
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA safety-catch protecting group strategy compatible with Boc-chemistry for the synthesis of peptide nucleic acids (PNAs)†
K. P.
Nandhini
ab,
Sikabwe
Noki
 ab,
Edikarlos
Brasil
b,
Fernando
Albericio
*bc and
Beatriz G.
de la Torre
*a
ab,
Edikarlos
Brasil
b,
Fernando
Albericio
*bc and
Beatriz G.
de la Torre
*a
aKwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban 4041, South Africa. E-mail: garciadelatorreb@ukzn.ac.za; Tel: +27614 475528
bPeptide Science Laboratory, School of Chemistry and Physics, University of KwaZulu-Natal, Westville, Durban 4000, South Africa. E-mail: albericio@ukzn.ac.za; Tel: +27 614009144
cCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, and Department of Organic Chemistry, University of Barcelona, Martí i Franqués 1-11, 08028 Barcelona, Spain
First published on 25th September 2023
Abstract
Peptide Nucleic Acids (PNAs) are an intriguing class of synthetic biomolecules with great potential in medicine. Although PNAs could be considered analogs of oligonucleotides, their synthesis is more like that of peptides. In both cases, a Solid-Phase Synthesis (SPS) approach is used. Herein, the advantage using Boc as a temporal protecting group has been demonstrated to be more favored than Fmoc. In this context, a new PNA SPS strategy has been developed based on a safety-catch protecting group scheme for the exocyclic nitrogen of the side-chain bases and the linker. Sulfinyl (sulfoxide)-containing moieties are fully stable to the trifluoroacetic acid (TFA) used to remove the Boc group, but they can be reduced to the corresponding sulfide derivatives, which are labile in the presence of TFA. The efficiency of this novel synthetic strategy has been demonstrated in the synthesis of the PNA pentamer H–PNA(TATCT)–βAla–OH.
1. Introduction
Eleven oligonucleotide-based drugs with U.S. Food and Drug Administration (FDA) approval are currently in the market.1 Peptide Nucleic Acids (PNAs) are modified forms of oligonucleotides in which the phosphate-sugar backbone is initially replaced by a neutrally charged N-(2-aminoethyl)glycine (AEG) linkage. In addition, achirality of their AEG linkage facilitates their selective hybridization to a specific DNA or mRNA strand, forming a PNA–DNA or PNA–RNA complex. Given the remarkable properties of PNAs, they have found extensive applications in the fields of gene therapeutics2,3 and biomedical diagnostics.4From a chemistry perspective, PNAs are considered more advantageous than natural oligonucleotides because they have a peptide-like AEG backbone, forming highly stable amide/peptide bonds between the monomeric units. This property protects these molecules from depurination, a common phenomenon that occurs in oligonucleotide molecules under acidic conditions.5 Despite the numerous benefits of PNAs, few studies have explored these molecules because of the ongoing challenges regarding their synthesis. Although PNAs are analogues of oligonucleotides, they are prepared following a Solid-Phase Synthesis (SPS) strategy similar to that used for peptides.6 In this regard, two temporary protecting groups for the amino terminal of the PNA monomer are mainly used, namely the tert-butyloxycarbonyl (Boc) and the fluorenylmethoxycarbonyl (Fmoc) groups. One of the main challenges in PNA SPS is the aggregation of the PNA chain during their elongation, which jeopardizes their synthesis. On the other hand, under basic conditions used in the Fmoc strategy for the removal of the N-temporary protecting group, the free N-terminal of PNAs tends to react, causing two kinds of side products. In the first, the transacylation of the nucleobase leads to the capping of the growing chain, while the second one leads to ketopiperazine formation by cyclization, which results in a n-1 deletion (Fig. 1).
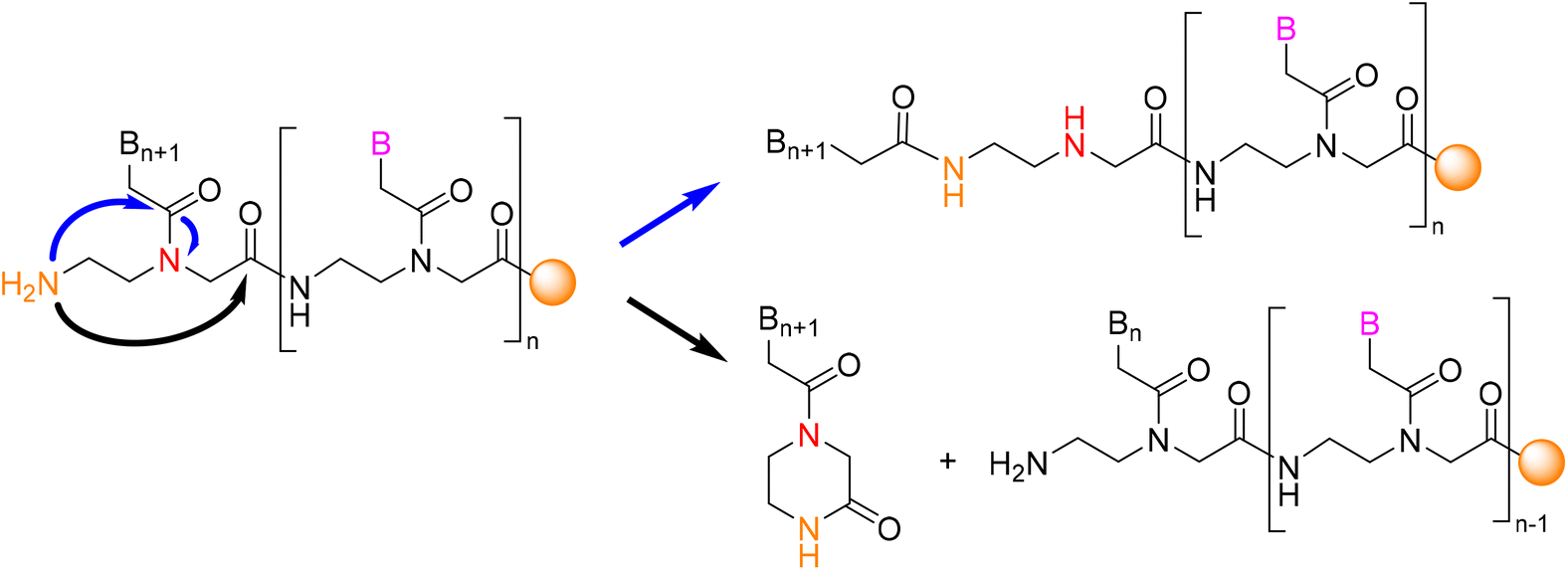 | ||
| Fig. 1 Side reactions during PNA SPS under basic conditions (Fmoc chemistry). | ||
In Boc SPS, the side reaction in basic conditions is minimized. However, this approach involves the use of strong acids such as TFMSA7 or HF8–11 for the final cleavage of the PNA chain from the solid support and also for the removal of the Z-protecting group from the nucleobases. These strong acids can impede progress towards the development of PNA–peptide12 and PNA–oligonucleotide conjugates.13 In this context, there is a need for protecting group strategies other than the commercially available Boc/benzyloxycarbonyl (Z) and Fmoc/Benzhydryloxycarbonyl (Bhoc).
