
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-phase synthesis of oligodeoxynucleotides using nucleobase N-unprotected oxazaphospholidine derivatives bearing a long alkyl chain†
Kiyoshi
Kakuta
 ,
Ryouta
Kasahara
,
Kazuki
Sato
and
Takeshi
Wada
*
,
Ryouta
Kasahara
,
Kazuki
Sato
and
Takeshi
Wada
*
Department of Medicinal and Life Sciences, Faculty of Pharmaceutical Sciences, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: twada@rs.tus.ac.jp; kazuki_sato@rs.tus.ac.jp
First published on 1st September 2023
Abstract
In this study, we developed a new approach for the solid-phase synthesis of oligodeoxynucleotides (ODNs) using nucleobase-unprotected oxazaphospholidine derivatives. We tackled the problem of the difficult purification of N-unprotected monomers due to their high affinity to silica gel by introducing a tetrahydrogeranyl group into the oxazaphospholidine monomers, thereby enhancing the lipophilicity and facilitating the isolation. In addition, the cyclic structure of oxazaphospholidine enabled a hydroxy-group-selective condensation with sufficient efficiency. Unmodified and boranophosphate/phosphate chimeric ODNs were successfully synthesized using this strategy. This synthetic method can be expected to afford ODNs containing base-labile functional groups.
Introduction
The phosphoramidite approach is considered the definitive means for the synthesis of oligonucleotides.1 In this method, chain elongation is conducted via condensation of a phosphoramidite monomer and a 5′-hydroxy group of a nucleotide under mild acidic conditions. The amino groups of nucleobases are generally protected by acyl groups to prevent undesired nucleobase phosphitylation during the condensation.2 However, avoiding the nucleobase protection is highly desirable because it has three advantages. First, skipping nucleobase protection and deprotection processes allows reducing the number of steps in the synthesis of the phosphoramidite monomer.3 Second, the synthesis of the oligomer without nucleobase protection considerably diminishes the risk of depurination of deoxyadenosine derivatives in the process of acid treatment, which has been reported to occur under the acidic conditions applied for the removal of the DMTr group in N6-acyl-protected derivatives.4 Third, the synthesis without nucleobase protection allows obtaining chemically modified oligodeoxynucleotides (ODNs), which are unstable under basic conditions, such as alkylphosphonate,5 because the base deprotection process usually conducted by treating with aqueous ammonia is unnecessary.Hayakawa et al. reported the synthesis of oligonucleotides using nucleobase-unprotected phosphoramidite derivatives.6,7 They found that using imidazolium triflate as an acidic activator can reduce the nucleobase phosphitylation to some extent. However, a part of the amino groups is still phosphitylated during the condensation reaction, and treatment with benzimidazolium triflate–methanol to cleave the P–N bond(s) is necessary after each condensation.6,7 An improved version of the nucleobase-unprotected approach was the proton-block method reported by Sekine et al.8 In this method, the amino groups of nucleobases are temporary protected as unreactive protonated forms during condensation to prevent the reaction with an activated monomer using an acidic activator having relatively low pKa values, affording 20-mer ODNs.
Meanwhile, we previously reported that the condensation reaction using nucleobase-unprotected oxazaphospholidine derivatives proceeded with a hydroxy group in a chemoselective manner even under mild acidic conditions.9–11 Oxazaphospholidines are cyclic phosphoramidite derivatives that were originally developed to synthesize P-stereodefined oligonucleotides.12 The chemoselectivity brought by the oxazaphospholidine derivatives can be explained in terms of an intramolecular recyclization that regenerates the oxazaphospholidine derivatives and the nucleobase having a free amino9,10 or imide11 group (Scheme 1).
 | ||
| Scheme 1 Mechanism of the intermolecular nucleophilic addition. | ||
In such reports, we synthesized 5-H/N-Me- and 5-Ph/N-Me-substituted oxazaphospholidine derivatives with unprotected nucleobases (Scheme 2) and obtained unmodified ODNs up to tetramers and boranophosphate (PB) DNAs up to dimers using the former and the latter monomers, respectively. PB DNAs, in which one of the nonbridging oxygen atoms of the phosphodiester is substituted with a borano group (BH3), have attractive properties as antisense oligonucleotides, such as high nuclease resistance and lipophilicity and low cytotoxicity.13,14 When acyl-type amino protecting groups, which are widely used for nucleobase protection, are employed for the synthesis of PB DNAs, irreversible reduction of amide groups to alkylamino groups occurs as a serious side reaction during the boronation reaction.15 This problem can be overcome by using synthetic strategies without nucleobase protection.9,16 However, when using 5-H/N-Me- or 5-Ph/N-Me-substituted monomers, the synthesis of longer oligomers was unsuccessful, which was attributed to insufficient condensation efficiency, especially in the case of the deoxyguanosine monomer. Moreover, the purity of the isolated unprotected deoxyguanosine monomer was not satisfactory (ca. 81% for the 5-H/N-Me-substituted derivative and ca. 71% for the 5-Ph/N-Me-substituted derivative according to a 31P NMR spectroscopic analysis), most likely due to the high affinity of the monomer to silica gel, which results in a longer elution and, consequently, in the decomposition of the monomer.
 | ||
| Scheme 2 Comparison of the monomer unit structure used in our previous study and this work. | ||
To address this issue, in this study, we introduced a long alkyl chain into the oxazaphospholidine monomer to endow it with lipophilicity. The tetrahydrogeranyl (Thg) group, which has a branched structure, was selected as the long alkyl group because it would also improve the monomer solubility in reaction solvents compared with linear alkyl groups due to diminished interactions between branched alkyl chains.17 Herein, we demonstrate that the monomers were applicable to the synthesis of ODNs up to dodecamers and oligonucleotides contain PB linkages.
Results and discussion
Synthesis of oxazaphospholidine monomers
First, an amino alcohol bearing an N-Thg group was synthesized using 2-amino-1-phenyl ethanol 1 as a substrate (Scheme 3). The hydroxy group of 1 was protected using trimethylsilyl chloride (TMSCl) followed by the introduction of a 2-nitrobenzenesulfonyl (Ns) group into the amino group. The Thg group was introduced into the sulfonamide using a Mitsunobu reaction with tetrahydrogeraniol.18 Then, the TMS group was removed by treating with trifluoracetic acid (TFA), and the resulting crude product was purified by silica gel column chromatography to obtain compound 2 (84% isolated yield over four steps). The Ns group was removed via treatment with 4-mercaptobenzoic acid, which is an odorless thiol,19 to afford amino alcohol 3 in a 56% isolated yield from 1.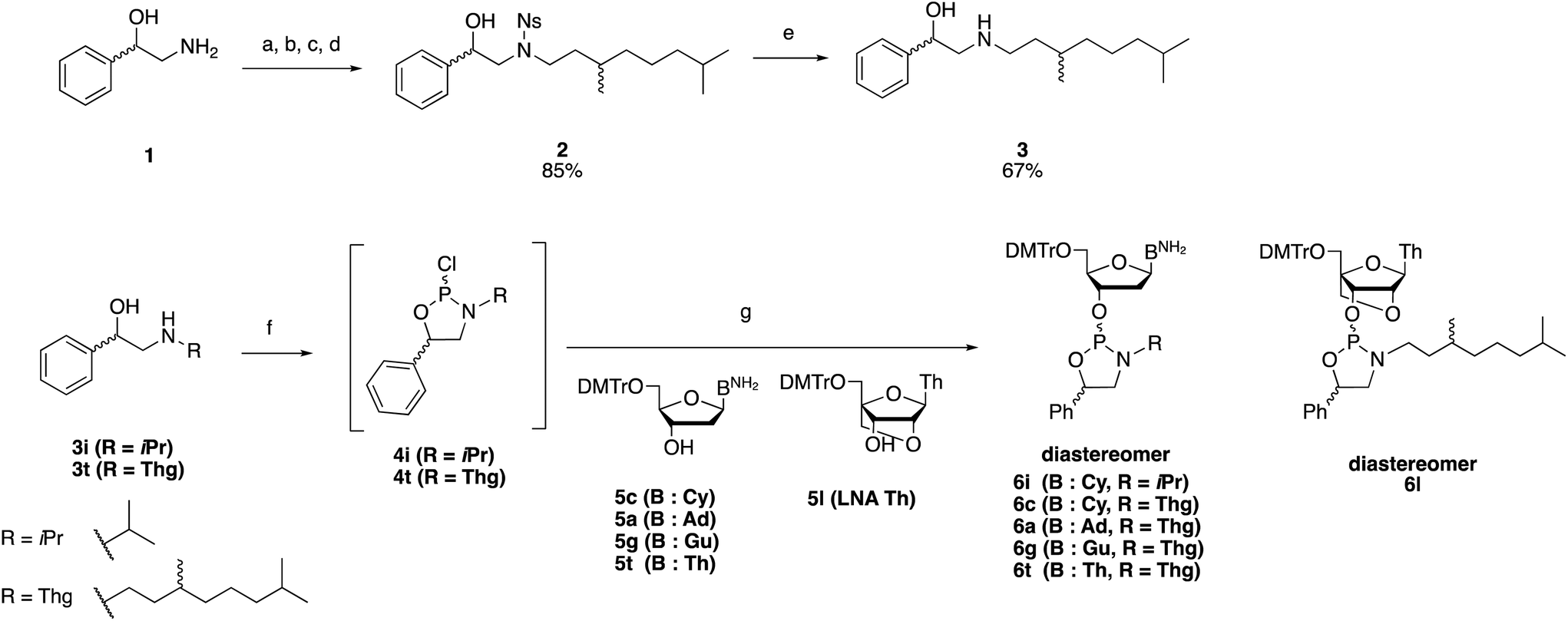 | ||
| Scheme 3 Synthesis of N-Thg and nucleobase-unprotected oxazaphospholidine derivatives 6a–l. Reagents and conditions: (a) TMSCl (1.5 equiv.), Et3N (1.5 equiv.), THF, room temperature (rt), 2 h; (b) NsCl (1.05 equiv.), Et3N (2.0 equiv.), CH2Cl2, 0 °C to rt, 2 h; (c) tetrahydrogeraniol (1.4 equiv.), PPh3 (2.0 equiv.), DIAD (2.0 equiv.), THF, 0 °C to rt, 24 h; (d) 3% TFA, CH2Cl2, rt, 18 h, 85% over four steps; (e) 4-mercaptobenzoic acid (2.0 equiv.), K2CO3 (4.0 equiv.), DMF, 40 °C, 12 h, 67%; (f) PCl3 (1.1 equiv.), N-methylmorpholine (2.1 equiv.), toluene, 0 °C to rt, 2 h; (g) 4 (2.5 equiv.), Et3N (7.0 equiv.), THF, −78 °C (6i, 6a, 6c), −40 °C (6g), −78 °C to rt (6t, 6l), 2 h, 27% (6i), 34% (6a), 39% (6c), 30% (6g), 55% (6t), 32% (6l). | ||
