomers of [6]helicenes is known and even when unsubstituted, the half-life is higher than a year at room temperature.14 However, the half-life of methylated [4]helicenes are much shorter (closer to one hour) and it was necessary to check the potential stability of the enantiomers of compound 8 by variable temperature NMR, before starting a tedious preparative HPLC separation of the enantiomers.
The 1H NMR spectra of compound 8 were recorded at different temperatures in D2-tetrachloroethane (from 25 °C to 100 °C, see Fig. S05 in ESI†). The two aromatic protons of each meso-aryl groups (noted HAr and HAr′), located above or below the mean plane of the porphyrin, are not equivalent because the fused dimethyl-helicene differentiates the two faces. Six signals were observed at room temperature and even heating up to 100 °C did not modify the spectrum. By assuming a coalescence temperature of 150 °C, the enantiomerisation barrier would be approximatively 85 kJ mol−1, a value close to the one reported for 1-methyl-[4]helicene.14 After chiral HPLC separation of the enantiomers, this value was determined more precisely (vide infra).
Separation of the enantiomers by analytical and preparative HPLC
The newly prepared helicene-porphyrin derivatives 8 and 11, and the helicene-bodipy product 12, together with a series of additional compounds already reported previously27 (compound 14 and methoxy-substituted systems 13 and 15, see Fig. 2 for the structures) were also subjected for chiral HPLC separation. | ||
| Fig. 2 Chemical formula of the compounds submitted for chiral HPLC separation. Porphyrins 13, 14, and 15 were described earlier.27 | ||
The separation of compound 8 enantiomers was studied in detail (see ESI† for experimental conditions). Even if baseline peak separation was observed in analytical separations, it was not possible to isolate the two enantiomers in high purity, due to relatively rapid racemisation. After isolation of the almost pure enantiomers, the racemisation rate of these metastable compounds was determined by following the enantiomeric ratio as a function of time. At 25 °C and in a mixture heptane/dichloromethane 80/20, the enantiomerisation barrier was equal to 93.4 kJ mol−1 and a half-life of 21 minutes was determined. This half-life is still notably too short for accurate ECD measurements. The isolated enantiomers of the five other compounds containing a [6]helicene were stable and their ECD spectra were determined; while for three of them CPL measurements were carried out. The enantiomers of the two nickel porphyrins 11 and 14 were obtained in purities close to 99% after separations using a (S,S)-Whelk-O1 HPLC column. The UV-vis and ECD spectra of these pairs of enantiomers are shown in Fig. 3.
 | ||
| Fig. 3 UV-visible absorption (b and d) and CD (a and c) spectra of enantiomers of nickel porphyrins 11 and 14 recorded in dichloromethane. | ||
Very similar signatures are observed in the UV-vis spectra of the Ni complexes 11 and 14, with (i) a set of high energy bands below 400 nm (33 × 103 < ε < 70 × 103 M−1 cm−1), corresponding to the absorption region of the helicene, (ii) the highly intense Soret band at 416–418 nm (ε > 2 × 105 M−1 cm−1), and (iii) two small Q bands at 528–563 nm (ε ∼17 × 103 and 3 × 103 M−1 cm−1 respectively). In the ECD spectra, the helicenic region displays the intense classical negative-positive signature for the P-enantiomers at around 250 and 330 nm, respectively, thus enabling clear assignment of the absolute configuration, while the Soret band also displays a negative–positive signature at around 390 and 420 nm, respectively, but of lower intensities compared to the helicenic ones (see Table 1 for the ECD intensities). The Q bands do not show any ECD activity. Note that nickel(II) porphyrins are non-emissive, and thus not CPL-active.