To address the above-mentioned challenges in PNA SPS, here we report on the development of new PNA monomers and novel strategic methodologies based in the safety-catch concept. First developed by Kenner,14 it implies the use of a protecting group stable to a determined conditions, which after modification it becomes labile to the same conditions that before was stable. Our strategy is based on the application of the safety-catch strategy based on the 4-(methylsulfinyl/thio)benzyloxycarbonyl (Msz/Mtz) protecting group and the linker 2-methoxy-4-methylsulfinyl/thiobenzyl alcohol (Mmsb/Mmtb). The pair Msz/Mtz was previously described by Kiso et al.15 and others for the side-chain protection in a Nα-Boc strategy and by our group as backbone protecting group (Fig. 2). Furthermore, the Mmsb/Mmtb linker and other related linkers has been used by several groups in Nα-Boc strategies16–18 and by our group as semipermanent protecting group in a Nα-Fmoc strategy.19
 | ||
| Fig. 2 Safety-catch protecting groups, based on the sulfinyl/sulfide pair. | ||
The above described safety-catch protecting groups and linker are fully stable to acid and base conditions and are thus compatible with Fmoc and Boc chemistry. Therefore, at the end of the PNA chain elongation, the reduction of these groups to the 4-(methylthio)benzyloxycarbonyl (Mtz) or 2-methoxy-4-(methylthio)benzyl (Mmtb) moieties makes them labile in trifluoroacetic (TFA) conditions (Fig. 2). Herein we describe a suitable method for the synthesis of the Msz-protected PNA monomers Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2) (Fig. 3A). In addition, we also demonstrate the use of the Boc/Msz strategy for the preparation of PNA pentamers in combination with the safety-catch linker Mmsb (Fig. 3B).
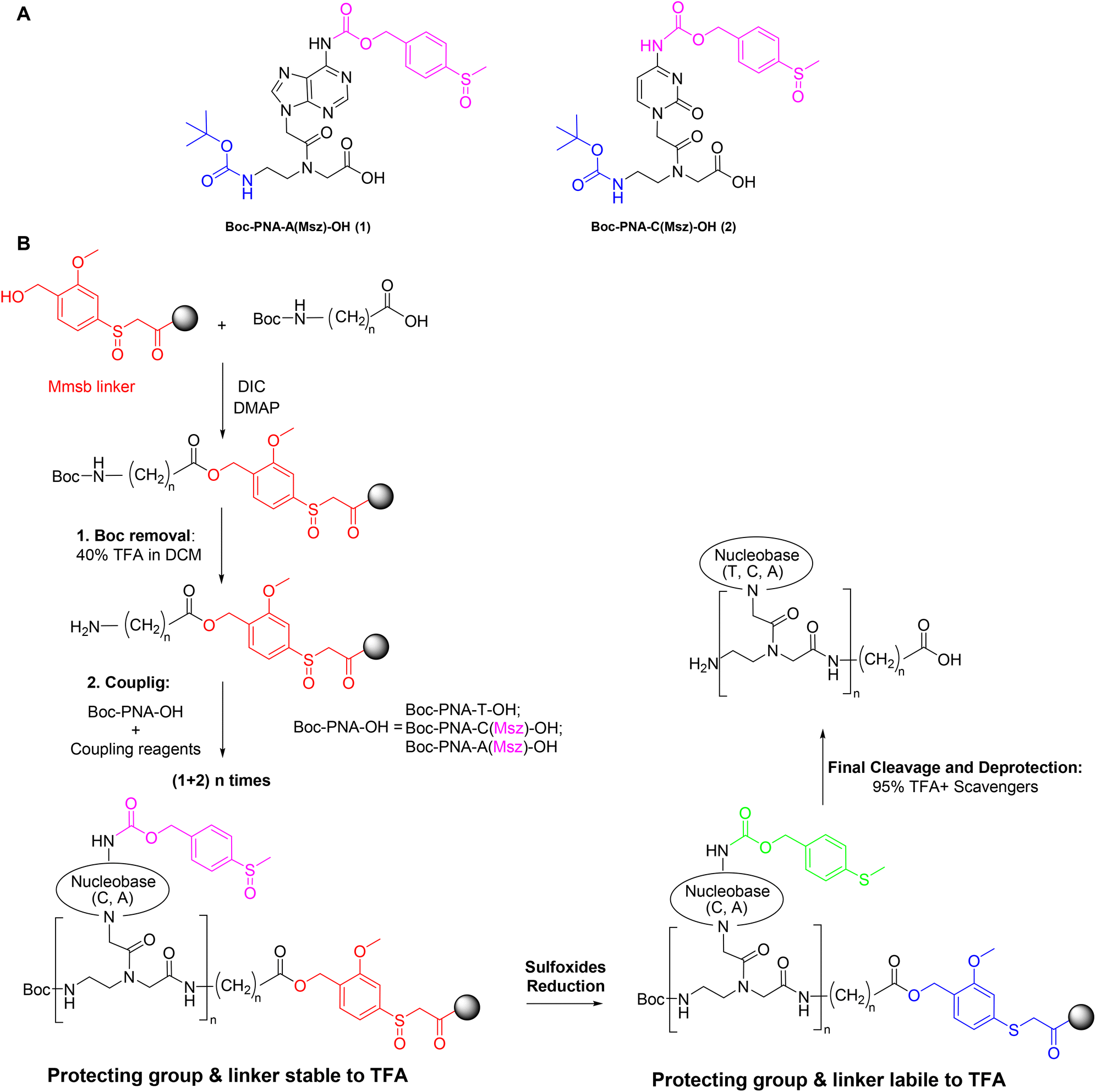 | ||
| Fig. 3 (A) Structure of new Boc PNA monomers (A and C). (B) General solid-phase PNA synthesis used in this work. | ||
2. Results and discussion
First, we studied the compatibility of the Mmsb linker with PNA synthesis. To this end, we prepared T4–PNA–βAla–OH and T6–PNA βAla–OH on HO–Mmsb–resin. The synthesis of polyT–PNA sequences is challenging, especially in the Fmoc strategy because aggregation is favored.20 Thus, the syntheses were run from commercially available Boc–PNA–T–OH and Fmoc–PNA–T–OH monomers (in the latter case on Wang resin) for comparison purposes.To obtain the HO–Mmsb–resin, the initial loading of the aminomethyl (AM)–resin was reduced from 0.92 to 0.4 mmol g−1 by incorporating Fmoc–Ala–OH in deficit to the AM–resin, followed by acetylation of the unreacted amino groups. The HO–Mmsb linker was then introduced using DIC and N-hydroxysuccinimide (HOSu), which render a poor reactive active ester, to avoid the double incorporation of the bidentate linker was performed. This HO–Mmsb–resin was used in all the synthesis described in this work. The βAla residue was introduced to prevent the ketopiperazine formation during the deprotection step when the first PNA monomer is incorporated directly on a resin–OH. The incorporation of Fmoc–βAla–OH as a spacer was done with DIC and N-dimethylaminopyridine (DMAP).