Next, the synthesis of oxazaphospholidine monomers with a iPr or Thg group introduced into the N-atom of the oxazaphospholidine ring was carried out. These monomer units were synthesized using unprotected nucleosides including deoxyribose and 2′-O-4′-C-locked nucleoside (LNA)20 derivatives and phosphitylating reagents 4i (R = iPr) or 4t (R = Thg), which was prepared from phosphorus trichloride and amino alcohols 3i (R = iPr) or 3t (R = Thg). The compounds 4 were not isolated considering the chemical instability and used after only removal of insoluble salts by filtration in the following reaction. For the synthesis of the deoxyadenosine and deoxycytidine derivatives, the reactions were conducted at −78 °C to prevent the phosphitylation of the unprotected amino groups. In the case of the deoxyguanosine derivative, the reaction was performed at −40 °C to enhance the conversion rate. Meanwhile, the phosphitylation of the thymidine and LNA thymidine derivatives was conducted initially at −78 °C and finally at room temperature (rt) through gradual warming. The deoxyadenosine, deoxycytidine, thymidine, and LNA thymidine oxazaphospholidine monomers were isolated with high purity and moderate yields (6i, 27%; 6a, 34%; 6c, 39%; 6t, 55%; 6l, 32%) via silica gel column chromatography. It is worth noting that t-butyl alcohol was effective as the eluent of silica gel chromatography in the case of the deoxycytidine and deoxyguanosine derivatives. When methanol was used as an eluent, the purified oxazaphospholidine monomer decomposed to phosphitetriester during the solvent removal process. Thus, an alcohol with low nucleophilicity was more suitable as a polar eluent. In the case of the deoxyguanosine monomer, the purity was about 90% (determined via31P NMR spectroscopy) and its separation from oxidized compounds via silica gel column chromatography was troublesome. Therefore, reversed-phase high-performance liquid chromatography (RP-HPLC) was used to purify the monomer after a silica gel column chromatography which roughly removed highly polar compounds and reduced the burden on HPLC columns. As a result, the deoxyguanosine monomer was isolated with a high purity and moderate yield (6g, 30%).
Although the isolated yields of N-Thg monomers were in the range of 30%–55%, these yields were considerably higher than those of 5-Ph/N-Me-substituted unprotected monomers, especially deoxycytidine and deoxyguanosine derivatives (17% and 3%, respectively). In addition to this, the isolated yield of N-Thg monomer was higher than that of N-iPr-substituted monomer (6i; 27% vs.6c; 39%). TLC monitoring of the reaction mixtures indicated that the conversion rates of 5c and 5g to 6c and 6g were about 50%, which were low compared with those of their N-Me-substituted counterparts probably due to the steric hindrance of the alkyl group. However, the higher isolation yields of the N-Thg derivatives compared with those of the N-Me derivatives can be attributed to their easier purification and higher chemical stability. Although a greater excess of compound 4t could lead to the completion of the reaction, no optimization of equivalents of phosphitylating reagent was conducted.
Solid-phase synthesis of oligonucleotide phosphates
With the nucleobase-unprotected oxazaphospholidine monomers in hand, the solid-phase synthesis of the dinucleoside phosphates was conducted using highly crosslinked polystyrene (HCP) as a solid support.21 The 5′-hydroxy group of 7 was condensed with monomer 6 in the presence of N-(phenyl)imidazolium triflate (PhIMT)22 as an acidic activator in CH3CN–iPrCN (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The resultant phosphite 8 was oxidized using t-butyl hydroperoxide (TBHP) to afford a phosphotriester. Moreover, the DMTr group on the 5′-hydroxy group was removed under acidic conditions with 3% dichloroacetic acid (DCA), affording 9. The cleavage of the succinyl linker using an aqueous NH3–EtOH (3:1, v/v) solution yielded dinucleoside phosphate derivatives 10a–l (Scheme 4). It is worth noting that PhIMT was found to be effective for the activation of oxazaphosholidine monomers in the synthesis of oligoribonucleotide derivatives. Thus, PhIMT was chosen as an optimal activator in the current study.23,24
:3). The resultant phosphite 8 was oxidized using t-butyl hydroperoxide (TBHP) to afford a phosphotriester. Moreover, the DMTr group on the 5′-hydroxy group was removed under acidic conditions with 3% dichloroacetic acid (DCA), affording 9. The cleavage of the succinyl linker using an aqueous NH3–EtOH (3:1, v/v) solution yielded dinucleoside phosphate derivatives 10a–l (Scheme 4). It is worth noting that PhIMT was found to be effective for the activation of oxazaphosholidine monomers in the synthesis of oligoribonucleotide derivatives. Thus, PhIMT was chosen as an optimal activator in the current study.23,24
 | ||
| Scheme 4 Solid-phase synthesis of NPOT dimers and oligonucleotide phosphate. | ||
To begin with, the condensation efficiency was compared using N-Me-, N-iPr-, and N-Thg-substituted monomers. The HPLC yields of dinucleotide phosphates other than the deoxyguanosine derivative were 95%–97% when the N-Me-substituted monomers were used, whereas N-iPr- and N-Thg-substituted monomers afforded the corresponding products in HPLC yields higher than 99% (Table 1, entries 1–3 vs. 4–7), as confirmed by the area ratio defined by product/(product + Th). This result suggested that the N-iPr- and N-Thg-substituted monomers exhibited higher coupling efficiencies. In general, the phosphoramidite reactivity is known to be largely affected by steric hindrance.25 However, in this case, the steric hindrance of the N-Me-substituted monomers around the N-atom are small, resulting in immediate hydrolysis during condensation. Thus, the introduction of the iPr and Thg groups improved the stability of the monomer while maintaining satisfactory condensation efficiency. The N-Thg-substituted monomer was chosen as an optimal monomer unit due to the higher isolated yield of the monomer.
| Entry | Producta | R | Yieldb (%) |
|---|---|---|---|
| a Subscript PO = phosphate, superscript L = LNA. b Determined via RP-HPLC. c Isolated yield, determined according to the UV absorbance at 260 nm. | |||
| 1 | dCPOT 10c | Me | 97 |
| 2 | dAPOT 10a | Me | 95 |
| 3 | TPOT 10t | Me | 97 |
| 4 | dCPOT 10c | iPr | >99 |
| 5 | dCPOT 10c | Thg | >99 |
| 6 | dAPOT 10a | Thg | >99 |
| 7 | TPOT 10t | Thg | >99 |
| 8 | dGPOT 10g | Thg | >99 |
| 9 | LTPOT 10l | Thg | >99 |
| 10 | d(CPOAPOGPOTPOCPOAPOGPOTPOCPOAPOGPOT) (11) | Thg | 14c |
Then, the deoxyguanosine monomer was examined for the synthesis of dimers (Table 1, entry 7). The dGPOT dimer was obtained in more than 99% HPLC yield. No phosphitylation on the nucleobase amino groups was observed using oxazaphospholidine monomers bearing the N-Thg group. In addition, the synthesis was performed using LNA thymidine monomer 6l, obtaining the LTPOT dimer (the superscript L indicates LNA nucleoside) in more than 99% HPLC yield. The LNA monomer also exhibited high condensation efficiency, although the reactivity of LNA derivatives is typically low due to the steric hindrance caused by the 2′ and 4′ locked structure.
Next, the synthesis of a dodecamer of deoxyribonucleotide d(CPOAPOGPOTPOCPOAPOGPOTPOCPOAPOGPOT) 11 bearing four nucleobases was investigated. Fig. S2† shows the UPLC profile of crude 11, which indicates that the desired product was obtained as the main product. The dodecamer was isolated in 14% yield, identified via mass spectrometry, and analyzed using 1H and 31P NMR spectroscopies (Fig. S7 and S8†). The 1H NMR spectrum revealed the presence of signals stemming from the sugar backbone and the nucleobases. Meanwhile, in the 31P NMR spectrum, only signals in the PO region were observed (Fig. S8†).
Finally, the purity of the synthesized oligomer was compared with that of a commercial one via RP-UPLC. The peaks of both products appeared at the same retention time, confirming the successful synthesis of the oligomer with similar purity to that of the commercial product and demonstrating that the newly designed monomers enable the synthesis of a PO ODN without nucleobase protection.