| Compound (1st eluted) | Solvent | λ (ε) (nm/M−1 cm−1) | λ (Δε) (nm/M−1 cm−1) |
|---|---|---|---|
| P-11 | CH2Cl2 | 237 (64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400), 270 (48000), 322 (33000), 418 (212000), 528 (17900), 561 (3000). 400), 270 (48000), 322 (33000), 418 (212000), 528 (17900), 561 (3000). |
222 (−29), 250 (−191), 276 (−51), 298 (+8), 318 (+36), 336 (+117), 390 (−8), 421 (+57). |
| P-12 | CH3CN | 199 (39200), 232 (43000), 260 (45200), 320 (26900), 344 (15000), 474 (17500), 499 (37200). |
194 (+86.2), 210 (−37.4), 219 (−49), 230 (+18.6), 248 (−212.1), 308 (+54.5), 321 (+181.8), 347 (+67), 430 (+15.5), 500 (−16.5). |
| P-13 | CH2Cl2 | 244 (67000), 279 (49200), 323 (44300), 403 (89300), 420 (350000), 515 (22800), 550 (6300), 646 (4500). |
228 (+23), 246 (−202), 278 (−72.5), 294 (+28), 310 (+71.1), 333 (+162), 375 (+37.2), 398 (−15.5), 419 (−67.3), 432 (+21.6), 545 (+2.5), 646 (+4.6). |
| P-14 | CH2Cl2 | 239 (69000), 278 (45000), 322 (33400), 416 (202000), 528 (17500), 563 (2400). |
230 (+32), 246 (−180), 277 (−70), 315 (+59), 333 (+125), 391 (−7), 408 (−12), 425 (+20). |
| P-15 | CH2Cl2 | 250 (66000), 275 (44700), 326 (37600), 405 (94400), 422 (330000), 518 (22600), 550 (6800), 593 (6500), 650 (5350). |
230 (+19.4), 250 (−164), 273 (−58.5), 291 (+23.8), 311 (+84.8), 330 (+218), 354 (+53.3), 403 (−41), 421 (−66.5), 518 (−6). |
After HPLC separations (using a chiralpak IG column), the two enantiomers of bodipy 12 were isolated in very good purities (>99.5%). The enantiomers of porphyrin free bases 13 and 15 were obtained in purities higher than 99% by chiral HPLC separations (again using analytical and preparative (S,S)-Whelk-O1 HPLC columns). UV-vis and ECD spectra of all isolated compounds are shown in Fig. 4. Similarly to the Ni complexes, free porphyrin-helicenes 13 and 15 display three distinct sets of bands, corresponding to the helicenic part, the intense Soret band and a set of four Q bands. In the ECD spectra, the Soret band signatures appear slightly differently, i.e. displaying only one negative band in the case of 15. Interestingly, in the two systems, some Q bands are slightly ECD-active (see the exact intensities in Table 1). Here again, the typical ECD signature of the helicenic part enables the assignment of the absolute configuration. Overall, compared to the nickel complexes, the free systems show similar signature, except that some of the Q bands are slightly ECD-active, while the Soret band shows slightly different features. These features also deserve to be compared to previously reported helicene-porphyrins, especially to a helicene-bis-porphyrin system displaying a strong excitonic coupling signature in the porphyrin Soret band (Δε −82 at 430 nm and +393 M−1 cm−1 at 446 nm) and ECD-active Q bands (Δε +18 M−1 cm−1 at 570 nm and +6 M−1 cm−1 at 654 nm) for the P enantiomer.24 In the case of bodipy-helicene 12 enantiomers, aside from the classical helicenic UV-vis bands below 400 nm, a set of strong absorption bands is found in the visible region around 500 nm, typical of the Bodipy chromophore. This set of bands appears moderately ECD-active, with a positive Cotton effect at 430 nm and a negative one at 500 nm for the P enantiomer (Δε ∼ 15 M−1 cm−1, 1st eluted). In addition, the strong ECD-active helicenic bands are present (see Table 1 for the exact intensities).