For the PNA synthesis in both resins, the coupling of the protected-PNA(T) was carried out with 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)]methanaminium hexafluorophosphate (COMU) and N,N-diisopropylethylamine (DIEA). In the case of the Boc synthesis, the coupling was carried out using in situ neutralization, which implies that after Boc removal with TFA and washings, the PNA–resin was not neutralized (to avoid aggregation) and the next Boc–PNA–T–OH, already activated with COMU, was incorporated with a slight excess of DIEA. Thus, once the amine was released, it could be acylated with the next Boc–PNA–T–OH. In the case of Fmoc chemistry, the protecting group was removed with piperidine. In this Fmoc synthesis, the final PNA was cleaved from the resin by TFA treatment. For the Boc synthesis, before the TFA treatment, the sulfinyl group (Mmsb) was reduced to the sulfide (Mmtb) moiety using Me3SiCl and Ph3P. The LC-MS of both the tetramer and the hexamer synthesized using Boc chemistry on HO–Mmsb–resin showed a major peak with the expected mass (Fig. 4A).
 | ||
| Fig. 4 Chromatographic traces of PNA tetramers and hexamers using (A) Boc chemistry on OH–Mmsb–resin and (B) Fmoc chemistry on Wang resin. Coupling reagents used: COMU and DIEA. Mobile phase A: 0.1% HCOOH in H2O; mobile phase B: 0.1% HCOOH in CH3CN. Gradient elution: 5–60% of B into A in 15 min. | ||
In contrast, the LC-MS corresponding to the synthesis of the tetramer using Fmoc chemistry showed two major peaks, one corresponding to the target PNA and the other much more hydrophobic, corresponding to the protected tetramer (Fig. 3B). This finding reveals that the Fmoc group from the last T monomer was not fully removed, presumably due to the aggregation of the PNA chains. The PNA–resin was then treated again with piperidine to achieve Fmoc removal. However, the signal of the protected tetramer was still present in the chromatogram (Fig. S1†). Next, the PNA chain was further elongated to couple two more PNA monomers to obtain PNA hexamers. Analysis of the LC-MS corresponding to this stage (Fig. 4B) clarified the PNS SPS issues encountered using Fmoc chemistry. Thus, the most important products corresponded to H–PNA(T4)–βAla–OH and Fmoc–PNA(T4)–βAla–OH with the presence of some pentamers and hexamers. These results indicate that most of the synthetic process stopped at the tetramer.
Having compared Boc and Fmoc chemistry in the simple H–PNA(T6)–βAla–OH, we proceeded to synthesize a more complex sequence, namely H–PNA(T–A(Z)–T–A(Z)–T)–βAla–OH, from commercially available Boc PNA monomers [Boc–PNA–T–OH; Boc–PNA–A(Z)–OH] and HO–Mmsb–resin. The LC-MS chromatogram shows that the target product was obtained with excellent purity (Fig. 5). However, there was still a major issue here. In the case of Boc chemistry, removal of the commercially available protecting group (Z in this case) required the use of strong acids such as HF8,9 or BBr3.21
 | ||
| Fig. 5 Chromatographic traces of H–PNA(T–A(Z)–T–A(Z)–T)–βAla–OH. Mobile phase A: 0.1% HCOOH in H2O; mobile phase B: 0.1% HCOOH in CH3CN. Gradient elution: 5–60% of B into A in 15 min. | ||
The above experiments supported the need for new protecting groups for nucleobases in PNA SPS that are compatible with Boc as temporary protecting group and therefore, the repetitive treatment with TFA as well as the removal process can be performed under friendly conditions. Previously, our group successfully introduced a new safety-catch protecting group, namely Msz, in SPPS to facilitate the synthesis and manipulation of difficult peptide sequences.22 Like the HO–Mmsb linker, this protecting group also allows the use of Boc and Fmoc chemistry. In this regard, the protecting group in the form of the Msz carbamate was introduced on nucleobases [A and C (G was left of this initial study)], which are TFA-stable, and in reduced form (Mtz) are TFA-labile. This flexibility makes them highly compatible with the Boc strategy.
