
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of amphiphilic hydantoin-based universal peptidomimetics as antibiotic agents†
Alessio M.
Caramiello
 a,
Maria Cristina
Bellucci
b,
Emerenziana
Ottaviano
c,
Silvia
Ancona
c,
Elisa
Borghi
c and
Alessandro
Volonterio
*a
a,
Maria Cristina
Bellucci
b,
Emerenziana
Ottaviano
c,
Silvia
Ancona
c,
Elisa
Borghi
c and
Alessandro
Volonterio
*a
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, via Mancinelli 7, 20131 Milano, Italy. E-mail: alessandro.volonterio@polimi.it
bDepartment of Food, Environmental and Nutritional Sciences, Università degli Studi di Milano, via Celoria 2, 20131 Milano, Italy
cDepartment of Health Sciences, Università degli Studi di Milano, via Di Rudinì 8, 20142, Milan, Italy
First published on 31st August 2023
Abstract
Three model hydantoin-based universal peptidomimetics were designed and synthetized. Their preferred amphiphilic β-turn conformation was assessed using molecular modeling and NMR experiments, and their antibacterial activity was tested against Gram-positive and Gram-negative bacteria strains, which demonstrated that these compounds could be a captivating class of antibiotics to fight emergent drug resistance.
One of the most serious public health issues of the last four decades is antibiotic resistance.1 Indeed, the capability of microbes to evolve mechanisms that protect them from the effects of antimicrobials, along with the misuse and over-prescription of antibiotics, are among the most important factors responsible for the alarming increase in resistant strains.2 Moreover, despite the fact that the research activity oriented toward the discovery of new antibiotics is on the rise in both academia and industry, the discovery of new effective antibiotics is dramatically decreasing.3 For these reasons, there is a great interest in the search for new scaffolds that can overcome multidrug-resistant infections in the antibacterial research community. In medicinal chemistry, nature is often the first source of inspiration in the search for active scaffolds.4 Among the different classes of natural antibiotics, such as aminoglycosides, β-lactams, and macrolides, among others, antimicrobial peptides (AMPs) have attracted significant research interest to fight drug resistance.5 AMPs are associated with various advantages, the most important being their range of activities,6 the possibility of varying their chemical functionalities using natural (or even unnatural) amino acids, and the lower capacity of bacteria to develop resistance toward them.7 Nevertheless, AMPs have experienced little clinical success, mainly due to their low enzymatic proteolytic stability and bioavailability, high toxicity, and high cost of production.8 In order to overcome these disadvantages, the design of small-molecule peptidomimetics or short di- or tripeptides has recently emerged as a promising strategy