 | ||
| Fig. 4 UV-visible absorption (b, d and f) and ECD (a, c and e) spectra of the enantiomers of bodipy-helicene 12 (recorded in acetonitrile) and free porphyrin-helicenes 13 and 15 (recorded in dichloromethane). | ||
Interestingly, the free helicene-porphyrin 13, 15 and the helicene-bodipy 12 enantiomers are emissive and their circularly polarized emission can be studied. The non-polarized and circularly polarized emission properties of compounds 12, 13 and 15 were studied in solution. In dichloromethane, 2-Bodipy-substituted [6]helicene 12 exhibits two emission bands at 525 and 608 nm (excitation wavelength λexc at 490 nm, C ≈ 10−5 M) most probably corresponding to typical fluorescence signals. A low quantum yield was found for compound 12 (2% in dichloromethane using rhodamine 6G as standard). The latter nevertheless also displayed CPL activity, with a negative signal at 525 nm and a positive one at 608 nm for the 1st eluted enantiomer (P enantiomer). At 608 nm, the dissymmetry factors glum were found to be +2.3 × 10−3 for the first eluted (P-12) and −1.9 × 10−3 for the second eluted (M-12). These values fall within the classical range for this type of Bodipy substituted [5] or [6]helicene derivatives.32,34,35 Note however that the CPL signal found at 525 nm may be due to residual reabsorption phenomenon in the Soret band. Regarding porphyrin-helicenes 13 and 15, they exhibit two emission bands at 652/717 nm and 654/720 nm (in dichloromethane, excitation wavelength λexc at 420 nm, C ∼2.5–3 × 10−5 M), with respective quantum yields of 9% and 11% (with H2TPP used as standard). Compound 13 also shows CPL activity with a positive signal for the first eluted (P enantiomer) and a negative one for the second eluted (M enantiomer) at around 655 nm, with respective emission dissymmetry factors glum of around +1 × 10−3 and −0.7 × 10−3 (noisy signals, see Fig. 5).
 | ||
| Fig. 5 Fluorescence (b and d) and CPL (a and c) spectra of compounds 12 and 13 (λexc at 490 and 420 nm, respectively, recorded in dichloromethane). | ||
Note that the CPL signals were found noisy. In order to get reasonable signal to noise ratios, it was necessary to irradiate at the Soret band rather than the lower energy Q bands. Nevertheless, CPL-active porphyrin derivatives still remain quite rare and are thus of interest in the area of far-red CPL emitters.24,25 Compound 15 was found to display no clear CPL activity in dichloromethane solution.
Experimental part
General information
The HRMS ESI and MALDI were obtained on a Bruker MicrOTOF. UV-vis spectra were recorded in dichloromethane with a Cary 5000 UV-Vis-NIR double-beam instrument. ECD and UV spectra were measured on a JASCO J-815 spectrometer equipped with a JASCO Peltier cell holder PTC-423 to maintain the temperature at 25.0 ± 0.2 °C. A CD quartz cell of 1 mm of optical pathlength was used. The CD spectrometer was purged with nitrogen before recording each spectrum, which was baseline subtracted. UV-Visible (UV-vis, in M−1 cm−1) absorption spectra were recorded on a UV-2401PC Shimadzu spectrophotometer. Steady-state fluorescence measurements were performed on dilute solutions (ca. 10−5 M, optical density <0.1) contained in standard 1 cm quartz cuvettes using an Edinburgh Instrument (FLS920) spectrometer in photon-counting mode. The circularly polarized luminescence (CPL) measurements were performed using a home-built CPL spectrofluoropolarimeter (constructed with the help of the JASCO Company). The samples were excited using a 90° geometry with a Xenon ozone-free lamp 150 W LS. The following parameters were used: emission slit width ≈10 mm, integration time = 8 s, scan speed = 50 nm min−1, accumulations = 5. The concentration of all the samples was ca. 10−5 M. Chromatographic separations were performed with silica gel (Merck, 40–63 μm). Dichloromethane was distilled over calcium hydride and THF from sodium/benzophenone ketyl. NMR spectra were recorded on Bruker Avance 400 and 500 spectrometers equipped with a cryoprobe for 13C. Chemical shifts are given in parts per million (ppm) by taking the solvent signals as a reference (δ = 7.26 ppm or δ = 77.16 ppm). All other solvents and reagents were used without further purification. Porphyrins 13, 14, and 15 were prepared as previously described.25Syntheses
A solution of the intermediate stilbene (1.0 g, 2.29 mmol, 1 eq.) and iodine (≃0.015 eq.) in cyclohexane (400 mL) was irradiated in a photoreactor equipped with an immersion lamp (150 W) for 10 h. Sodium thiosulfate (5 g) was added and the solution was stirred overnight. The solvent was evaporated, and the crude product was purified by column chromatography (SiO2, CH2Cl2/cyclohexane 40/60) to afford compound 4 as a yellow solid (678 mg, 1.56 mmol, 68%). 1H NMR (CDCl3, 500 MHz, 25 °C): δH (ppm) = 8.08 (d, J = 8.1 Hz, 1H, H7–12), 8.07–8.02 (m, 2H, H5 + H7–12), 8.01 (d, J = 8.1 Hz, 1H, H7–12), 7.97 (d, J = 8.2 Hz, 1H, H7–12), 7.93 (d, J = 8.7 Hz, 1H, H6), 7.91–7.88 (m, 2H, H7–12), 7.86 (dd, J = 8.1, 1.3 Hz, 1H, H13), 7.30 (s, 1H, H3), 7.19 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H, H14), 6.81 (dd, J = 8.5, 1.2 Hz, 1H, H16), 6.47 (ddd, J = 8.5, 6.8, 1.3 Hz, 1H, H15), 2.78 (s, 3H, CH3out), 0.88 (s, 3H, CH3in). 13C NMR (126 MHz, CDCl3): δC (ppm) = 133.3, 132.8, 132.1, 131.5, 131.0, 130.8 (CH), 130.5, 128.49, 128.48, 128.0 (CH), 127.5 (CH), 127.3 (CH), 127.2 (CH), 126.8 (CH), 126.4 (CH), 126.3 (CH), 125.9 (CH), 125.80, 125.77 (CH), 125.4 (CH), 124.6, 123.7 (CH), 123.5 (CH), 24.0 (CH3), 19.4 (CH3). ESI-TOF-HR-MS (m/z): Calcd for ([M˙+]) 434.0665; found 434.0640.