Solid-phase synthesis of dinucleoside and trinucleoside boranophosphates
Next, the synthesis of the dinucleoside boranophosphates was studied as an example of P-modifications using the nucleobase-unprotected approach. The succinyl sarcosyl linker26 tethered between thymidine and HCP was selected to gain stability under the DBU treatment conditions required to remove the protecting group of the PO moiety.27,28 The succinyl linker has been reported to be gradually cleaved by DBU to form a five-membered ring.26 After the condensation reaction, the resultant phosphite was boronated with 1.0 M BH3·SMe2/toluene for 15 min to afford boranophosphotriester 12. Moreover, the DMTr group on the 5′-hydroxy group was cleaved under acidic conditions with DCA using Et3SiH as a DMTr cation scavenger to prevent the side reaction on the borano group.29 To avoid an intramolecular attack to the internucleotidic linkage of the boranophosphotriester by the 5′-hydroxy group,30 the solid support was treated with DBU to afford the desired boranophosphodiester 13 after capping the 5′-hydroxy group with acetic anhydride (Ac2O). Finally, the resulting dimers 14 were cleaved from the solid support after ammonia treatment (Scheme 5). | ||
| Scheme 5 Solid-phase synthesis of NPBT dimers and NPBNPBT trimer. | ||
First, the dinucleoside boranophosphate was synthesized using the deoxycytidine monomer under boronation conditions with 1.0 M BH3·SMe2/toluene. The HPLC yield of the target compound was 97% as confirmed by the product/product + Th area ratios. This result prompted us to synthesize the trinucleoside boranophosphate. Since our group found that the boronation reagent and/or its residue(s) inhibited the subsequent condensation reaction in the synthesis of tetrasaccharide boranophosphate,31 a washing step with EtOH was conducted after the boronation reaction to remove the reagent. However, the coupling yield of the second condensation reaction was not satisfactory; specifically, dCPBT and d(CPBCPBT) were obtained in 12% and 85% HPLC yields, respectively (Table 2, entry 2). To address this issue, the boronation conditions were investigated, finding that boronation using 0.05 M BH3·THF/THF for 2 min were the optimal conditions (see ESI† for details), which provided dCPBT in a comparable yield to that obtained when using harsher conditions. With regard to the synthesis of trimers, the HPLC yield of d(CPBCPBT) was improved to 94%.
| Entry | Producta | Boronation conditions | T:dimer or T:dimer:trimerb |
|---|---|---|---|
| a Subscript PB = boranophosphate. b Determined via RP-HPLC. c Ammonia treatment was conducted at rt, for 3 h and then at 50 °C for 17 h. | |||
| 1 | dCPBT 14c | 1.0 M BH3·SMe2/toluene | 3:97 |
| 2 | d(CPBCPBT) 15 | 1.0 M BH3·SMe2/toluene | 3:12:85 |
| 3 | dCPBT 14c | 0.05 M BH3·THF/THF | 3:97 |
| 4 | d(CPBCPBT) 15 | 0.05M BH3·THF/THF | 1:5:94 |
| 5 | dAPBT 14ac | 0.05 M BH3·THF/THF | 1:99 |
| 6 | dGPBT 14g | 0.05 M BH3·THF/THF | 1:99 |
| 7 | TPBT 14t | 0.05 M BH3·THF/THF | 2:98 |
When other nucleobase monomers were used for the synthesis of dimers, the dAPBT, dGPBT, and TPBT dimers were obtained in 97%–99% HPLC yields (Table 2, entries 5–7). In the case of dAPBT, the removal of the borane adducts on the N1 and/or N7 positions of an adenine29 required ammonia treatment for longer time and higher temperature, whereas other nucleobases did not require high-temperature conditions.32 As summarized in Table 2, the dinucleoside and trinucleoside boranophosphates were synthesized in satisfactory HPLC yields without considerable amounts of byproducts.
Solid-phase synthesis of oligonucleotides bearing boranophosphates
On the basis of these results, the synthesis of all-PB and PB/PO chimeric tetramers was attempted according to Scheme 6. The synthesis cycle consisted of condensation of 6 in the presence of PhIMT, boronation or oxidation, and removal of the DMTr group. Capping of the 5′-hydroxy group and deprotection of PO or PB moieties followed by ammonia treatment afforded the tetramers. | ||
| Scheme 6 Solid-phase synthesis of oligonucleotides bearing boranophosphates. | ||
Fig. S5† shows the HPLC profiles of all-PB and PB/PO chimeric tetramers, i.e., d(CPBAPBGPBT) 16 and d(CPBAPOGPBT) 17, respectively, which indicate that the desired products were obtained as the main products. Tetramers 16 and 17 were isolated in 28% and 33% yields, respectively (Table 3, entries 1 and 2). The isolated compounds were analyzed via1H NMR spectroscopy. The corresponding NMR spectra showed the characteristic signals of nucleobases and the borano groups (Fig. S9 and S10†). The successful synthesis of the PB/PO chimeric tetramer indicated that the PB linkages were stable under the oxidation conditions using TBHP.
In a similar manner, PB/PO chimeric dodecamer 18 containing alternate PB moieties was synthesized. Considering the number of protecting groups on the PO and PB moieties, a longer DBU treatment was performed.27,28 After RP-HPLC purification, dodecamer 18 was obtained in a 3% yield (Table 3, entry 3). The oligomer was identified via mass spectrometry. Peaks corresponding to deboronated and nucleotide deletion compounds were not detected. Unfortunately, the low yield of 18, which can be attributed to the inferior coupling efficiency after boronation, prevented us from recording a 1H NMR spectrum. Nevertheless, although there is room for improvement, the synthesis of chimeric oligonucleotides was achieved.
Conclusion
An efficient synthetic strategy for oligonucleotides was developed according to a nucleobase-unprotected approach using oxazaphospholidine monomers bearing a Thg group. One of the major obstacles of the nucleobase-unprotected synthesis is the difficult purification of the deoxyguanosine monomer. The introduction of a Thg group into the oxazaphospholidine ring facilitated the purification by increasing lipophilicity. Nucleobase phosphitylation during the condensation reaction was not observed, unlike the synthesis using nucleobase-unprotected phosphoramidite monomers.This synthetic strategy could be also applicable to the synthesis of stereocontrolled PB ODNs because the oxazaphospholidine method was originally developed to synthesize stereopure P-modified oligonucleotides. Further investigation on the synthesis of stereopure oligonucleotides containing PB, PS, and PO linkages based on this concept is now in progress.
Experimental section
General information
All the reactions were conducted under Ar atmosphere. Organic solvents were dried according to the relevant procedures. 1H NMR spectra were recorded at 400 MHz using tetramethylsilane (δ 0.0 ppm) as the internal standard in CDCl3 or at 500 MHz or 600 MHz with CH3CN (δ 2.06 ppm) as the internal standard in D2O. 13C NMR spectra were recorded at 101 MHz in CDCl3, which was used as the internal standard (δ 77.0 ppm). COSY, HMQC, and HMBC spectra were recorded on a 400 MHz spectrometer. The 31P NMR spectra were recorded at 162 MHz using H3PO4 (δ 0.0 ppm) as the external standard in CDCl3 or at 201 MHz using H3PO4 (δ 0.0 ppm) as the external standard in D2O. Analytical TLC was performed on commercial glass-coated 0.25 mm-thick silica gel plates. Silica gel column chromatography was performed using spherical, neutral, 63–210 μm silica gel unless otherwise noted. The solid-phase synthesis was conducted manually using a glass filter (10 × 50 mm) with a stopper at the top and a stopcock at the bottom as a reaction vessel, and the obtained compounds were analyzed and purified via RP-HPLC or RP-UPLC and identified via ESI MS.The RP-HPLC analysis was performed at 260 nm, 30 °C, and a flow rate of 0.5 mL min−1 using a C18 column (100 Å, 3.9 mm × 150 mm) unless otherwise noted. The RP-UPLC measurements were performed at 260 nm, 50 °C, and a flow rate of 0.5 mL min−1 using a C18 column 1.7 μm (2.1 mm × 50 mm) unless otherwise noted.
The amount of a loaded nucleoside on solid-support was estimated by the calculation of released 4,4′-dimethoxytrityl cation by a solution of 0.1 M TsOH/CH3CN using a molar absorption constant at 498 nm (ε = 76000 L mol−1 cm−1).33
The isolated yields of the synthesized oligomers were estimated via UV–vis spectroscopy. The PB/PO chimeric dodecamer was only characterized via ESI MS owing to the small quantities obtained. The isolation of the oxazaphospholidine monomer via RP-HPLC was conducted with detection at 260 nm, at rt, and a flow rate of 10 mL min−1 using a C18 column (120 Å) unless otherwise noted.
N-(3,7-Dimethyloctyl)-N-(2-hydroxy-2-phenylethyl)-2-nitrobenzenesulfonamide (2)
2-Amino-1-phenyl ethanol 1 (11.3 g, 83 mmol) was dried by performing repeated coevaporations with pyridine and toluene and then dissolved in dry THF (500 mL). Triethylamine (17.5 mL, 125 mmol) and TMSCl (15.7 mL, 124 mmol) were added to the mixture, which was then stirred for 2 h. Subsequently, the reaction was quenched by adding MeOH (5.5 mL), and the mixture was concentrated under reduced pressure to give a mixture of colorless crystals. The residue was dissolved in dry CH2Cl2 (160 mL) and cooled to 0 °C. After the addition of triethylamine (29.0 mL, 209 mmol), NsCl (19.3 g, 87.1 mmol) was added in ten installments, and the mixture was stirred for 2 h. The reaction was then quenched by adding MeOH (5 mL), and the mixture was concentrated under reduced pressure. CH2Cl2 (150 mL) was added to the residue, and the organic layer was washed with saturated aqueous solutions of NaHCO3 (3 × 300 mL). The aqueous layers were combined and extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a mixture of colorless crystals.The crude products were dissolved in dry THF (260 mL), and tetrahydrogeraniol (22.3 mL, 116 mmol) and PPh3 (44.0 g, 168 mmol) were then added. To this solution, a diisopropyl azodicarboxylate solution in dry THF (33.0 mL, 168 mmol per 150 mL) was added at 0 °C, and the mixture was then warmed to rt. After 24 h, the reaction was quenched by adding MeOH (7 mL) and concentrated under reduced pressure. A mixed solution of hexane–EtOAc (6:1, v/v 350 mL) was added to the residue and then cooled at 0 °C. Triphenyl phosphine oxide was removed by means of repeated precipitation, filtration, evaporation, dilution, and cooling. These operations were conducted three times. EtOAc (500 mL) was added to the residue, and the organic layer was washed with saturated aqueous solutions of NaCl (3 × 200 mL). The aqueous layers were combined and extracted with EtOAc (2 × 200 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated to give a light-yellow oil.