to fight bacterial resistance.9 Indeed, taking into consideration the general proposed mechanism of action of AMPs, which relies on an initial electrostatic interaction between the negatively charged bacterial membrane and the cationic moieties of the peptide, followed by the intercalation of the hydrophobic residues into the lipophilic interior of the cell,6 it is possible to rationally design small-molecule peptidomimetics with the right balance and orientation of hydrophilic and hydrophobic residues. An outstanding example is brilacidin 1 (Fig. 1), a defensin peptidomimetic that has completed phase II clinical trials for the treatment of bacterial skin infection, and whose activity probably depends on its planar structure, which is able to project the cationic moieties (guanidino groups) and the lipophilic substituents (trifluoromethyl groups) in a position mimicking the secondary structure of defensins.10 On the other hand, short tripeptides able to assume turn conformations could cross Gram-negative bacterial membranes thanks to oligopeptide transporters.11 For instance, short arginine-based tripeptides with a central hydrophobic residue (2, Fig. 1) showed selective activity against Pseudomonas aeruginosa thanks to their ability to exist in a β-turn conformation.12
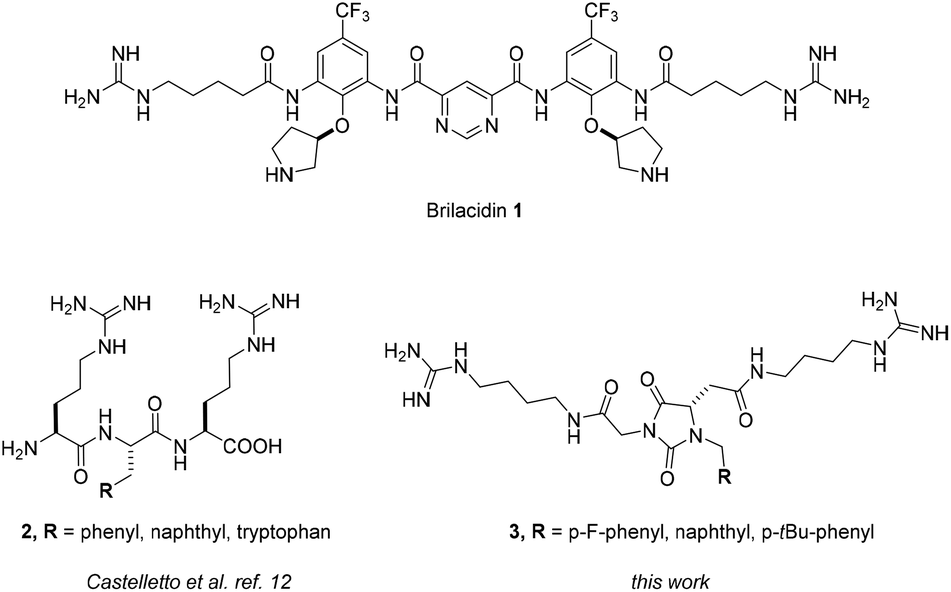 | ||
| Fig. 1 Structures of Brilacidin 1, β-turn tripeptides 2 and hydantoin-based universal peptidomimetics 3. | ||
Recently, we have introduced a hydantoin-based framework as a novel universal peptidomimetic scaffold able to mimic different protein secondary structures through rotation around a few significant degrees of freedom.13 The planar hydantoin framework is able to project the substituents into a position strictly related to the α-helix and β-turn conformations, with the latter being favoured both in solution and in the solid state, as evidenced in NMR and X-ray experiments. Taking inspiration from the structure and features of tripeptides 2, we present herein the synthesis and antibacterial activity of three model hydantoin-based universal peptidomimetics 3 (Fig. 1) presenting two guanidino groups and a hydrophobic aromatic residue in positions related to the classical i + n side chain positions of typical protein secondary structures.
It is well recognized that the activity of large AMPs depends on the fact that they often adopt protein secondary structures, such as α-helix and β-sheets, in solution or during the interaction with the cell membrane.14 Small tripeptides 2 were also designed to be able to form turn structures.12 This characteristic inspired us to exploit the conformational features of hydantoin-based universal peptidomimetics to synthesize three model peptidomimetics (3) that would be able to project two hydrophilic guanidino substituents and a hydrophobic moiety at the opposite face of the β-turn-mimicking secondary structure.