2.1. Synthesis of Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2)
The protected PNA monomers were prepared following the most widely used protocol in the literature.6 It consists of a convergent synthetic scheme in which the two moieties, i.e., the protected aminoethylglycine backbone and the protected nucleobase acetic acid derivative, are synthesized independently and then made to react to obtain the final PNA monomer. Thus, Boc-N-2-aminoethylglycine with the carboxylic group protected as ester and the nucleobases acetic acid (N6-Msz-adenine-9-yl)acetic acid (4) and (N4-Msz-cytosine-1-yl)acetic acid (5) were synthesized.For the preparation of the Boc-N-2-aminoethylglycine ester, several groups, such as ethyl (Et), methyl (Me), and cyanoethyl ester, were explored. However, we found that the benzyl ester was the most effective in terms of preventing dialkylation of the amino group of Boc–ethylenediamine (Boc–EDA) and it was also higher yielding. Moreover, UV absorption of the benzyl group helped the monitoring of the reaction. Thus, Boc–Aeg–OBn (3) was efficiently prepared as shown in Scheme 1. First, following the procedure described by Grate et al.,23 ethylenediamine was reacted with (Boc)2O to render Boc–EDA in quantitative yield. The product obtained was reacted with benzyl bromoacetate in the presence of triethylamine (TEA), following the protocol described by Feagin et al. with minor modifications,24 rendering Boc–Aeg–OBn (3) in a yield of 56%.
 | ||
| Scheme 1 Synthesis of Boc–Aeg–OBn (3). | ||
For the synthesis of (N6-Msz-adenine-9-yl)acetic acid (4) and (N4-Msz-cytosine-1-yl)acetic acid (5), a two-step procedure, namely alkylation followed by protection, described by Pothukanuri et al.25 with minor modifications, was followed. Adenine alkylation using tert-butyl bromoacetate in DMF in the presence NaH rendered the product in a moderate yield of 44%. However, in the case of cytosine, the attempt to use potassium tert-butoxide, as mentioned in the procedure,25 yielded a very low amount of product. The excess of potassium tert-butoxide led to di-alkylation on N4 position of cytosine. After several attempts, the use of NaH gave rise to the target compound in ∼40% yield. At this point, no further attempts were made to optimize the reaction yield of this step. Next, the alkylated bases were protected in the form of carbamate using 4-methylthiobenzyl alcohol (Mtb–OH), by first activating the exocyclic N4 (in cytosine) and N6 (in adenine) using carbonyldiimidazole (CDI) to obtain tert-butyl (N6-Mtz-adenine-9-yl)acetate and tert-butyl (N4-Mtz-cytosine-1-yl)acetate in good yields (Scheme 2). Further conversion of the Mtz-protecting group into an acid-stable Msz-protecting group is crucial step because it involves the oxidation of the sulfur atom to sulfinyl, a process in which overoxidation to sulfonyl as well as the oxidation of the nitrogen atom in the heterocycles must be avoided. Hence, after several attempts, the oxidation step was carried out smoothly with meta-chloroperbenzoic acid (mCPBA) in dichoromethane (DCM) to give Msz-protected adenine and cytosine in excellent yields (>90%). Finally, the tBu ester was removed efficiently using 60% TFA in DCM for 2 h to obtain the final compounds in quantitative yields (Scheme 2).
 | ||
| Scheme 2 Synthetic steps for the preparation of (N6-Msz-adenine-9-yl)acetic acid (4) and (N4-Msz-cytosine-1-yl)acetic acid (5). | ||
The last step to obtain the respective adenine and cytosine PNA monomers involved the condensation reaction of (N6-Msz-adenine-9-yl)acetic acid (4) or (N4-Msz-cytosine-1-yl)acetic acid (5) with the backbone moiety, Boc–Aeg–OBn (3). Although this appears to be a simple reaction, there are many precedents in the literature studying the acylation of the secondary amine of the Aeg backbone with the protected nucleobase derived as carboxylic acid. Some of the these reagents include the following: carbodiimides such as dicyclohexylcarbodiimide (DCC),8,10,11,26–31 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide·HCl (EDC·HCl),9,32–34 and diisopropylcarbodiimide (DIC);35,36 aminium salts such as N-[(1H-benzotriazol-1-yloxy)(dimethylamino)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HBTU)37 and N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]-pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU);38,39 and some phosphonium salts such as benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP)9,38,40 and bromotri(pyrrolidino) phosphonium hexafluorophosphate (PyBroP).8,26,27,29,41 Considering all the above-cited coupling reagents, we decided to first use COMU/DIEA to activate the free carboxylic acids of the protected nucleobases, followed by the slow addition of Boc–Aeg–OBn (3). However, after the work-up, a sticky compound was obtained, which, after purification, only gave a 20% yield. Next, we tested other coupling reagents and activators. In our hands, the best protocol proved to be first the activation of (N6-Msz-adenine-9-yl)acetic acid (4) or (N4-Msz-cytosine-1-yl)acetic acid (5) using EDC·HCl in the presence of 1-hydroxybenzotriazole (HOBt), followed by the reaction with Boc–Aeg–OBn (3). The condensation products were obtained with an improved yield of ∼55% after column purification (Scheme 3). Finally, the hydrolysis of corresponding benzyl esters was carried out using LiOH in H2O to give the final monomers Boc–Aeg–A(Msz)–OH or Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2) as white solids with ∼55–65% yields (Scheme 3).