MeOH (41.5 mL) and a 3% TFA/CH2Cl2 solution (360 mL) were added to the crude product at rt. After 18 h, the mixture was washed with saturated aqueous solutions of NaHCO3 (3 × 200 mL). The aqueous layers were combined and extracted with CH2Cl2 (2 × 200 mL). The organic layers were combined and dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (neutral silica gel) using CHCl3–hexane (5:5–10:0, v/v) as an eluent to afford 2 as light yellow oil (32.4 g, 70 mmol, 85% from 1).
1H NMR (400 MHz, CDCl3) δ 8.05–8.02 (m, 1H, Ar), 7.72–7.60 (m, 3H, Ar), 7.36–7.26 (m, 5H, Ar), 4.90 (dt, J = 8.5, 3.5 Hz, 1H, Ph–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 3.56–3.29 (m, 4H, H-2, NC2), 2.77–2.75 (m, 1H, O, mixtures of diastereomers), 1.60–1.03 (m, 10H, C2 × 4, C × 2), 0.90–0.81 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 148.0, 141.1, 141.1, 133.6, 133.2, 131.6, 130.9, 128.6, 128.1, 125.8, 124.2, 79.0, 72.5, 72.3, 54.8, 54.7, 47.2, 47.0, 39.1, 36.9, 36.9, 34.8, 34.7, 30.6, 30.5, 28.0, 27.9, 24.5, 22.6, 22.5, 19.4, 19.3.
), 3.56–3.29 (m, 4H, H-2, NC2), 2.77–2.75 (m, 1H, O, mixtures of diastereomers), 1.60–1.03 (m, 10H, C2 × 4, C × 2), 0.90–0.81 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 148.0, 141.1, 141.1, 133.6, 133.2, 131.6, 130.9, 128.6, 128.1, 125.8, 124.2, 79.0, 72.5, 72.3, 54.8, 54.7, 47.2, 47.0, 39.1, 36.9, 36.9, 34.8, 34.7, 30.6, 30.5, 28.0, 27.9, 24.5, 22.6, 22.5, 19.4, 19.3.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C24H35N2O5S+, 463.2262; found 463.2262.
2-((3,7-Dimethyloctyl)amino)-1-phenylethan-1-ol (3)
Compound 2 (32.4 g, 70.0 mmol) was dissolved in dry DMF (350 mL), and 18-crown 6-ether (37.0 g, 140 mmol), K2CO3 (37.9 g, 274 mmol), and 4-mercaptobenzoic acid (19.0 g, 126 mmol) were added at rt. After 12 h, half of the solvent was removed under reduced pressure. The mixture was diluted with hexane–EtOAc (4:1, v/v, 300 mL) and then washed with 1.0 M NaOH aqueous solutions (5 × 300 mL). The aqueous layers were combined and extracted with hexane–EtOAc (4:1, v/v, 1 × 300 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified via silica gel column chromatography (neutral silica gel) using CHCl3–MeOH (100:0–97:3, v/v) as an eluent to afford 3 as a light-yellow oil (12.99 g, 46.8 mmol, 67% from 2).
1H NMR (400 MHz, CDCl3) δ 7.39–7.25 (m, 5H, Ar), 4.75 (dd, J = 9.1, 3.7 Hz, 1H, H-1), 2.93 (dt, J = 11.9, 3.2 Hz, 1H, H-2), 2.78–2.60 (m, 3H, H-2, NC2), 2.6–2.1 (br, 2H, O, N), 1.58–1.45 (m, 3H, C2 × 0.5, C × 2), 1.37–1.19 (m, 4H, C2 × 2), 1.18–1.05 (m, 3H, C2 × 1.5), 0.87 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 142.5, 128.4, 127.5, 125.8, 71.4, 71.4, 57.1, 57.0, 47.4, 47.3, 39.2, 37.3, 37.2, 30.8, 28.0, 24.7, 22.7, 22.6, 19.7.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C18H32NO+, 278.2479; found 278.2474.
General procedure for the synthesis of the 2-chloro-1,3,2-oxazaphospholidine derivative 4i and 4t
Compound 3i (0.67 g, 3.8 mmol) or compound 3t (13.9 g, 50 mmol) was dried by performing repeated coevaporations with toluene and dissolved in toluene (4.0 mL for the synthesis of 4i, 40.0 mL for 4t). N-Methylmorpholine (0.9 mL, 8.2 mmol for 4i, 11.5 mL, 105 mmol for 4t) was added to the solution, and the resulting solution was then added dropwise to a solution of phosphorus trichloride (0.9 mL, 3.9 mmol for 4i, 4.8 mL, 55 mmol for 4t) in toluene (2.5 mL for 4i, 40 mL for 4t) at 0 °C over 10 min. The mixture was warmed to rt, stirred for 2 h, and then filtered under Ar atmosphere at −78 °C. The filtrate was then concentrated to afford compound (4i; 1.18 g, 4t; 18.2 g) as a light-yellow oil, which was used for the phosphitylation without further purification.General procedure for the synthesis of oxazaphospholidine monomers 6a, 6c, 6i and 6g
5′-O-DMTr-deoxyadenosine (2.2 g, 4.0 mmol for the synthesis of 6a), 5′-O-DMTr-deoxycytidine (3.2 g, 6.0 mmol for the synthesis of 6c, 0.82 g, 1.5 mmol for 6i), or 5′-O-DMTr-deoxyguanosine (2.6 g, 4.5 mmol for the synthesis of 6c) was dried by performing repeated coevaporations with pyridine, toluene, and THF and then dissolved in dry THF (10 mL for the synthesis of 6a, 15 mL for 6c, 3.8 mL for 6i, and 10 mL for 6g). Triethylamine (3.9 mL, 28 mmol for 6a, 5.8 mL, 42 mmol for 6c, 1.5 mL, 10.8 mmol for 6i, and 4.4 mL, 32 mmol for 6g) was added, and the mixture was cooled to −78 °C for 6a and 6c, and 6i and to −40 °C for 6g. A 0.6 M solution of compound 4t in dry THF (17 mL for 6a, 25 mL for 6c, and 20 mL for 6g) or a 0.6 M solution of compound 4i in dry THF (6.3 mL for 6i) was added dropwise over 5 min and the mixture was stirred at −78 °C for 6a and 6c, and 6i and at −40 °C for 6g. After 2 h, the mixture was diluted with CHCl3 (20 mL) and the reaction was quenched by adding a saturated aqueous solution of NaHCO3 (5 mL). CHCl3 and a saturated aqueous of NaHCO3 were added to the mixture, and the organic layer was collected and washed with a saturated aqueous solution of NaHCO3 (3 × 50 mL). The aqueous layers were combined and extracted with CHCl3 (3 × 50 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was dried by performing coevaporations with toluene.Seven tenths of the crude product (2.75 g out of 3.92 g) obtained in the synthesis of 6a was purified via silica gel column chromatography (NH-silica gel) using toluene–EtOAc (10:0–8:2, v/v, containing 0.5% triethylamine) as an eluent to afford 6a as a colorless foam (0.82 g, 0.95 mmol, 34%).
1H NMR (400 MHz, CDCl3) δ 8.31, 8.30 (s, s, 1H, H-2, diastereomers), 7.99 (s, 1H, H-8), 7.41–7.16 (m, 14H, Ar), 6.80–6.74 (m, 4H, Ar), 6.48–6.44 (m, 1H, H-1′), 5.67 (brs, 2H, N2), 5.59–5.54 (m, 1H, 5-position of oxazaphospholidine), 5.01 (qd, J = 5.8, 2.9 Hz, 1H, H-3′), 4.23 (q, J = 3.4 Hz, 1H, H-4′), 3.77, 3.77, 3.74 (s, s, s, 6H, OC3 × 2, diastereomers), 3.58–3.34 (m, 3H, H-5′, 5′′, 4-position of oxazaphospholidine), 3.09–2.97 (m, 2H, NC2), 2.95–2.85 (m, 2H, 4-position of oxazaphospholidine, H-2′), 2.61–2.54 (m, 1H, H-2′′), 1.62–1.04 (m, 10H, C2 × 4, C × 2), 0.88–0.82 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 158.5, 158.5, 155.4, 153.0, 149.7, 149.7, 144.5, 144.5, 140.5, 139.0, 135.7, 130.0, 128.5, 128.2, 128.0, 127.8, 127.8, 126.9, 126.8, 125.8, 125.8, 120.1, 113.1, 86.5, 86.0 (d, 3JPC = 4.8 Hz), 85.8, 84.3, 81.5, 81.4, 81.2, 72.9 (d, 2JPC = 10.6 Hz), 72.8 (d, 2JPC = 9.6 Hz), 63.3, 54.5 (d, 2JPC = 4.8 Hz), 43.7 (d, 2JPC = 22.2 Hz), 43.3 (d, 2JPC = 21.2 Hz) 40.1, 39.8, 39.2, 37.3, 37.1, 37.0, 36.8, 36.7, 36.7, 36.6, 36.4, 36.4, 30.5, 30.5, 30.4, 27.9, 24.7, 22.7, 22.6, 19.6, 19.5, 19.4. 31P {1H} NMR (162 MHz, CDCl3) δ 142.7, 142.1, 141.9, 141.5.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C49H60N6O6P+, 859.4306; found 859.4292.