The synthetic pathway to synthesize 3 is very straightforward and is depicted in Scheme 1. Isocyanates 4a,b reacted smoothly with aspartic acid 5 having a carboxylic functional group at the α-position and the lateral chain protected as methyl ester and tert-butyl ester, respectively, producing urea derivatives 6a,b. We have previously shown that upon treatment with a base, urea derivatives of type 6 with a further alkyl substituent tethered to the nitrogen of the aspartic acid cyclize regioselectively, with the carboxylic ester in the α-position forming the five-member hydantoin ring rather than the cyclisation at the carboxylic ester on the lateral chain, which would form a six-member dihydrouracil ring.13b In the present case, in which the two nitrogen atoms on the urea moiety are mono-substituted, two cyclization reactions could occur, one on the methyl ester of the aspartic acid and the other on the benzyl or ethyl ester of the glycine, leading to the formation of two different regioisomeric hydantoins. To our delight, upon treatment with a 1M solution of NaOH, after 5 minutes we observed the selective formation of hydantoins 7a,b in very good yields.15 Alkylation promoted by K2CO3 in DMF with benzyl halides such as p-tert-butyl benzyl bromide 8a, p-F-benzyl bromide 8b and 1-(bromomethyl)naphthalene 8c yielded fully substituted hydantoins 9a–c, respectively, in high efficiency. The tert-butyl ester functional group of hydantoins 9a–c was selectively hydrolysed under acid conditions (TFA/DCM), and the resulting free carboxylic acids coupled with mono-Boc-1,4-butandiamine 10 to produce hydantoin derivatives 11a–c in very good yields. Diamine 10, which has two amino moieties separated by four methylene groups, was chosen based on its similarity with the arginine side chain of tripeptides 2. Next, the ethyl ester of compounds 11a,b was hydrolysed under basic conditions (NaOH 1M solution), and the resulting carboxylic acids were coupled with a second molecule of 10, which led to the formation of “symmetric” hydantoins 12a,b with the same arm attached to the imide-nitrogen and to the carbon of the ring. It is worth noting that hydantoins 9a–c have two orthogonally protected carboxy functionalities and, therefore, functionalization with different substituents would be feasible in future studies. The benzyl ester of intermediate 11c was cleaved by catalytic hydrogenation, and after coupling with 10 and deprotection of the Boc protecting group, we recovered the free diamine 13c, which was used in the biological test for comparison between the amino groups and guanidino groups (see below). Finally, after Boc-deprotection of hydantoins 12a,b, the free amino groups of the resulting compounds, as well as the amino groups of 13c, were transformed into guanidino groups by reaction with 1-[N,N′-(di-Boc)amidino]pyrazole 14, which provided the final peptidomimetics 3a–c after Boc-deprotection.
 | ||
| Scheme 1 Synthesis of hydantoin-based universal peptidomimetics 3. | ||
In analogy with what we have done for the hydantoin-based peptidomimetics having hydrophobic substituents,13 we used a combined Monte Carlo/Molecular Mechanics (MM) analysis on amino and guanidino derivatives 13c and 3c as model compounds to obtain insight into their propensity to adopt secondary structures, with particular interest in the β-turn conformation triggered by an intramolecular hydrogen bond involving the amidic NHB hydrogen (Fig. 2). An interatomic distance of dα < 7 Å and an absolute value of the dihedral angle C1–C2–C3–N4 β < 30° were considered as the conditions for the β-turn conformation, while the interatomic distances between Ci, Ci + 4 and Ci + 7, related to the substituents i, i + 4 and i + 7 of an ideal α-helix, namely i − i + 4 = 6.2 Å, i − i + 7 = 10.3 Å and i + 4 − i + 7 = 5.8 Å, were used as references for the α-helix conformation. In both cases, we considered the conformers within a 10 kcal mol−1 range from the conformer at minimum energy.16 In Table 1 we report the results as the percentage of conformers satisfying the requirements. For diamine derivative 13c, the α-helix conformation was slightly preferred, even though the minimum conformation was found to be a β-turn, while for the bis-guanidino peptidomimetic 3c, the β-turn geometry was the most favoured both at the ground-state and in the 10 kcal mol−1 range.