 | ||
| Scheme 3 PNA monomer subunits coupling and hydrolysis of the ester. | ||
2.2. Solid phase synthesis of PNA pentamer H–PNA(TATCT)–βAla–OH
Although PNA SPS is normally carried out using a large excess of the protected monomers (4 to 10 equiv.),8,30,41 we opted to design a more conservative strategy to optimize the use of protected monomers synthesized in-house. In this regard, we used only 3 equiv. of Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2) in conjunction with a low-loading resin to reduce intra- and interchain aggregation. The HO–Mmsb–resin prepared as discussed above was used for this synthesis. The introduction of the protected nucleobases (equiv. 3) was performed in the presence of slight deficit of COMU (equiv. 2.9), to avoid the capping of the resin in the form of guanidylation,42 and 9 equiv. of DIEA. After each coupling, Boc was removed using 50% TFA in DCM (1 min + 15 min) and the next coupling of protected monomer was carried out with in situ neutralization. Once the last monomer had been introduced, the desired PNA pentamer was obtained by performing a ‘two-step’ (reduction and cleavage) protocol. To this end, the sulfinyl groups (Mmsb and Msz) were reduced with 20 equiv. TMSCl and 10 equiv. Ph3P in THF (3 × 1 h), and the pentamer was cleaved from the resin using (TFA/TIS/H2O-95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5) for 1 h. The crude PNA was then analyzed by LC-MS, which revealed an acceptable purity profile and the mass of the main peak corresponding to the expected mass of the target compound (Fig. 6).
:2.5:2.5) for 1 h. The crude PNA was then analyzed by LC-MS, which revealed an acceptable purity profile and the mass of the main peak corresponding to the expected mass of the target compound (Fig. 6).
 | ||
| Fig. 6 Chromatographic traces of H–PNA(TATCT)–βAla–OH using Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2) on HO–Mmsb–resin. Mobile phase A: 0.1% HCOOH in H2O; mobile phase B: 0.1% HCOOH in CH3CN. Gradient elution: 0–25% of B into A in 15 min at λ = 260 nm. | ||
3. Conclusions
PNAs are a promising class of biomolecule mimetics formed by a concatenation of monomers bearing carboxylic acid and amino functions, together with the nucleobases. These molecules were first synthesized employing the SPS approach used to prepare peptides. However, contrary to peptides, the SPS of PNAs using the Fmoc strategy has not achieved the same results as those obtained when using the Boc protection. Fmoc is a more hydrophobic protecting group, which can favour inter/intra chain aggregation. On the other hand, the Boc group is removed by TFA, which shows excellent capacity to solvate the resin. Furthermore, the incorporation of the next Boc-monomer is carried out using an in situ neutralization protocol, with a well-solvated PNA–resin. The main advantage of the Fmoc strategy is that it is based on the use of a linker and base protecting group, which are conveniently removed with TFA at the end of the sequence elongation. In contrast, the Boc strategy requires strong acids such as HF or TFMSA in the last step.Herein, we describe a novel strategy that combines the advantages of the two protecting groups: firstly, Boc as temporary protection of the amino group and their final cleavage using TFA. To prevent premature cleavage of the PNA from the resin and the removal of the nucleobase-protecting groups, we have developed a safety-catch strategy. This approach benefits their use in both the linker and the nucleobase base protecting groups, based on the methylsulfinylbenzyl moiety, which are stable to TFA, and in the prior to last step (cleavage from resin), the reduction to the thio derivatives allows their removal with TFA.
The HO–Mmsb linker, which has previously been used by our group for the synthesis of peptides, allowed the preparation of H–PNA(T)6–βAla–OH using Boc-protected monomers. In contrast, the synthesis of the same PNA using Fmoc chemistry rendered a mixture of several unprotected and protected oligomers. In contrast, the use of Boc strategy allowed the preparation of H–PNA(T–A(Z)–T–A(Z)–T)–βAla–OH [the PNA(A) was incorporated with the TFA stable, Z group] in excellent purity.
In a key step, novel monomers were prepared using Msz for the protection of the nucleobases: Boc–PNA–A(Msz)–OH (1) and Boc–PNA–C(Msz)–OH (2). The use of these two monomers in combination with the HO–Mmsb linker allowed us to prepare H–PNA(TATCT)–βAla–OH in good purity. These results thus demonstrate that the HO–Mmsb linker and Msz protecting group tandem fulfils the requirements of our safety-catch approach.
We envisage that this strategy will be key to widening the applications of PNAs in several fields of biomedicine.
