
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved synthesis of DA364, an NIR fluorescence RGD probe targeting αvβ3 integrin†‡
Rachele
Maschio
 a,
Federica
Buonsanti
b,
Federico
Crivellin
b,
Fulvio
Ferretti
b,
Luciano
Lattuada
*b,
Federico
Maisano
b,
Laura
Orio
b,
Lorena
Pizzuto
b,
Raphael
Campanella
c,
Anthony
Clouet
c,
Camilla
Cavallotti
d and
Giovanni B.
Giovenzana
*ad
a,
Federica
Buonsanti
b,
Federico
Crivellin
b,
Fulvio
Ferretti
b,
Luciano
Lattuada
*b,
Federico
Maisano
b,
Laura
Orio
b,
Lorena
Pizzuto
b,
Raphael
Campanella
c,
Anthony
Clouet
c,
Camilla
Cavallotti
d and
Giovanni B.
Giovenzana
*ad
aDipartimento di Scienze del Farmaco, Università del Piemonte Orientale, Largo Donegani 2/3, 28100 Novara, Italy. E-mail: giovannibattista.giovenzana@uniupo.it
bBracco Imaging Spa, Bracco Research Centre, Via Ribes 5, 10010 Colleretto Giacosa, TO, Italy. E-mail: luciano.lattuada@bracco.com
cBracco Suisse SA, Route de la Galaise 31, 1228 Plan le Ouates, Switzerland
dCAGE Chemicals Srl, Via Bovio 6, 28100 Novara, Italy
First published on 12th October 2023
Abstract
Optical imaging (OI) is gaining increasing attention in medicine as a non-invasive diagnostic imaging technology and as a useful tool for image-guided surgery. OI exploits the light emitted in the near-infrared region by fluorescent molecules able to penetrate living tissues. Cyanines are an important class of fluorescent molecules and by their conjugation to peptides it is possible to achieve optical imaging of tumours by selective targeting. We report here the improvements obtained in the synthesis of DA364, a small fluorescent probe (1.5 kDa) prepared by conjugation of pentamethine cyanine Cy5.5 to an RGD peptidomimetic, which can target tumour cells overexpressing integrin αvβ3 receptors.
Introduction
Medicine can rely nowadays on a vast array of diagnostic imaging technologies, such as X-ray computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT), ultrasound (US) and, more recently, optical imaging (OI). All these technologies are routinely employed in clinical practice and in molecular imaging (MI), which is defined as the non-invasive, real-time visualization of biochemical events at the cellular and molecular levels within living organisms.1,2In particular, OI has experienced in the last few years an exponential growth in interest and applications due to its high sensitivity and safety.3,4 In fact, OI does not employ ionizing radiation and relies just on light in the spectral range spanning from the visible to the near-infrared (NIR) region.5 Moreover, OI has been extensively used in image-guided surgery, allowing surgeons to monitor precisely and in real time the resection of tumours and metastases.6–8 OI is usually performed by taking advantage of fluorescent molecules (or probes) emitting in the optical window between 650 and 900 nm (NIR-I region), thus limiting the absorption of light by the haemoglobin and water present in tissues.9 This spectral window represents an optimal compromise between image resolution and tissue penetration for in vivo imaging, although probes with higher emission wavelengths (1000–1700 nm, NIR-II region) are attracting increasing attention owing to their improved image quality.10 Among the huge variety of NIR fluorescent probes for bioimaging purposes,11–14 the class of cyanines15–18 is one of the most studied and extensively applied classes of fluorescent compounds; a relevant example being indocyanine green (ICG), the first optical contrast agent approved for human use.19,20 The synthesis of cyanines is rather simple and their structure can be easily modified to tune properties such as emission wavelength, brightness, stability, solubility, and pharmacokinetics.15,21–30 Many cyanines are commercially available and can be conjugated to biological vectors, providing tools to target in vivo specific receptors or tissues.31–33
An important and widely studied class of receptors is represented by integrins, a family of heterodimeric glycoproteins arranged in dimeric pairs of α- and β-subunits.34–36 These transmembrane receptors show affinities for extracellular matrix proteins and immunoglobulins and they play a key role in important regulation processes such as cell proliferation and migration, apoptosis, inflammation, and angiogenesis. One important member of this protein family is integrin αvβ3, which is the receptor of proteins having an arginine–glycine–aspartic acid (RGD) sequence and serves as a marker of angiogenesis, tumour development and metastasis. For these reasons, αvβ3 integrin is an attractive target for imaging probes and therapeutic drugs.37–39
Many linear and cyclic peptides having the RGD sequence have been evaluated to target αvβ3 integrin,40 and among them, we focused our attention on the rigid peptidomimetic 8 (Scheme 1).41,42 Compound 8 has been proved to be a useful targeting vector, and to such extent, it was conjugated to gold nanoparticles for tumour targeting,43 to Gd3+ and Ga3+ complexes to achieve probes for MRI and PET imaging,44 and to the water soluble pentamethine sulfo-cyanine Cy5.5 obtaining DA364 (Scheme 1), an NIR probe that can target tumour cells overexpressing integrin αvβ3 receptors.45,46 The safety and imaging efficacy of the NIR probe DA364 have been recently assessed during fluorescence-guided surgical resection of spontaneous solid tumours in dogs.47 To support this preclinical study, a batch of about 1 gram of DA364 was required but its first synthesis (Scheme 1, route a) was not amenable for scale-up to a multi-gram scale.46 First of all, sulfo-cyanine Cy5.5 activated as succinimidyl ester (sulfo-Cy5.5-NHS) is an extremely expensive reagent (6700 € for 50 mg)48 and is very hygroscopic, thus preventing its storage for a long period of time. Moreover, the purification of DA364, performed with semipreparative HPLC, required large volumes of eluent, long times and was rather inefficient due to the similar retention times of DA364 and the unreacted sulfo-Cy5.5-COOH. Last but not least, also the intermediate 8 required a long and tedious preparative HPLC purification. For all these reasons and to better support preclinical studies, we wanted to explore different synthetic strategies and we wish to report in this article on the improvements achieved in the synthesis of DA364 both with a traditional homogeneous methodology and with a new solid phase peptide synthesis (SPPS) approach.