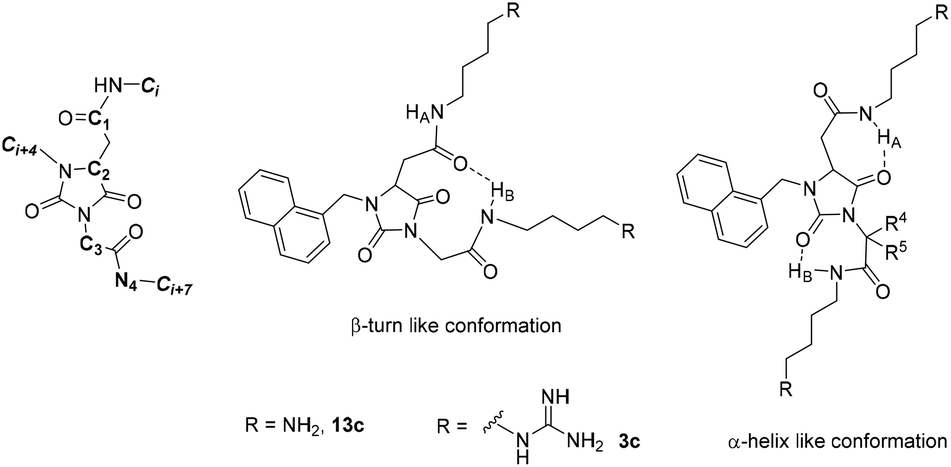 | ||
| Fig. 2 β-Turn and α-helix conformation of peptidomimetics 13c and 3c. | ||
| Compound | β-Turn (%) | α-Helix % | Global minimum |
|---|---|---|---|
| a Results are reported as a percentage of conformers satisfying the geometrical requirements adopted. | |||
| 13c | 18 | 27 | β-Turn |
| 3c | 26 | 17 | β-Turn |
This can be rationalized by considering that the hydrogen bond network is denser in 3c due to the involvement of the guanidino groups.
Fig. 3 presents the low-energy β-turn conformations and the fitting plots. Notably, the plot for 3c shows a higher number of conformations due to the very dense hydrogen bond network, which gives rise to many conformational possibilities. In the same images, the hydrogen bonds are also highlighted (dotted blue lines), showing that together with the known C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HB-N contacts, new guanidino H–N⋯H–N connections also further stabilize the conformations (Fig. 3).
O⋯HB-N contacts, new guanidino H–N⋯H–N connections also further stabilize the conformations (Fig. 3).
 | ||
| Fig. 3 (a) Low-energy β-turn conformation of 13c; (b) low-energy β-turn conformation of 3c; (c) fitting plots for 13c; (d) fitting plots for 3c. | ||
To check for the presence of secondary structures in solution, we investigated the strength of possible intramolecular hydrogen bonds involving the amidic protons NHA and NHB through 1H NMR experiments. The first parameter to be taken into consideration was the chemical shift in a relatively nonpolar solvent; NH amidic protons resonating at around 8.0 ppm are likely to be involved in intramolecular hydrogen bonding. Unfortunately, guanidino derivatives 3a–c were not soluble in deuterated nonpolar solvents, whereas diamine 13c was soluble in CDCl3. The NHB proton of 13c (2.5 nM solution) resonates at 8.04 ppm, while NHA proton resonates at 6.35 ppm (a copy of the spectrum is presented in the ESI†), indicating the involvement of NHB in the intramolecular hydrogen bond that triggered the β-turn conformation.17 Moreover, for guanidino derivative 3c, the rate of exchange of NHB with deuterium was very slow, as evidenced in the 1H NMR spectrum recorded in deuterated methanol. Actually, the proton NHB resonating at 7.95 ppm integrated to one hydrogen (see spectrum in ESI†), while NHA and the hydrogens belonging to the guanidino groups were not clearly detectable because, on the contrary, they did exchange quickly with deuterium. This evidence further reflects the involvement of the proton NHB in intramolecular hydrogen bonding. To investigate the possible presence of intramolecular hydrogen bonds in greater depth, we performed variable-temperature (VT) and DMSO titration 1H NMR experiments using 13c (Fig. 4). As evidenced in Fig. 4a and S1,† with increasing temperature, the proton shifts for NHA and NHB were quite similar, with Δδ/ΔT values of 9.0 and 5.9 ppb K−1, respectively. These values are not very indicative, since they are neither lower nor much higher than 2.4 ppb K−1.18 However, a DMSO titration experiment performed by gradually adding small aliquots (5 μL) of DMSO to a 2.0 mM CDCl3 solution of 13c showed that the chemical shift of NHB essentially did not change upon dilution because it was already involved in intramolecular hydrogen bonding, while we registered a sizable increase in the chemical shift of the amidic hydrogen NHA with increasing DMSO concentration due to the formation of intermolecular hydrogen bonding between the “free” NHA with the titrating solvent (Fig. 4b and S2†).19 All these experiments also proved that when the substituents on the hydantoin scaffold are polar, such as substituents bearing amino or guanidino moieties, the peptidomimetics show a great tendency to adopt secondary structures in solution, in particular, a β-turn conformation triggered by the formation of a quite strong intramolecular hydrogen bond.