4. Experimental section
4.1. General information
All the chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich, Germany). The solvents used were of analytical and HPLC reagent grade. Commercial Boc and Fmoc PNA monomers (T, C, A, and G) were purchased from PolyOrg. Inc, USA. Fmoc-L-amino acids were from Iris Biotech Gmbh, Germany. DIC and OxymaPure were gifts from Luxembourg Industries Ltd, Tel Aviv, Israel. All the chemicals and solvents were used without any further purification. AM–resin (0.92 mmol g−1) was a gift from Purolite. Analytical HPLC and mass spectra were obtained using a Thermo Fisher Scientific UltiMate 3000 UHPLC-ISQTM EC single quadrupole mass spectrometer in positive ion mode over a 5–95% gradient of MeCN (0.1% HCOOH)/H2O (0.1% HCOOH) for 15 min, if not stated otherwise.4.2. Protected backbone synthesis: Boc–Aeg–Bn (3)
A solution of ethylenediamine (30 mL, 450 mmol) in DCM (350 mL) was cooled in ice at 0 °C before the dropwise addition of Boc anhydride (11 mL, 48 mmol) in DCM (250 mL) for over 3 h with vigorous stirring. After completion of dropwise addition, the reaction was left to stir for 24 h at room temperature (RT). Next, the solvent was evaporated from the reaction mixture. The crude product was then dissolved in sat. NaHCO3 and washed with DCM (x3). The organic layer was dried over MgSO4 and evaporated to dryness to give the compound in quantitative yield (7.7 g, 48 mmol). The obtained compound was dissolved in DCM was cooled in an ice bath to 0 °C, and triethylamine (10 mL, 72 mmol) was slowly added. Benzyl bromoacetate (7.5 mL, 48 mmol) was then added dropwise for over 3 h with vigorous stirring. After this time, the reaction mixture was removed from the ice bath and the reaction was allowed to run overnight at RT. Then, the solvent was removed, water was added and the pH was adjusted to 4 using 1 N HCl solution. The aqueous mixture was extracted with DCM (x3), the organic layers were collected, dried over MgSO4 and evaporated to dryness to give 8.3 g (26.9 mmol, 56% yield) of the pure compound. HRMS (ES+): m/z calcd for C16H25N2O4 [M + H+] 309.1809, found 309.1814; m/z calcd for C16H25NaN2O4 [M + Na+] 331.1628, found 331.1631. 1H NMR ([d6]DMSO, 400 MHz, 25 °C): δ = 7.38 ppm (m, 5 H, ArH), 6.73 (t, J = 5.49 Hz, 1 H, NH), 5.12 (s, 2 H), 2.97 (q, J = 6.22 Hz, 2 H), 2.55 (t, J = 6.48 Hz, 2 H), 1.37 (s, 9 H); 13C NMR ([d6]DMSO, 100 MHz, 25 °C): δ = 172.6, 136.5, 128.9, 128.5, 128.4, 77.9, 65.8, 50.5, 48.7, 28.7.4.3. Synthesis of (N6-Msz-adenine-9-yl)acetic acid (4)
:30 h. Next, the reaction temperature was reduced to 95 °C to add Mtb–OH (4.16 g, 26.96 mmol, equiv. 2) in 5 mL DMF. The heat source was then removed, and the reaction was left to stir overnight while allowing it to cool to RT. Next, the reaction mixture was poured onto the ice cubes while stirring vigorously and allowed to stir for 2 h. After this time, the precipitates obtained were filtered, washed with water (×5) and dried under high vacuum to give 4.1 g (69% yield) of pure compound. 1H NMR ([d6]DMSO, 600 MHz, 25 °C): δ = 10.7(s, NH, 1 H), 8.62 (s, 1 H), 8.42 (s, 1 H), 7.41 (d, J = 8.3 Hz, 2 H), 7.29 (d, J = 8.3 Hz, 2 H), 5.17 (s, 2 H), 5.08 (s, 2 H), 2.48(s, 3 H) 1.42 (s, 9 H); 13C NMR ([d6]DMSO, 100 MHz, 25 °C): δ = 167.2, 152.6, 152.2, 149.9, 145.2, 138.5, 133.3, 129.2, 126.3, 123.4, 82.8, 66.4, 45.3, 28.1, 15.2.
4.4. Synthesis of (N4-Msz-cytosine-1-yl)acetic acid (5)
4.5. Synthesis of Boc–PNA–A(Msz)–OH (1)
:1), and the reaction was stirred at RT for 30 min. The reaction was monitored by TLC. After completion, more H2O was added to the mixture and THF was evaporated from it. The aqueous phase was then washed with DCM (×2). Next, the aqueous layer was acidified using 0.1 N HCl to pH 3–4 in an ice bath. It was then saturated using NaCl (while maintaining the pH 3–4) and extracted using EtOAc (×5) to get 0.55 g (63% yield). 1H NMR ([d6]DMSO, 600 MHz, 25 °C): δ = 10.76 (s, 1 H, NH), 8.61 (s, 1H), 8.32 (s, 1H), 7.72 (d, J = 8.19 Hz, 2 H), 7.67 (d, J = 8.21 Hz, 2 H), 7.05 (t, J = 5.26 Hz, 1 H), 5.34 (s, 1 H), 5.29 (s, 2 H), 5.12 (s, 1 H), 4.27 (s, 1H), 3.98 (s, 2H) 3.53 (t, J = 6.29 Hz, 2 H), 3.26 (q, J = 6.17 Hz, 2 H), 2.74 (s, 3 H), 1.38–1.36 (s, 9H); 13C NMR ([d6]DMSO, 100 MHz, 25 °C): δ = 170.8, 167.4, 166.9, 156.3, 152.9, 152.5, 151.9, 149.8, 149.7, 146.3, 145.8, 145.7, 139.7, 128.8, 124.2, 123.3, 78.6, 78.3, 66.1, 48.1, 47.5, 44.3, 43.6, 28.7, 28.6.
4.6. Synthesis of Boc–PNA–C(Msz)–OH (2)
:1), and the reaction was stirred at RT for 3 h. The progress of the reaction was monitored by TLC. After completion, more H2O was added to the reaction mixture and THF was evaporated from the mixture. The mixture was then washed with DCM (×2). Next, the aqueous layer was acidified using 0.1 N HCl to pH 3–4 in an ice bath. The aqueous layer was then saturated using NaCl (while maintaining the pH 3–4) and extracted using EtOAc (×5) to get 0.48 g (56% yield). 1H NMR ([d6]DMSO, 600 MHz, 25 °C): δ = 10.85 (s, 1 H), 7.91 (d, J = 7.37 Hz, 1 H), 7.71 (d, J = 8.23 Hz, 2 H, ArH), 7.62 (d, J = 8.22 Hz, 2 H, ArH), 7.01 (d, J = 7.28 Hz, 1 H), 6.94 (t, J = 5.71 Hz, 1 H), 5.27 (s, 2 H), 4.82 (s, 1 H), 4.62 (s, 1 H), 4.17 (s, 1 H), 3.98 (s, 2 H), 3.42 (t, J = 6.99 Hz, 2 H), 3.19 (q, J = 6.35 Hz, 2 H), 2.75 (s, 3 H), 1.38–1.36 (s, 9H); 13C NMR ([d6]DMSO, 100 MHz, 25 °C): δ = 170.9, 167.9, 167.5, 163.6, 155.4, 146.6, 146.0, 144.8, 139.3, 128.8, 127.5, 124.3, 123.9, 94.4, 78.5, 66.3, 62.8, 48.2, 47.4, 43.7, 43.6, 28.7, 28.6.