For 6c, forty-one hundredths of the crude product (2.50 g out of 6.11 g) was purified via silica gel column chromatography (NH-silica gel) using toluene–EtOAc–tBuOH (99:1, v/v, containing 0.5% triethylamine) as an eluent to afford 6c as a colorless foam (0.80 g, 0.96 mmol, 39%).
1H NMR (400 MHz, CDCl3) δ 7.92, 7.92, 7.92, 7.92 (d, d, d, d, J = 7.3 Hz, 1H, H-6, mixtures of diastereomers), 7.43–7.17 (m, 14H, Ar), 6.84–6.77 (m, 4H, Ar), 6.33–6.28 (m, 1H, H-1′), 5.51 (qd, J = 7.3, 2.4 Hz, 1H, (5-position of oxazaphospholidine), 5.42–5.34 (m, 1H, H-5, diastereomers), 4.90–4.81 (m, 1H, H-3′), 4.07–4.05 (m, H-4′), 3.77, 3.73, 3.71 (s, s, s, 6H, OC3 × 2, diastereomers), 3.50–3.36 (m, 3H, H-5′, 5′′, 4-position of oxazaphospholidine), 3.09–2.84 (m, 3H, 4-position of oxazaphospholidine, NC2), 2.60–2.53 (m, 1H, H-2′), 2.33–2.21 (m, 1H, H-2′′), 1.58–1.02 (m, 10H, C2 × 4, C × 2), 0.88–0.80 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 165.6, 158.5, 158.5, 158.5, 155.8, 155.7, 144.5, 144.4, 141.3, 141.2, 140.7 (d, 3JPC = 8.7 Hz), 140.6 (d, 3JPC = 9.6 Hz), 140.5 (d, 3JPC = 9.6 Hz), 140.4 (d, 3JPC = 9.6 Hz), 135.5, 135.4, 135.4, 130.1, 130.1, 130.0, 128.4, 128.3, 128.1, 127.9, 126.9, 126.9, 125.8, 125.8, 113.2, 94.0, 93.8, 86.7, 85.7, 85.2, 85.1, 85.0, 81.3 (d, 2JPC = 9.6 Hz), 81.2 (d, 2JPC = 9.6 Hz), 81.1 (d, 2JPC = 9.6 Hz), 81.0 (d, 2JPC = 9.6 Hz), 71.3 (d, 2JPC = 17.3 Hz), 71.2 (d, 2JPC = 18.3 Hz), 71.0, 70.9, 62.1, 61.9, 61.9, 55.2, 55.1, 54.6 (d, 2JPC = 3.9 Hz), 54.5 (d, 2JPC = 4.8 Hz), 54.3 (d, 2JPC = 4.8 Hz), 54.2 (d, 2JPC = 4.8 Hz), 43.6 (d, 2JPC = 22.2 Hz), 43.5 (d, 2JPC = 20.2 Hz), 43.1 (d, 2JPC = 20.2 Hz), 43.1 (d, 2JPC = 19.3 Hz), 39.2, 39.1, 37.3, 37.0, 37.0, 36.7, 36.6, 36.6, 36.4, 36.3, 36.3, 30.5, 30.4, 30.3, 30.3, 27.9, 24.6, 24.6, 22.7, 22.6, 19.7, 19.6, 19.5, 19.4. 31P {1H} NMR (162 MHz, CDCl3) δ 144.6, 144.0, 142.1, 141.6.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C48H60N4O7P+, 835.4194; found 835.4200.
All the crude product obtained in the synthesis of 6i was purified via silica gel column chromatography (NH-silica gel) using CHCl3–tBuOH (100:0–89:11, v/v, containing 0.5% triethylamine) as an eluent to afford 6i as a colorless foam (0.30 g, 0.41 mmol, 27%).
1H NMR (400 MHz, CDCl3) δ 8.04, 8.02 (d, d, J = 7.3 Hz, 1H, H-6, mixtures of diastereomers), 7.43–7.17 (m, 14H, Ar), 6.85–6.75 (m, 4H, Ar), 6.31, 6.28 (dd, J = 5.0 Hz, H-1′, 1H, mixtures of diastereomers) 5.49 (q, J = 7.0 Hz, 1H, 5-position of oxazaphospholidine), 5.30, 5.30 (d, J = 7.3 Hz, 1H, H-5, mixtures of diastereomers), 4.93–4.84 (m, 1H, H-3′), 4.07–4.05 (m, 1H, H-4′), 3.79, 3.74, 3.71 (s, s, s, 6H, OC3 × 2, diastereomers), 3.54–3.31 (m, 4H, H-5′, H-5′′ NC(CH3)2, 4-position of oxazaphospholidine), 2.89–2.82 (m, 1H, 4-position of oxazaphospholidine), 2.65–2.55 (m, 1H, H-2′), 2.38–2.26 (m, 1H, H-2′′), 1.21 (dd, J = 6.4, 4.1 Hz, 3H, NCH(C3)2), 1.14 (t, J = 6.9 Hz, 3H, NCH(C3)2). 13C {1H} NMR (101 MHz, CDCl3) δ 165.6, 165.5, 158.6, 158.5, 158.5, 155.8, 155.7, 144.5, 144.4, 141.4, 141.3, 140.5, 140.5, 140.3, 140.2, 135.5, 135.4, 135.4, 130.1, 130.0, 128.4, 128.2, 128.2, 128.0, 127.9, 127.9, 127.0, 126.9, 125.9, 113.2, 93.8, 93.7, 86.7, 86.7, 85.8, 85.7, 85.0, 84.9, 80.7 (d, 2JPC = 9.6 Hz), 80.6 (d, 2JPC = 8.7 Hz), 71.1, 71.0, 70.6, 70.4, 61.8, 55.2, 55.2, 51.2 (d, 2JPC = 4.8 Hz), 51.1 (d, 2JPC = 4.8 Hz), 46.5 (d, 2JPC = 9.6 Hz), 46.3 (d, 2JPC = 9.6 Hz), 41.3, 41.2, 41.0, 22.8 (d, 2JPC = 8.7 Hz), 22.7 (d, 2JPC = 8.7 Hz), 22.6 (d, 2JPC = 6.7 Hz), 22.5 (d, 2JPC = 6.7 Hz). 31P {1H} NMR (162 MHz, CDCl3) δ 143.8, 141.6.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C41H46N4O7P+, 737.3099; found 737.3100.
In the case of 6g, all the crude product (4.2 g) was purified via silica gel column chromatography (NH-silica gel) using CHCl3–tBuOH (100:0–93:7, v/v, containing 0.5% triethylamine) as an eluent to afford 6g as a colorless foam (1.59 g) containing 9% oxide and 1% hydrolysate. Afterward, the obtained foam (0.82 g out of 1.59 g) was purified via RP-HPLC (ODS column) using CH3CN–CHCl3 (85:15, v/v) as an eluent to afford 6g as a colorless foam (0.61 g, 0.70 mmol, 30%).
1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H, H-8), 7.43–7.15 (m, 14H, Ar), 6.81–6.75 (m, 4H, Ar), 6.27–6.23 (m, 1H, H-1′), 6.2–6.1 (br, 2H, N2), 5.57–5.54 (m, 1H, 5-position of oxazaphospholidine), 5.02–4.95 (m, 1H, H-3′), 4.21 (q, J = 3.5 Hz, 1H, H-4′), 3.74, 3.73, 3.71 (s, s, s, 6H, OC3 × 2, diastereomers), 3.55–3.31 (m, 3H, H-5′, 5′′, 4-position of oxazaphospholidine), 3.07–2.98 (m, 2H, NC2), 2.96–2.89 (m, 1H, 4-position of oxazaphospholidine), 2.86–2.77 (m, 1H, H-2′), 2.52–2.45 (m, 1H, H-2′′), 1.61–1.08 (m, 10H, C2 × 4, C × 2), 0.88–0.80 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 159.2, 158.5, 158.4, 153.6, 151.5, 144.5, 144.5, 140.6, 140.5, 135.6, 135.6, 130.1, 128.7, 128.4, 128.3, 128.2, 128.0, 127.9, 127.8, 126.9, 126.9, 126.7, 125.8, 125.8, 117.6, 113.1, 86.4, 85.8 (d, 3JPC = 3.9 Hz), 85.6, 83.9, 81.4, 81.3 (d, 2JPC = 10.6 Hz), 73.0 (d, 2JPC = 12.5 Hz), 63.2, 55.2, 54.8, 54.5 (d, 2JPC = 5.8 Hz), 54.5 (d, 2JPC = 4.8 Hz), 43.7 (d, 2JPC = 22.2 Hz), 43.3 (d, 2JPC = 20.2 Hz), 43.3 (d, 2JPC = 20.2 Hz), 39.6, 39.4, 39.2, 37.3, 37.1, 37.0, 36.8, 36.7, 36.6, 36.5, 36.4, 36.3, 30.7, 30.5, 30.5, 30.3, 27.9, 24.7, 22.7, 22.6, 22.6, 19.6, 19.5, 19.4, 19.4. 31P {1H} NMR (162 MHz, CDCl3) δ 142.8, 142.3, 142.2, 141.8.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C49H60N6O7P+, 875.4256; found 875.4256.