 | ||
| Fig. 4 (a) VT and (b) DMSO titration 1H NMR experiments using 13c. | ||
To assess the antimicrobial activity of the synthesized compounds, we used four bacterial species belonging to the ESKAPE group: the reference strains Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 25293, Acinetobacter baumannii A17, and Pseudomonas aeruginosa.20 Compounds 3a–c bearing guanidino groups as terminal moieties of the extended arms and three different hydrophobic moieties, namely, a p-tert-butyl benzyl, p-F-benzyl, and 1-naphthyl-methylene group, respectively, and 13c bearing two amino groups and a 1-naphthyl-methylene group were tested at concentrations ranging from 4–0.06 μg mL−1. The obtained results are summarized in Table 2.
Although designed as antibacterial agents against Gram-negative bacteria, compound 3a with a 4-tert-butylbenzyl group as hydrophobic moiety was found to be the most potent peptidomimetic, showing remarkable activity against the Gram-positive bacteria Staphylococcus aureus (MIC = 0.25 μg mL−1), and very good activity against the Gram-negative bacteria E. coli, P. aeruginosa, and A. baumannii. The substitution on the benzene ring of the hydrophobic moiety exerts an important influence on the antibacterial activity of the peptidomimetics. Indeed, derivative 3b with a less-hydrophobic fluoro substituent compared to the tert-butyl group of 3a was found to be completely inactive against all the strains tested in the measured concentration range. Interestingly, derivative 3c with a bulky, planar and enhanced π-stacking 1-naphthyl-methylene group appeared to be effective only against Staphylococcus aureus, while the corresponding diamino derivative 13c was inactive against the three strains tested.
Conclusions
In conclusion, we designed three model hydantoin-based universal peptidomimetics with two strands bearing terminal hydrophilic guanidino groups and differing in the hydrophobic aromatic moiety tethered to the N-1 of the heterocycle. Through molecular modelling and 1H NMR experiments, we demonstrated that these peptidomimetics can project the hydrophilic guanidino groups and the hydrophobic moiety at the opposite face of the β-turn-mimicking secondary structure. This feature is probably very important to the broad activity that compound 3a showed against Gram-positive and Gram-negative bacterial strains and for the selective activity of compound 3c toward the Gram-positive Staphylococcus aureus. Together with their facile synthesis, the results obtained demonstrate that these peptidomimetics are promising candidates for further structure–activity relationship studies to shed light on their mechanism of action and toxicity, and to optimize their activity/selectivity in vitro and in vivo. These studies are ongoing in our laboratories.Author contributions
A.C. and M.C.B. performed the chemical experiments. A.C. performed the computational modelling and the NMR studies. E.O. and S.A. performed the antibacterial tests. E.B. supervised the antibacterial tests. A.V. designed the scaffold, conceived the experiments and wrote the paper.Conflicts of interest
There are no conflicts to declare.References
- (a) A. Coates, Y. M. Hu, R. Bax and C. Page, Nat. Rev. Drug Discovery, 2002, 1, 895 CrossRef CAS PubMed; (b) M. E. de Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184 CrossRef PubMed; (c) C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski and G. Robles Aguilar, et al. , Lancet, 2022, 399, 629 CrossRef CAS PubMed.
- Editorial, Nature, 2013, 495, 141 Search PubMed.
- (a) C. Nathan, Nature, 2004, 431, 899 CrossRef CAS PubMed; (b) M. S. Butler, I. R. Henderson, R. J. Capon and M.-A. T. Blaskovich, J. Antibiot., 2023, 76, 431 CrossRef CAS PubMed.