000 L cm−1 mol−1), 519 (17700), 550 (6700), 592 (6100), 648 (4300).
000 L cm−1 mol−1), 529 (11000), 561 (2100).
000 L cm−1 mol−1), 584 (7900), 627 (6300), 694 (1900).
000 L cm−1 mol−1), 522 (13100), 557 (7500), 597 (4200), 654 (4000).
A solution of the porphyrin free base (80 mg, 86 mmol, 1 eq.) and Ni(acac)2 (110 mg, 0.43 mmol, 5 eq.) in toluene (40 mL) were heated to reflux overnight. Filtration of the reaction mixture through a pad of alumina, evaporation of the solvent and crystallization afforded the desired porphyrin 10 (85 mg, 86 mmol, quant.) as a red solid. 1H NMR (CDCl3, 500 MHz, 25 °C): δH (ppm) 9.49 (d, J = 4.9 Hz, 2H, Hpyrr), 8.60 (d, J = 4.9 Hz, 2H, Hpyrr), 8.51 (br s, 4H, Hpyrr), 7.43–7.38 (m, 6H, HAr), 1.89 (s, 12H, CH3Ar), 1.88 (s, 6H, CH3Ar), 1.57 (s, 9H, HtBu), 1.56 (s, 18H, HtBu). 13C NMR (126 MHz, CDCl3): δC (ppm) = 151.1, 144.5, 143.4, 142.8, 142.7, 138.6, 138.5 (CH), 136.8, 132.6 (CH), 132.20 (CH), 132.15 (CH), 124.1 (CH), 118.0, 117.9, 34.80, 34.75, 31.78 (tBu), 31.76 (tBu), 22.0 (CH3), 21.9 (CH3). ESI-TOF-HR-MS (m/z): Calcd for ([M + H+]) 972.3132; found 972.3126. UV-vis (CH2Cl2): λmax = 419 nm (ε = 258000 L cm−1 mol−1), 531 (18500), 564 (4400).
000 L cm−1 mol−1), 523 (13000), 553 (4100).
1H NMR (500 MHz, CDCl3, 25 °C) δH (ppm) = 8.08 (d, J = 8.5 Hz, 1H, Hhel), 7.92–8.05 (m, 5H, Hhel), 7.82 (d, J = 8.5 Hz, 1H, Hhel), 7.73–7.80 (m, 3Hhel + 2Hpyr), 7.72 (dd, J = 8.0 and 1.4 Hz, 1H, Hhel), 7.66 (dd, J = 8.5 and 1.1 Hz, 1H, Hhel), 7.36 (dd, J = 8.5 and 1.1 Hz, 1H, Hhel), 7.21 (ddd, J = 8.0, 6.8 and 1.1 Hz, 1H, Hhel), 6.79 (ddd, J = 8.5, 6.8 and 1.4 Hz, 1H, Hhel), 6.30 (br s, 2H, Hpyr), 5.85 (br s, 2H, Hpyr). 13C NMR (125 MHz, CDCl3, 25 °C): δC (ppm) = 156.2, 154.9, 147.2, 143.7, 144.9, 133.5, 132.7, 132.6, 131.8, 131.7, 130.7, 130.1, 129.8 (CH), 129.4, 128.41, 128.40 (CH), 128.38 (CH), 128.37 (CH), 128.21, 128.19 (CH), 127.84, 127.79 (CH), 127.7, 127.5 (CH), 127.44 (CH), 127.43 (CH), 127.2 (CH), 126.9 (CH), 126.7 (CH), 126.1 (CH), 124.9 (CH), 123.8, 118.1 (CH), 114.5 ppm. 19F NMR (282 MHz, CDCl3, 25 °C): δ = −145.3 ppm. UV-Vis (CH2Cl2): λmax = 261 nm (ε = 45000 L cm−1 mol−1), 321 (27000), 345 (14700), 497 (82300), 475 (17700), 498 nm (37100 M−1 cm−1). MS, EI: m/z = 518.3 Calcd for (M˙+): 518.1766.