General procedure for the synthesis of oxazaphospholidine monomers 6t and 6l
5′-O-DMTr-thymidine (1.4 g, 2.5 mmol) or 5′-O-DMTr-2′-O-4′-C-locked-thymidine (1.7 g, 3.0 mmol) was dried by performing repeated coevaporations with pyridine, toluene, and THF and then dissolved in dry THF (6.3 mL for the synthesis of 6t and 7.5 mL for 6l). Triethylamine (2.4 mL, 28 mmol for 6t and 2.9 mL, 21 mmol for 6l) was added, and the mixture was cooled to −78 °C. A 0.6 M solution of compound 4t in dry THF (10 mL for 6t and 13 mL for 6l) was added dropwise over 5 min, and the mixture was stirred at rt. After 2 h, the mixture was diluted with CHCl3 (20 mL) and the reaction was quenched by adding a saturated aqueous solution of NaHCO3 (5 mL). CHCl3 and a saturated aqueous of NaHCO3 were added to the mixture, and the organic layer was collected and washed with a saturated aqueous solution of NaHCO3 (3 × 50 mL). The aqueous layers were combined and extracted with CHCl3 (3 × 50 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was dried by performing coevaporations with toluene.All the crude product (2.32 g) obtained in the synthesis of 6t was purified via silica gel column chromatography (NH-silica gel) using toluene–EtOAc (10:0–8:2, v/v, containing 0.5% triethylamine) as an eluent to afford 6t as a colorless foam (1.02 g, 1.20 mmol, 55%).
1H NMR (400 MHz, CDCl3) δ 8.7–8.3 (br, 1H, N-3), 7.63–7.61 (m, 1H, H-6), 7.42–7.19 (m, 14H, Ar), 6.85–6.79 (m, 4H, Ar), 6. 42 (dd, J = 7.5, 6.2 Hz, 1H, H-1′), 5.51–5.45 (m, 1H, 5-position of oxazaphospholidine), 4.92–4.86 (m, 1H, H-3′), 4.11–4.07 (m, 1H, H-4′), 3.77, 3.75, 3.74 (s, s, s, 6H, OC3 × 2, diastereomers), 3.53–3.33 (m, 3H, H-5′, 5′′, 4-position of oxazaphospholidine), 3.07–2.95 (m, 2H, NC2), 2.92–2.83 (m, 1H, 4-position of oxazaphospholidine), 2.47–2.29 (m, 2H, H-2′, 2′′), 1.60–1.01 (m, 13H, C2 × 4, C × 2, thymine-5-C3), 0.88–0.80 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 163.6, 158.7, 150.3, 144.3, 144.2, 140.4, 135.6, 135.3, 135.3, 130.1, 130.1, 128.5, 128.2, 128.0, 128.0, 127.2, 127.1, 125.9, 113.2, 111.2, 86.9, 85.8 (d, 3JPC = 4.8 Hz), 85.7, 84.6, 81.3 (d, 2JPC = 9.6 Hz), 81.3 (d, 2JPC = 10.1 Hz), 72.9 (d, 2JPC = 15.4 Hz), 72.6 (d, 2JPC = 13.5 Hz), 63.1, 63.0, 54.7 (d, 2JPC = 4.8 Hz), 54.6 (d, 2JPC = 4.8 Hz), 54.4 (d, 2JPC = 4.8 Hz), 54.3 (d, 2JPC = 5.8 Hz), 43.6 (d, 2JPC = 21.2 Hz), 43.3 (d, 2JPC = 20.2 Hz), 40.5, 40.3, 39.2, 37.3, 37.1, 37.0, 36.7, 36.4, 30.5, 30.4, 30.4, 30.4, 27.9, 24.6, 22.7, 22.6, 19.6, 19.5, 19.4, 11.7, 11.7. 31P {1H} NMR (162 MHz, CDCl3) δ 143.9, 143.4, 142.5, 142.1.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C49H61N3O8P+, 850.4191; found 850.4203.
For 6l, ninety-five hundredths of the crude product (2.95 g out of 3.10 g) was purified via silica gel column chromatography (NH-silica gel) using toluene–EtOAc (10:0–8:2, v/v, containing 0.5% triethylamine) as an eluent to afford 6l as a colorless foam (0.82 g, 0.93 mmol, 32%).
1H NMR (400 MHz, CDCl3) δ 9.1–8.7 (br, 1H, N-3), 7.74–7.73 (m, 1H, H-6), 7.48–7.45 (m, 2H, Ar), 7.37–7.17 (m, 12H, Ar), 6.85–6.82 (m, 2H, Ar), 6.78–6.73 (m, 2H, Ar), 5.66 (d, J = 9.6 Hz, 1H, H-1′), 5.54 (t, J = 7.3 Hz, 0.5 H, 5-position of oxazaphospholidine), 5.40 (t, J = 7.3 Hz, 0.5 H, 5-position of oxazaphospholidine), 4.61–4.58 (m, 1H, H-3′), 4.53, 4.47 (s, s, 1H, H-2′, diastereomers), 3.86–3.68 (m, 8H, OC3 × 2, LNA–C2), 3.61–3.49 (m, 1.5H, H-5′, 5′′), 3.41–3.38 (m, 1.5H, H-5′′, 4-position of oxazaphospholidine), 3.12–2.79 (m, 3H, 4-position of oxazaphospholidine, NC2), 1.59–1.00 (m, 13H, C2 × 4, C × 2, thymine-5-C3), 0.89–0.73 (m, 9H, C3 × 3). 13C {1H} NMR (101 MHz, CDCl3) δ 163.9, 163.8, 158.7, 158.6, 158.5, 149.8, 149.7, 144.4, 144.1, 140.5 (d, 3JPC = 2.9 Hz), 140.4 (d, 3JPC = 2.9 Hz), 140.1 (d, 3JPC = 2.9 Hz), 139.9 (d, 3JPC = 2.9 Hz), 135.4, 135.3, 135.2, 134.6, 134.5, 130.2, 130.1, 128.5, 128.4, 128.3, 128.2, 128.0, 127.9, 127.1, 127.0, 126.8, 125.9, 125.9, 125.7, 113.2, 113.2, 113.2, 113.1, 110.5, 110.5, 88.0, 87.9 (d, 3JPC = 2.9 Hz), 87.2, 86.8, 86.7, 81.7 (d, 2JPC = 9.6 Hz), 81.6 (d, 2JPC = 10.6 Hz), 81.2 (d, 2JPC = 9.6 Hz), 81.2 (d, 2JPC = 9.6 Hz), 78.9, 78.8, 78.8, 78.7, 71.9, 70.8, 70.7, 69.9, 69.8, 69.8, 69.7, 58.0, 57.8, 55.1, 55.1, 54.3 (d, 2JPC = 5.8 Hz), 54.2 (d, 2JPC = 3.9 Hz), 53.9 (d, 2JPC = 4.8 Hz), 53.9 (d, 2JPC = 4.8 Hz), 43.3 (d, 2JPC = 19.3 Hz), 43.1 (d, 2JPC = 21.1 Hz), 42.9 (d, 2JPC = 19.3 Hz), 42.8 (d, 2JPC = 20.3 Hz), 39.1, 37.4, 37.3, 37.1, 37.0, 36.6, 36.5, 36.4, 36.3, 36.3, 36.2, 30.4, 30.3, 30.2, 27.9, 24.6, 24.6, 22.7, 22.6, 22.5, 19.5, 19.4, 19.3, 12.4, 12.3. 31P {1H} NMR (162 MHz, CDCl3) δ 145.3, 144.9, 142.3, 141.7.
HRMS (ESI-QTOF) m/z: [M + H]+ calcd for C50H61N3O9P+, 878.4140; found 878.4141.
General procedure for the synthesis of dinucleoside phosphates 10a–l
The HCP-loaded 5′-O-DMTr-Th (30.3 μmol g−1, 0.50 μmol), via a succinyl linker, was treated in a reaction vessel with 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each) and washed with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL). The product was dried in vacuo for 5 min. Then, the oxazaphospholidine monomer (6a, 6c, 6g, 6t, 6i, or 6l, 30 μmol), which was dried in vacuo overnight, was added to the reaction vessel and dried in vacuo for 5 min. A 1.0 M solution of PhIMT (44.1 mg, 150 μmol) in dry CH3CN–iPrCN (7:3, v/v, 150 μL), which was dried over MS 3 Å overnight, was added under Ar atmosphere to the reaction vessel. After 10 min, the HCP was washed with dry CH3CN (3 × 1 mL) and dry CH2Cl2 (3 × 1 mL) and dried in vacuo for 5 min. The resultant phosphite was oxidized via treatment with a 1.0 M solution of TBHP (500 μL, 500 μmol) in dry toluene, and the reaction vessel was shaken for 5 min. Then, the HCP was washed with dry CH2Cl2 (6 × 1 mL) and the detritylation reaction was conducted using 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each). The HCP was then washed with dry CH2Cl2 (3 × 1 mL) and dry CH3CN (3 × 1 mL), treated with a 25% NH3 aqueous solution–EtOH (3:1, v/v, 5 mL) at rt for 3 h, filtered, and washed with CH3CN. The filtrate and the washings were combined, concentrated under reduced pressure, and the obtained residue was analyzed via RP-HPLC, which was performed with a linear gradient of 0%–20% CH3CN for 60 min in a 0.1 M TEAA buffer (pH 7.0).