4.7. General protocol for the solid-phase synthesis of PNAs
The PNAs were synthesized on AM resin (0.92 mmol g−1) for Boc chemistry and on Wang resin (0.43 mmol g−1) for Fmoc chemistry. The resins were first conditioned by washing with DMF (2 × 1 min), DCM (2 × 1 min), and DMF (2 × 1 min). In the case of the AM resin, the loading was reduced from 0.92 to 0.4 mmol using Fmoc–Ala–OH. Fmoc–Ala–OH (equiv. 1) was coupled to the AM resin using DIC (equiv. 1) and OxymaPure (equiv. 1) in DMF for 45 min. The acetylation in the free N-terminal was performed using acetic anhydride (10 equiv.), DIEA (20 equiv.) in DMF for 45 min. Next, Fmoc was removed by treatment with 20% piperidine/DMF (1 × 1 min and 1 × 7 min), followed by washing the resin with DMF, DCM, and DMF. The resin was then functionalized by coupling the HO–Mmsb linker. HO–Mmsb (equiv. 1.5) was incorporated on the AM resin using N-hydroxysuccinimide (HOSu) (equiv. 1.5) and DIC (equiv. 1.5) in DMF for 1 h. The coupling of the first amino acid onto the HO–Mmsb–Ala–AM–resin (this resin is generally referred to as HO–Mmsb–resin throughout this article) was achieved through esterification using Fmoc–βAla–OH (equiv. 4), DIC (equiv. 2) and a catalytic amount of DMAP (equiv. 0.2) in DMF for 3 h, followed by an extra coupling overnight using the same conditions. Fmoc was removed using 20% piperidine/DMF (1 × 1 min and 1 × 7 min), followed by washing the resin and coupling of the next PNAs were carried out using COMU (equiv. 2.9) and DIEA (equiv. 9) in DMF, as a coupling system, for 30 min at RT. These steps were repeated until the final PNAs were achieved. Washes between couplings and deprotections were performed with DMF (3 × 1 min), DCM (3 × 1 min), and DMF (3 × 1 min). The Boc group was removed using 50% TFA in DCM (1 min + 15 min). Boc removal was followed by coupling of the next Boc–PNA monomer and so on. The final PNA–resin was then washed and dried well to continue with the reduction and total cleavage of the desired PNAs.In the case of Wang resin, the loading was reduced from 0.83 mmol to 0.4 mmol using Fmoc–βAla–OH (equiv. 1), DIC (equiv. 2.5), and DMAP (equiv. 0.25) in DCM for 1 h. After 1 h, capping was performed using acetic anhydride (equiv. 10) and DIEA (equiv. 20) in DMF for 30 min. Fmoc was removed using 20% piperidine/DMF (1 × 1 min and 1 × 7 min), followed by washing the resin with DMF, DCM, and DMF. Couplings were performed using Fmoc–PNA monomer (equiv. 3), COMU (equiv. 2.9), and DIEA (equiv. 6) in DMF for 30 min. Fmoc was then removed, followed by the coupling of the next Fmoc–PNA monomer and so on. The final PNA–resin was then washed and dried well to continue with total cleavage (TFA/TIS/H2O-95:2.5:2.5) of the desired PNAs from the Wang resin.
4.8. ‘Two-step’ (reduction and cleavage) protocol for HO–Mmsb–resin
After the last synthetic step, the PNA–resin was reduced by treatment with 20 equiv. Me3SiCl and 10 equiv. Ph3P in THF (3 × 1 h). The reduced PNA–resin was then washed well with THF (×3), DMF (×3), and DCM (×3), and dried to continue with the cleavage from resin. This process was performed using (TFA/TIS/H2O-95:2.5:2.5) for 1 h. After cleavage, the mixture was precipitated with chilled Et2O and centrifuged. The pellet was resuspended in H2O for analysis by LC-MS.
Author contributions
The idea was conceived by BGT and FA, the most part of the experimental pat was carried out by NKP with the aid of SN and EB. All authors have contributed to the preparation of the manuscript and agreed with the final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work in the laboratory of the authors is being partially funded by the National Research Foundation (NRF), South Africa. Thomas Bruckdorfer (Iris Biotech GmbH, Marktredwitz, Germany) for encouraging this work.References
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2022, 15, 222 CrossRef CAS PubMed.
- P. E. Nielsen, Perspect. Drug Discovery Des., 1996, 4, 76–84 CrossRef CAS.
- S. Montazersaheb, M. S. Hejazi and H. Nozad Charoudeh, Adv. Pharm. Bull., 2018, 8, 551–563 CrossRef CAS PubMed.
- K. R. Singh, P. Sridevi and R. P. Singh, Eng. Rep., 2020, 2, e12238 CrossRef CAS PubMed.
- R. An, Y. Jia, B. Wan, Y. Zhang, P. Dong, J. Li and X. Liang, PLoS One, 2014, 9, e115950 CrossRef PubMed.
- K. P. Nandhini, D. A. Shaer, F. Albericio and B. G. de la Torre, Chem. Soc. Rev., 2023, 52, 2764–2789 RSC.
- L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M. Egholm, O. Buchardt, P. E. Nielsen, J. Coull and R. H. Berg, J. Pept. Sci., 1995, 1, 175–183 CrossRef CAS PubMed.