Conclusions
Several nickel and free base porphyrins bearing helicene meso-groups were synthesized and their enantiomers separated by preparative chiral HPLC. CPL measurements for the free base porphyrins and one Bodipy derivative were realized and these new compounds showed glum values in the range of the previously described porphyrinoids. Since porphyrins presenting CPL activities are still quite rare, synthetic efforts to obtain new chiral extended porphyrins with absorbances reaching the NIR region are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Continuous financial support from the University of Strasbourg, the University of Rennes and the CNRS are acknowledged. V. S. thanks the French Ministry of Research for a PhD fellowship. The European Commission Research Executive Agency (Grant Agreement number: 859752 – HEL4CHIROLED – H2020-MSCA-ITN-2019) is thanked for financial support.References
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- C. F. Chen and Y. Shen, Helicene Chemistry, from Syntheses to Applications, Springer, Berlin, 2017 Search PubMed.
- E. S. Gauthier, R. Rodriguez and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 22840–22856 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, J. Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144–158 CrossRef PubMed.
- J. Crassous, I. G. Stará and I. Starý, Helicenes – Synthesis, Properties and Applications, Wiley, 2022 Search PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2262 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- S. Van Elshocht, B. Busson, T. Verbiest, M. Kauranen, J. Snauwaert, L. Hellemans, A. Persoons, C. Nuckolls and T. J. Katz, Mater. Res. Soc. Symp. Proc., 1999, 561, 15–20 CrossRef CAS.
- P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263–15278 RSC.
- P. Ravat, T. Solomek and M. Juricek, ChemPhotoChem, 2019, 3, 180–186 CrossRef CAS.
- M. Atzori, K. Dhbaibi, H. Douib, M. Grasser, V. Dorcet, I. Breslavetz, K. Paillot, O. Cador, G. L. J. A. Rikken, B. Le Guennic, J. Crassous, F. Pointillard and C. Train, J. Am. Chem. Soc., 2021, 143, 2671–2675 CrossRef CAS PubMed.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 5660–5664 CrossRef CAS PubMed.
- J. M. Fox, T. J. Katz, S. Van Elshocht, T. Verbiest, M. Kauranen, A. Persoons, T. Thongpanchang, T. Krauss and L. Brus, J. Am. Chem. Soc., 1999, 121, 3453–3459 CrossRef CAS.
- K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem. – Eur. J., 2018, 24, 572–575 CrossRef CAS PubMed.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- S. A. Balahoju, Y. K. Maurya, P. J. Chmielewski, T. Lis, M. Kondratowicz, J. Cybinska and M. Stepien, Angew. Chem., Int. Ed., 2022, 61, e202200781 CrossRef CAS PubMed.
- B. Szyszko, M. Przewoznik, M. J. Bialek, A. Bialonska, P. J. Chmielewski, J. Cichos and L. Latos-Grazynski, Angew. Chem., Int. Ed., 2018, 57, 4030–4034 CrossRef CAS PubMed.
- B. Szyszko, P. J. Chmielewski, M. Przewoznik, M. J. Bialek, K. Kupietz, A. Bialonska and L. Latos-Grazynski, J. Am. Chem. Soc., 2019, 141, 6060–6072 CrossRef CAS PubMed.
- S. Shimizu, N. Aratani and A. Osuka, Chem. – Eur. J., 2006, 12, 4909–4918 CrossRef CAS PubMed.