General procedure for the synthesis of oligonucleotide 11 bearing phosphate linkages
The HCP-loaded 5′-O-DMTr-Th (30.3 μmol g−1, 0.50 μmol), via a succinyl linker, was treated in a reaction vessel with 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each), washed with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL), and dried in vacuo for 5 min. Then, the oxazaphospholidine monomer (6a, 6c, 6g, or 6t, 30 μmol), which was dried in vacuo overnight, was added to the reaction vessel and dried in vacuo for 5 min. A 1.0 M solution of PhIMT (44.1 mg, 150 μmol) in dry CH3CN–iPrCN (7:3, v/v, 150 μL), which was dried over MS 3 Å overnight, was added under Ar atmosphere to the reaction vessel. After 10 min, the HCP was washed with dry CH3CN (3 × 1 mL) and dry CH2Cl2 (3 × 1 mL) and dried in vacuo for 5 min. The resultant phosphite was oxidized with a 1.0 M solution of TBHP (500 μL, 1000 μmol) in dry toluene, and the reaction vessel was shaken for 5 min. After washing the HCP with dry CH2Cl2 (6 × 1 mL), the detritylation reaction was conducted using 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each). Then, the HCP was washed with dry CH2Cl2 (3 × 1 mL) and dry CH3CN (3 × 1 mL).
The cycles of detritylation, condensation, and oxidation were repeated. After extending the chain length, the HCP was treated with a 25% NH3 aqueous solution–EtOH (3:1, v/v, 5 mL) at rt for 17 h, filtered, and washed with CH3CN. The filtrate and the washings were combined and concentrated under reduced pressure, and the obtained residue was analyzed via RP-UPLC, which was performed with a linear gradient of 5%–25% MeOH for 10 min in 0.4 M 1,1,1,3,3,3-hexafluoro-2-propanol and 16 mM triethylamine at 50 °C. The purification was conducted with four-fifths of the crude mixture, and the quantity of purified 11 was estimated by measuring the UV absorption at 260 nm. Isolated yield: 14% (11, 57 nmol).
11: HRMS (ESI-QTOF) m/z: [M − 4H]4− calcd for C117H143N45O70P114−, 727.9238; found 727.9209.
1H NMR (500 MHz, D2O) δ 8.19–7.41 (m, 15H, nucleobase), 6.20–5.89 (m, 15H, H-1′, nucleobase), 4.98–3.61 (m, 48H, H-3′, H-4′, H-5′), 3.22–3.18 (q, 66H, TEA), 2.69–2.63 (m, 12H, H-2′), 2.35–2.11 (m, 12H, H-2′), 1.72–1.67 (s, s, 9H, nucleobase), 1.29–1.26 (q, 99H, TEA). 31P {1H} NMR (201 MHz, D2O) −0.48 to −0.61.
General procedure for the synthesis of dinucleoside boranophosphates 14a–t
The HCP-loaded 5′-O-DMTr-Th (29.6 μmol g−1, 0.50 μmol), via a succinyl sarcosinyl linker, was treated in a reaction vessel with 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each), washed with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL), and dried in vacuo for 5 min. Then, the oxazaphospholidine monomer (6a, 6c, 6g, or 6t, 30 μmol), which was dried in vacuo overnight, was added to the reaction vessel and dried in vacuo for 5 min. A 1.0 M solution of PhIMT (44.1 mg, 150 μmol) in dry CH3CN–iPrCN (7:3, v/v, 150 μL), which was dried over MS 3 Å overnight, was added under Ar atmosphere to the reaction vessel. After 10 min, the HCP was washed with dry CH3CN (3 × 1 mL) and dry CH2Cl2 (3 × 1 mL) and dried in vacuo for 5 min.
The resultant phosphite was boronated using a 1.0 M solution of BH3·THF (50 μL, 50 μmol) and dry THF (950 μL), and the reaction vessel was shaken for 2 min. Afterward, the HCP was washed with dry THF (3 × 1 mL) and dry CH2Cl2 (3 × 1 mL) and the detritylation was performed using 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (4 × 5 s and 1 × 40 s, 1 mL each). After washing the HCP with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL) and drying in vacuo for 5 min, DMAP (10 mg, 82 μmol), dry 2,6-lutidine (450 μL), and Ac2O (50 μL, 529 μmol) were sequentially added, and the mixture was shaken for 1 min. Subsequently, the HCP was washed with dry CH2Cl2 (3 × 1 mL) and dry CH3CN (3 × 1 mL) and dried in vacuo for 5 min. Dry CH3CN (450 μL) and DBU (50 μL, 335 μmol) were added, and the reaction vessel was shaken for 1 h. The HCP was washed with dry CH3CN (6 × 1 mL), treated with a 25% NH3 aqueous solution–EtOH (3:1, v/v, 5 mL) at rt for 17 h (or at rt for 3 h and then 50 °C for 17 h for the synthesis of dAPBT), filtered, and washed with CH3CN. The filtrate and the washings were combined and concentrated under reduced pressure, and the obtained residue was analyzed via RP-HPLC with a linear gradient of 0%–20% CH3CN for 60 min in a 0.1 M TEAA buffer (pH 7.0).
General procedure for the synthesis of oligonucleotides bearing PB linkages: d(CPBCPBT) 15, d(CPBAPBGPBT) 16, d(CPBAPOGPBT) 17, and d(CPBAPOGPBTPOCPBAPOGPBTPOCPBAPOGPBT) 18
The HCP-loaded 5′-O-DMTr-Th (29.6 μmol g−1, 0.50 μmol), via a succinyl sarcosinyl linker, was treated in a reaction vessel with 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each), washed with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL), and dried in vacuo for 5 min. Then, the oxazaphospholidine monomer (6a, 6c, 6g, or 6t, 30 μmol), which was dried in vacuo overnight, was added to the reaction vessel and dried in vacuo for 5 min. A 1.0 M solution of PhIMT (44.1 mg, 150 μmol) in dry CH3CN–iPrCN (7:3, v/v, 150 μL), which was dried over MS 3 Å overnight, was added under Ar atmosphere to the reaction vessel. After 10 min, the HCP was washed with dry CH3CN (3 × 1 mL) and dry CH2Cl2 (3 × 1 mL) and dried in vacuo for 5 min. The resultant phosphite was boronated using a 1.0 M solution of BH3·THF (50 μL, 50 μmol) and dry THF (950 μL), and the reaction vessel was shaken for 2 min. After washing the HCP with dry THF (3 × 1 mL), dry EtOH (3 × 1 mL), and dry CH2Cl2 (3 × 1 mL), the detritylation was conducted using 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (4 × 5 s and 1 × 40 s, 1 mL each). The HCP was washed with dry CH2Cl2 (3 × 1 mL) and CH3CN (3 × 1 mL) and then dried in vacuo for 5 min. The cycles of detritylation, condensation, and boronation or oxidation were repeated. After extending the chain length, DMAP (10 mg, 82 μmol), dry 2,6-lutidine (450 μL), and Ac2O (50 μL, 529 μmol) were successively added and the vessel was shaken for 1 min. Subsequently, the HCP was washed with dry CH2Cl2 (3 × 1 mL) and dry CH3CN (3 × 1 mL) and then dried in vacuo for 5 min. Dry CH3CN (450 μL) and DBU (50 μL, 335 μmol) were added, and the reaction vessel was shaken for 1 h. The HCP was washed with dry CH3CN (6 × 1 mL), treated with a 25% NH3 aqueous solution–EtOH (3:1, v/v, 5 mL) at rt for 17 h (for the synthesis of 15) or at rt for 3 h and then 50 °C for 17 h (for the synthesis of 16–18), filtered, and washed with CH3CN. The filtrate and the washings were combined and concentrated under reduced pressure, and the obtained residue of trimer 15, tetramers 16 and 17, or dodecamer 18 was analyzed via RP-HPLC using a linear gradient of 0%–20% CH3CN for 60 min in a 0.1 M TEAA buffer (pH 7.0) for trimer 15 or 0%–60% CH3CN for 60 min in a 0.1 M TEAA buffer (pH 7.0) for tetramers 16 and 17. The purification of tetramers 16 and 17 was conducted with one-fifth of the crude mixture and the quantity of purified 16 and 17 was estimated by measuring the UV absorption at 260 nm. Isolated yield: 28% and 33% for 16 (25 nmol) and 17 (30 nmol), respectively.
16: HRMS (ESI-QTOF) m/z: [M − 2H]2− calcd for C39H57B3N15O19P32−, 582.6729; found 582.6742.
17: HRMS (ESI-QTOF) m/z: [M − 2H]2− calcd for C39H59B2N15O20P32−, 583.6540; found 583.6559.
The residue obtained in the synthesis of dodecamer 18 was analyzed via RP-HPLC using a linear gradient of 5%–40% MeOH for 20 min in 0.4 M 1,1,1,3,3,3-hexafluoro-2-propanol and 8 mM triethylamine at 60 °C. The purification was conducted with one-twentieth of the crude mixture, and the quantity of purified 18 was estimated by measuring the UV absorption at 260 nm. Isolated yield: 3% (18, 0.8 nmol).
18: HRMS (ESI-QTOF) m/z: [M − 4H]4− calcd for C117H6B2 N45O64P114−, 906.9642; found 906.9630.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Ms Noriko Sawabe (Tokyo University of Science) for her technical assistance with the NMR measurements. We also thank Dr Yayoi Yoshimura (Tokyo University of Science) for the mass spectra measurements. We would like to thank Enago (https://www.enago.jp) for the English language review. This research was supported by JSPS KAKENHI Grant Numbers JP21H02610.References
- S. L. Beaucage and M. H. Caruthers, Deoxynucleoside Phosphoramidites-A New Class of Key Intermediates for Deoxypolynucleotide Synthesis, Tetrahedron Lett., 1981, 22(20), 1859–1862, DOI:10.1016/S0040-4039(01)90461-7.
- J. B. Lambert, Tetrahedron Report Number 273, Tetrahedron, 1990, 46(8), 2677–2689, DOI:10.1016/s0040-4020(01)88362-9.
- S. M. Gryaznov and R. L. Letsinger, Synthesis of Oligonucleotides via Monomers with Unprotected Bases, J. Am. Chem. Soc., 1991, 113(15), 5876–5877, DOI:10.1021/ja00015a059.