- K. L. Dueholm, M. Egholm, C. Behrens, L. Christensen, H. F. Hansen, T. Vulpius, K. H. Petersen, R. H. Berg, P. E. Nielsen and O. Buchardt, J. Org. Chem., 1994, 59, 5767–5773 CrossRef CAS.
- S. A. Thomson, J. A. Josey, R. Cadilla, M. D. Gaul, C. Fred Hassman, M. J. Luzzio, A. J. Pipe, K. L. Reed, D. J. Ricca, R. W. Wiethe and S. A. Noble, Tetrahedron, 1995, 51, 6179–6194 CrossRef CAS.
- M. Egholm, O. Buchardt, P. E. Nielsen and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 1895–1897 CrossRef CAS.
- M. Egholm, P. E. Nielsen, O. Buchardt and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 9677–9678 CrossRef CAS.
- N. Brodyagin, Y. Kataoka, I. Kumpina, D. W. McGee and E. Rozners, Biopolymers, 2022, 113, e23484 CrossRef CAS PubMed.
- K. Klabenkova, A. Fokina and D. Stetsenko, Molecules, 2021, 26, 5420 CrossRef CAS PubMed.
- G. W. Kenner, J. R. McDermott and R. C. Sheppard, J. Chem. Soc. D, 1971, 636–637 RSC.
- Y. Kiso, T. Kimura, M. Yoshida, M. Shimokura, K. Akaji and T. Mimoto, J. Chem. Soc., Chem. Commun., 1989, 1511–1513 RSC.
- S. Thennarasu and C.-F. Liu, Tetrahedron Lett., 2010, 51, 3218–3220 CrossRef CAS.
- Y. Kiso, T. Fukui, S. Tanaka, T. Kimura and K. Akaji, Tetrahedron Lett., 1994, 35, 3571–3574 CrossRef CAS.
- K. P. Nandhini, F. Albericio and B. G. de la Torre, J. Org. Chem., 2022, 87, 9433–9442 CrossRef CAS PubMed.
- M. Paradís-Bas, J. Tulla-Puche and F. Albericio, Org. Lett., 2015, 17, 294–297 CrossRef PubMed.
- F. Altenbrunn and O. Seitz, Org. Biomol. Chem., 2008, 6, 2493–2498 RSC.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- S. Noki, E. Brasil, H. Zhang, T. Bruckdorfer, B. G. de la Torre and F. Albericio, J. Org. Chem., 2022, 87, 9443–9453 CrossRef CAS PubMed.
- J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925–3930 CrossRef CAS PubMed.
- T. A. Feagin, N. I. Shah and J. M. Heemstra, J. Nucleic Acids, 2012, 2012, 354549 Search PubMed.
- S. Pothukanuri, Z. Pianowski and N. Winssinger, Eur. J. Org. Chem., 2008, 3141–3148 CrossRef CAS.
- M. Egholm, C. Behrens, L. Christensen, R. H. Berg, P. E. Nielsen and O. Buchardt, J. Chem. Soc., Chem. Commun., 1993, 800–801 RSC.
- T. Kofoed, H. F. Hansen, H. Ørum and T. Koch, J. Pept. Sci., 2001, 7, 402–412 CrossRef CAS PubMed.
- O. Seitz and O. Köhler, Chem. – Eur. J., 2001, 7, 3911–3925 CrossRef CAS PubMed.
- J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef PubMed.
- H. Lee, J. H. Jeon, J. C. Lim, H. Choi, Y. Yoon and S. K. Kim, Org. Lett., 2007, 9, 3291–3293 CrossRef CAS PubMed.
- F. Wojciechowski and R. H. E. Hudson, J. Org. Chem., 2008, 73, 3807–3816 CrossRef CAS PubMed.
- B. Falkiewicz, A. Kozyra, A. S. Kolodziejczyk, B. Liberek and K. Wisniewski, Nucleic Acids Symp. Ser., 1999, 42, 9–10 CrossRef CAS PubMed.
- E. C. Browne, S. J. Langford and B. M. Abbott, Aust. J. Chem., 2012, 65, 539–544 CrossRef CAS.
- A. H. St. Amant and R. H. E. Hudson, Org. Biomol. Chem., 2012, 10, 876–881 RSC.
- D. W. Will, G. Breipohl, D. Langner, J. Knolle and E. Uhlmann, Tetrahedron, 1995, 51, 12069–12082 CrossRef CAS.
- Y.-C. Huang, C. Cao, X.-L. Tan, X. Li and L. Liu, Org. Chem. Front., 2014, 1, 1050–1054 RSC.
- S. R. Malamgari, P. Manikandan, P. Ramani and V. R. Katta, ChemistrySelect, 2018, 3, 3948–3951 CrossRef CAS.
- T. Sugiyama, A. Kittaka, Y. Takemoto, H. Takayama and R. Kuroda, Nucleic Acids Symp. Ser., 2002, 2, 145–146 CrossRef CAS PubMed.
- D. Chouikhi, M. Ciobanu, C. Zambaldo, V. Duplan, S. Barluenga and N. Winssinger, Chem. – Eur. J., 2012, 18, 12698–12704 CrossRef CAS PubMed.
- Z.-C. Liu, D.-S. Shin, K.-T. Lee, B.-H. Jun, Y.-K. Kim and Y.-S. Lee, Tetrahedron, 2005, 61, 7967–7973 CrossRef CAS.
- L. Bialy, J. J. Díaz-Mochón, E. Specker, L. Keinicke and M. Bradley, Tetrahedron, 2005, 61, 8295–8305 CrossRef CAS.
- R. Subirós-Funosas, L. Nieto-Rodriguez, K. J. Jensen and F. Albericio, J. Pept. Sci., 2013, 19, 408–414 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01348k |
| This journal is © The Royal Society of Chemistry 2023 |