- D. C. G. Götz, A. C. Gerhold, S. J. Dorazio, P. Daddario, L. Samankumara, G. Bringman, C. Brückner and T. Bruhn, Eur. J. Org. Chem., 2015, 3913–3922 CrossRef.
- I. Nishimura, T. Higashino and H. Imahori, Chem. Commun., 2021, 57, 9606–9609 RSC.
- K. Dhbaibi, P. Matozzo, L. Abella, M. Jean, N. Vanthuyne, J. Autschbach, L. Favereau and J. Crassous, Chem. Commun., 2021, 57, 10743–10746 RSC.
- C. Maeda, K. Ogawa, K. Sadanaga, K. Takaishi and T. Ema, Chem. Commun., 2019, 55, 1064–1067 RSC.
- S. Belviso, G. Marsico, R. Franzini, C. Villani, S. Abbate and G. Longhi, Dalton Trans., 2022, 51, 16453–16464 RSC.
- V. Silber, N. Gruber, M. Jean, N. Vanthuyne and R. Ruppert, Chem. Commun., 2022, 58, 6012–6015 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed.
- P. Moneva Lorente, A. Wallabregue, F. Zinna, C. Besnard, L. Di Bari and J. Lacour, Org. Biomol. Chem., 2020, 18, 7677–7684 RSC.
- Z. Wang, L. Huang, Y. Yan, A. M. El-Zhory, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed and G. Han, Angew. Chem., Int. Ed., 2020, 59, 16114–16121 CrossRef CAS PubMed.
- C. Maeda, K. Nagahata, T. Shirakawa and T. Ema, Angew. Chem., Int. Ed., 2020, 59, 7813–7817 CrossRef CAS PubMed.
- For a recent review see: M. Cei, L. Di Bari and F. Zinna, Chirality, 2023, 35, 192–210 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- Y. Yan, A. A. Sukhanov, M. H. E. Bousquet, Q. Guan, J. Zhao, V. K. Voronkova, D. Escudero, A. Barbon, Y. Xing, G. G. Gurzadyan and D. Jacquemin, J. Phys. Chem. B, 2021, 125, 6280–6295 CrossRef CAS PubMed.
- S. S. Sahoo and P. K. Panda, Chem. Commun., 2023, 59, 9603–9606 RSC.
- L.-Y. Wang, Z.-F. Liu, K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Commun., 2022, 58, 3807–3810 RSC.
- F. B. Mallory and C. W. Mallory, Photocyclizations of stilbenes and related molecules, in Organic Reactions, J. Wiley & Sons Inc., 1984, vol. 30, pp. 3 Search PubMed.
- M. Jakubec, T. Beranek, P. Jakubik, J. Sykora, J. Zadny, V. Cirkva and J. Storch, J. Org. Chem., 2018, 83, 3607–3616 CrossRef CAS PubMed.
- A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304–10321 CrossRef CAS PubMed.
- H. S. Gill, M. Harmjanz, J. Santamaria, I. Finger and M. J. Scott, Angew. Chem., Int. Ed., 2004, 43, 485–490 CrossRef CAS PubMed.
- M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069–2071 RSC.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS PubMed.
- J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069–10086 RSC.
- D. Bao, E. Sebai, O. Vakuliuk, M. Scigaj and D. T. Gryko, Org. Biomol. Chem., 2011, 9, 8178–8181 RSC.
- M. M. Martin, C. Oleszak, F. Hampel and N. Jux, Eur. J. Org. Chem., 2020, 6758–6762 CrossRef CAS.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- A. Cui, X. Peng, J. Fan, X. Chen, Y. Wu and B. Guo, J. Photochem. Photobiol., A, 2007, 186, 85–92 CrossRef CAS.
- Compound prepared from commercial 2,5-dibromo-p-xylene: formylation, reduction to the alcohol, bromination and Michaelis–Arbuzov reaction gave the desired functionalised phosphonate. Procedures according to: E. Diez-Barra, J. C. Garcia-Martinez, S. Merino, R. del Rey, J. Rodriguez-Lopez, P. Sanchez-Verdu and J. Tejeda, J. Org. Chem., 2001, 66, 5664–5670 CrossRef CAS PubMed; T.-C. Liang and H.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2734–2753 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01459b |
| This journal is © The Royal Society of Chemistry 2023 |