- L. A. Marky, J. G. Snyder and K. J. Breslauer, Volume 11 Number 16 1983 Nucleic Acids Research, Nucleic Acids Res., 1983, 11(16), 5701–5715 CrossRef CAS PubMed.
- P. S. Miller, J. Yano, E. Yano, C. Carroll, K. Jayaraman and P. O. P. Ts'o, Nonionic Nucleic Acid Analogues. Synthesis and Characterization of Dideoxyribonucleoside Methylphosphonates, Biochemistry, 1979, 18(23), 5134–5143, DOI:10.1021/bi00590a017.
- A. Ohkubo, Y. Kuwayama, T. Kudo, H. Tsunoda, K. Seio and M. Sekine, O-Selective, Condensation Using P-N Bond Cleavage in RNA Synthesis without Base Protection, Org. Lett., 2008, 10(13), 2793–2796, DOI:10.1021/ol800911b.
- Y. Hayakawa and M. Kataoka, Facile Synthesis of Oligodeoxyribonucleotides via the Phosphoramidite Method without Nucleoside Base Protection, J. Am. Chem. Soc., 1998, 120(48), 12395–12401, DOI:10.1021/ja973731g.
- M. Sekine, Proton-Block Strategy for the Synthesis of Oligodeoxynucleotides without Base Protection, Capping Reaction, and P-N Bond Cleavage Reaction, J. Org. Chem., 2003, 5478–5492 CrossRef CAS PubMed.
- T. Wada, Y. Maizuru, M. Shimizu, N. Oka and K. Saigo, Stereoselective Synthesis of Dinucleoside Boranophosphates by an Oxazaphospholidine Method, Bioorg. Med. Chem. Lett., 2006, 16(12), 3111–3114, DOI:10.1016/j.bmcl.2006.03.076.
- N. Oka, Y. Maizuru, M. Shimizu and T. Wada, Solid-Phase Synthesis of Oligodeoxyribonucleotides without Base Protection Utilizing o-Selective Reaction of Oxazaphospholidine Derivatives, Nucleosides, Nucleotides Nucleic Acids, 2010, 29(2), 144–154, DOI:10.1080/15257771003612839.
- Y. Nukaga, N. Oka and T. Wada, Stereocontrolled Solid-Phase Synthesis of Phosphate/Phosphorothioate (PO/PS) Chimeric Oligodeoxyribonucleotides on an Automated Synthesizer Using an Oxazaphospholidine-Phosphoramidite Method, J. Org. Chem., 2016, 81(7), 2753–2762, DOI:10.1021/acs.joc.5b02793.
- R. P. Iyer, D. Yu, N. H. Ho, W. Tan and S. Agrawal, A Novel Nucleoside Phosphoramidite Synthon Derived from 1R, 2S-Ephedrine, Tetrahedron: Asymmetry, 1995, 6(5), 1051–1054, DOI:10.1016/0957-4166(95)00122-6.
- I. H. Hall, B. S. Burnham, K. G. Rajendran, S. Y. Chen, A. Sood, B. F. Spielvogel and B. R. Shaw, Hypolipidemic Activity of Boronated Nucleosides and Nucleotides in Rodents, Biomed. Pharmacother., 1993, 47(2–3), 79–87, DOI:10.1016/0753-3322(93)90295-V.
- D. S. Sergueev and B. R. Shaw, H-Phosphonate, Approach for Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates and Their Characterization, J. Am. Chem. Soc., 1998, 120(37), 9417–9427, DOI:10.1021/ja9814927.
- Z. A. Sergueeva, D. S. Sergueev and B. R. Shaw, Rapid and Selective Reduction of Amide Group by Borane-Amine Complexes in Acyl Protected Nucleosides, Nucleosides, Nucleotides Nucleic Acids, 2000, 19(1–2), 275–282, DOI:10.1080/15257770008033009.
- D. S. Sergueev, Z. A. Sergueeva and B. R. Shaw, Synthesis of Oligonucleoside Boranophosphates via an H-Phosphonate Method without Nucleobase Protection, Nucleosides, Nucleotides Nucleic Acids, 2001, 20(4–7), 789–795, DOI:10.1081/NCN-100002431.
- D. Takahashi, T. Inomata and T. Fukui, AJIPHASE®: A Highly Efficient Synthetic Method for One-Pot Peptide Elongation in the Solution Phase by an Fmoc Strategy, Angew. Chem., Int. Ed., 2017, 56(27), 7803–7807, DOI:10.1002/anie.201702931.
- T. Kan and T. Fukuyama, Ns Strategies: A Highly Versatile Synthetic Method for Amines, Chem. Commun., 2004, 4(4), 353–359, 10.1039/b311203a.
- M. Matoba, T. Kajimoto and M. Node, Application of Odorless Thiols for the Cleavage of 2- and 4-Nitrobenzenesulfonamides, Synth. Commun., 2008, 38(8), 1194–1200, DOI:10.1080/00397910701866098.
- S. Obika, D. Nanbu, Y. Hari, K. I. Morio, Y. In, T. Ishida and T. Imanishi, Synthesis of 2′-O,4′-C-Methyleneuridine and -Cytidine. Novel Bicyclic Nucleosides Having a Fixed C3-Endo Sugar Puckering, Tetrahedron Lett., 1997, 38(50), 8735–8738, DOI:10.1016/S0040-4039(97)10322-7.
- C. McCollum and A. Andrus, An Optimized Polystyrene Support for Rapid, Efficient Oligonucleotide Synthesis, Tetrahedron Lett., 1991, 32(33), 4069–4072, DOI:10.1016/S0040-4039(00)79865-0.
- Y. Hayakawa, R. Kawai, A. Hirata, J. I. Sugimoto, M. Kataoka, A. Sakakura, M. Hirose and R. Noyori, Acid/Azole Complexes as Highly Effective Promoters in the Synthesis of DNA and RNA Oligomers via the Phosphoramidite Method, J. Am. Chem. Soc., 2001, 123(34), 8165–8176, DOI:10.1021/ja010078v.
- N. Oka, T. Kondo, S. Fujiwara, Y. Maizuru and T. Wada, Stereocontrolled Synthesis of Oligoribonucleoside Phosphorothioates by an Oxazaphospholidine Approach, Org. Lett., 2009, 11(4), 967–970, DOI:10.1021/ol802910k.
- K. Sato, Y. Nukaga and T. Wada, Solid-Phase Synthesis and Properties of Stereocontrolled Boranophosphate/Phosphate and Phosphorothioate/Phosphate Chimeric Oligouridylates, R. Soc. Open Sci., 2023, 10(4), 230095, DOI:10.1098/rsos.230095.
- A. Mathematics, Mechanistic Studies on the Phosphoramidite Coupling Reaction in Oligonucleotide Synthesis. I. Evidence for Nudeophilic Catalysis by Tetrazole and Rate Variations with the Phosphorus Substituents, Nucleic Acids Res., 1987, 15(4), 1–23 Search PubMed.
- T. Brown, C. E. Pritchard, G. Turner and S. A. Salisburyb, A New Base-Stable Linker for Solid-Phase Oligonucleotide Synthesis, J. Chem. Soc., Chem. Commun., 1989, 7–9 CAS.
- N. Oka, T. Wada and K. Saigo, Diastereocontrolled Synthesis of Dinucleoside Phosphorothioates Using a Novel Class of Activators, Dialkyl(Cyanomethyl)Ammonium Tetrafluoroborates, J. Am. Chem. Soc., 2002, 124(18), 4962–4963, DOI:10.1021/ja017275e.
- N. Oka, T. Wada and K. Saigo, An Oxazaphospholidine Approach for the Stereocontrolled Synthesis of Oligonucleoside Phosphorothioates, J. Am. Chem. Soc., 2003, 125(27), 8307–8317, DOI:10.1021/ja034502z.
- Z. A. Sergueeva, D. S. Sergueev and B. R. Shaw, Borane-Amine Complexes - Versatile Reagents in the Chemistry of Nucleic Acids and Their Analogs, Nucleosides, Nucleotides Nucleic Acids, 2001, 20(4–7), 941–945, DOI:10.1081/NCN-100002464.
- M. Shimizu, K. Saigo and T. Wada, Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates by the Boranophosphotriester Method, J. Org. Chem., 2006, 71(11), 4262–4269, DOI:10.1021/jo0603779.
- K. Sato, T. Hagio, M. Sano, K. Muramoto, A. Yaoita, M. Noro, R. I. Hara and T. Wada, Solid-Phase Stereocontrolled Synthesis of Oligomeric P-Modified Glycosyl Phosphate Derivatives Using the Oxazaphospholidine Method, ACS Omega, 2021, 6(30), 20026–20041, DOI:10.1021/acsomega.1c03058.
- R. I. Hara, T. Saito, T. Kogure, Y. Hamamura, N. Uchiyama, Y. Nukaga, N. Iwamoto and T. Wada, Stereocontrolled Synthesis of Boranophosphate DNA by an Oxazaphospholidine Approach and Evaluation of Its Properties, J. Org. Chem., 2019, 84(12), 7971–7983, DOI:10.1021/acs.joc.9b00658.
- U. A. Spitzer, T. W. Toone and R. Stewart, Aqueous Trifluoroacetic Acid as a Medium for Organic Reactions. I. Acidity Functions and the Identity of the Manganese(VII) Species Found in Powerfully Acidic Media, Can. J. Chem., 1976, 54(3), 440–447, DOI:10.1139/v76-060.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC profiles, copies of 1H, 13C, 31P NMR, COSY, HMQC and HMBC spectra. See DOI: https://doi.org/10.1039/d3ob01255g |
| This journal is © The Royal Society of Chemistry 2023 |