- (a) F. Peláez, Biochem. Pharmacol., 2006, 71, 981 CrossRef; (b) M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089 CrossRef CAS PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820 RSC.
- M. Zasloff, Nature, 2002, 415, 389 CrossRef CAS PubMed.
- (a) M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27 CrossRef CAS PubMed; (b) V. Nizet, Curr. Issues Mol. Biol., 2006, 8, 11 CAS.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- (a) C. Gosh and J. Haldar, ChemMedChem, 2015, 10, 1606 CrossRef PubMed; (b) P. Teng, H. Shao, B. Huang, J. Xie, S. Cui, K. Wang and J. Cai, J. Med. Chem., 2023, 66, 2211 CrossRef CAS PubMed; (c) D. Ding, S. Xu, E. Ferreira da Silva-Júnior, X. Liu and P. Zhan, Drug Discovery Today, 2023, 28, 103486 CrossRef PubMed.
- (a) R. P. Kowalsky, E. G. Romanowski, K. A. Yates and F. S. Mah, J. Ocul. Pharmacol. Ther., 2016, 32, 23 CrossRef PubMed; (b) B. Mensa, G. L. Howell, R. Scott and W. F. DeGrado, Antimicrob. Agents Chemother., 2014, 58, 5136 CrossRef PubMed.
- D. Weitz, D. Harder, F. Casagrande, D. Fotiadis, P. Obrdlik, B. Kelety and H. Daniel, J. Biol. Chem., 2007, 282, 2832 CrossRef CAS PubMed.
- V. Castelletto, C. J. C. Edwards-Gayle, I. W. Hamley, G. Barrett, J. Seitsonen, J. Ruokolainen, L. Rodrigues de Mello and E. Rodrigo da Silva, Chem. Commun., 2020, 56, 615 RSC.
- (a) M. C. Bellucci, M. Frigerio, C. Castellano, F. Meneghetti, A. Sacchetti and A. Volonterio, Org. Biomol. Chem., 2018, 16, 521 RSC; (b) A. M. Caramiello, M. C. Bellucci, G. Cristina, C. Castellano, F. Meneghetti, M. Mori, F. Secundo, F. Viani, A. Sacchetti and A. Volonterio, J. Org. Chem., 2023, 88, 10325 CrossRef PubMed.
- B. Ding, Q. Guan, J. P. Walsh, J. S. Boswell, T. W. Winter, E. S. Winter, S. S. Boyd, C. Li and P. B. Savage, J. Med. Chem., 2002, 45, 663 CrossRef CAS PubMed.
- The study of the regioselective cyclization of ureas of type 6 coming from two amino esters is underway, and the results will be reported in due course.
- (a) E. Ko, J. Liu, L. M. Perez, G. Lu, A. Schaefer and K. Burgess, J. Am. Chem. Soc., 2011, 133, 462 CrossRef CAS PubMed; (b) E. Ko, J. Liu and K. Burgess, Chem. Soc. Rev., 2011, 40, 4411 RSC.
- The chemical shifts of the NHA and NHB protons did not depend on the concentration for values below 4.0 mM.
- (a) E. S. Stevens, N. Suguwara, G. M. Bonora and C. Toniolo, J. Am. Chem. Soc., 1980, 102, 7048 CrossRef CAS; (b) C. André, B. Legrand, C. Deng, C. Didierjean, G. Pickaert, G. Martinez, M. C. Averlant-Petit, M. Amblard and M. Calmes, Org. Lett., 2012, 14, 960 CrossRef PubMed; (c) M. G. Memeo, M. Mella, V. Montagna and P. Quadrelli, Chem. – Eur. J., 2015, 21, 16374 CrossRef CAS PubMed.
- G. Wagner, A. Pardi and K. Wuethrich, J. Am. Chem. Soc., 1983, 105, 5948 CrossRef CAS.
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, Approved standard M07-A10, Clinical and Laboratory Standards Institute, Wayne, PA, 11th edn, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01247f |
| This journal is © The Royal Society of Chemistry 2023 |