 | ||
| Scheme 1 Syntheses of NIR probe DA364; route a: previously reported synthesis (ref. 46); route b: this article. Reagents and conditions: (a) TFA and CH2Cl2; (b) H-Arg(Pmc)-GlyOMe, DIC, HOBt, THF, and CH2Cl2; (c) H2, Pd/C, and MeOH; (d) Z-Asp(OtBu)OH, DIC, HOBt, TEA, and THF; (e) BnOH, Ti(iPrO)4, and THF; (f) HATU, HOAt, DIPEA, and DMF; (g) MsCl, TEA, and CH2Cl2; (h) NaN3 and DMF; (i) TFA, thioanisole, 1,2-ethanedithiol, and anisole; (j) sulfo-Cy5.5-NHS, borate buffer, pH 9; (k) sulfo-Cy5.5-COOH, TBTU, NMM, and DMF. | ||
Results and discussion
Peptidomimetic 8 was synthesized by following a previously reported synthetic strategy as shown in Scheme 1.41,42 Isoxazolidine 1,48 obtained from the known stereoselective 1,3-dipolar nitrone cycloaddition on a suitably protected allylproline, was deprotected with trifluoroacetic acid and coupled to the dipeptide H-Arg(Mtr)-GlyOMe by means of N,N′-diisopropylcarbodiimide (DIC) and 1-hydroxy-1H-benzotriazole (HOBt). Hydrogenation of the N-benzylisoxazolidine moiety of 2 and coupling with Z-Asp(OtBu)OH provided the intermediate compound 3. The reaction with benzyl alcohol and Ti(iPrO)4 allowed the transesterification of the methyl ester group in compound 3 into the corresponding benzyl ester group in compound 4, which was then hydrogenated and cyclized with O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluoro-phosphate (HATU) and 7-aza-1-hydroxy-1H-benzotriazole (HOAt) as condensing agents to give the peptidomimetic 5. Functional group interconversion of the hydroxyl group in 5 was accomplished by sequential treatment with mesyl chloride and then with sodium azide in DMF to obtain the azido derivative 6, which was finally hydrogenated to provide the primary amine of compound 7, strategic for its conjugation to other molecules. The deprotection with trifluoroacetic acid of all the protective groups in 7 gave crude 8, requiring a first preparative HPLC purification. This compound was reacted, as reported in the literature,46 with commercially available succinimidyl ester sulfo-Cy5.5 in borate buffer to give, after a second preparative HPLC purification, the final product DA364 in 24% overall yield (starting from azide 6). The final preparative HPLC purification was heavily demanding in terms of time and required large amounts of solvents since the compound DA364 and the unreacted sulfo-Cy5.5-COOH showed very similar retention times. In order to avoid the preparative HPLC purification of 8 and the use of the expensive succinimidyl ester of sulfo-Cy5.5, we opted for the direct coupling of the protected peptidomimetic 7 with the cheaper sulfo-Cy5.5-COOH,50 which can be purchased at lower prices (for example, 1200 € for 50 mg) or easily synthesized by following the procedures reported for the corresponding non-sulfonated derivative.51–53 The reaction was performed in DMF by employing a TBTU/N-methylmorpholine combination (Scheme 1, route b). The conjugated product was deprotected with trifluoroacetic acid in the presence of a cocktail of cation scavengers (thioanisole, 1,2-ethanedithiol and anisole). We found that the first elution of the crude on a Capto Q ion exchange chromatography resin allowed a fast and efficient purification from residual scavenger agents and, most importantly, from unreacted sulfo-Cy5.5-COOH, which was conveniently retained on the column. This compound can be later separately recovered and recycled. Then, with a much easier and quick preparative HPLC purification, pure DA364 was collected in 36% overall yield (starting from the azide 6).In order to speed up the preparation of DA364, we then studied the application of Fmoc solid phase peptide synthesis (SPPS)54,55 not only for the RGD sequence construction but also for the cyclization step and the final conjugation with sulfo-Cy5.5-COOH. Indeed, the automated SPPS strategy allows easier and faster operations and facilitates the cyclization thanks to the pseudo-dilution conditions promoting intramolecular resin-bound reactions over detrimental dimerization and/or oligomerization reactions.56
For this goal, we specifically designed and synthesized the new building block 17 from isoxazolidine 1, prepared with a different and improved synthetic sequence reported in Scheme 2. The trans-5-allylproline 9,57 prepared in four steps starting from (S)-pyroglutamic acid, was reacted with acetoxyacetyl chloride and then the acetate group was hydrolyzed by treatment with ammonia in methanol. The alcohol 11 was oxidized under Swern conditions with oxalyl chloride and the resulting unstable aldehyde 12 was immediately reacted with N-benzylhydroxylamine to give the isoxazolidine 1 in 64% overall yield, with a substantial improvement in the previously reported 45% yield.49
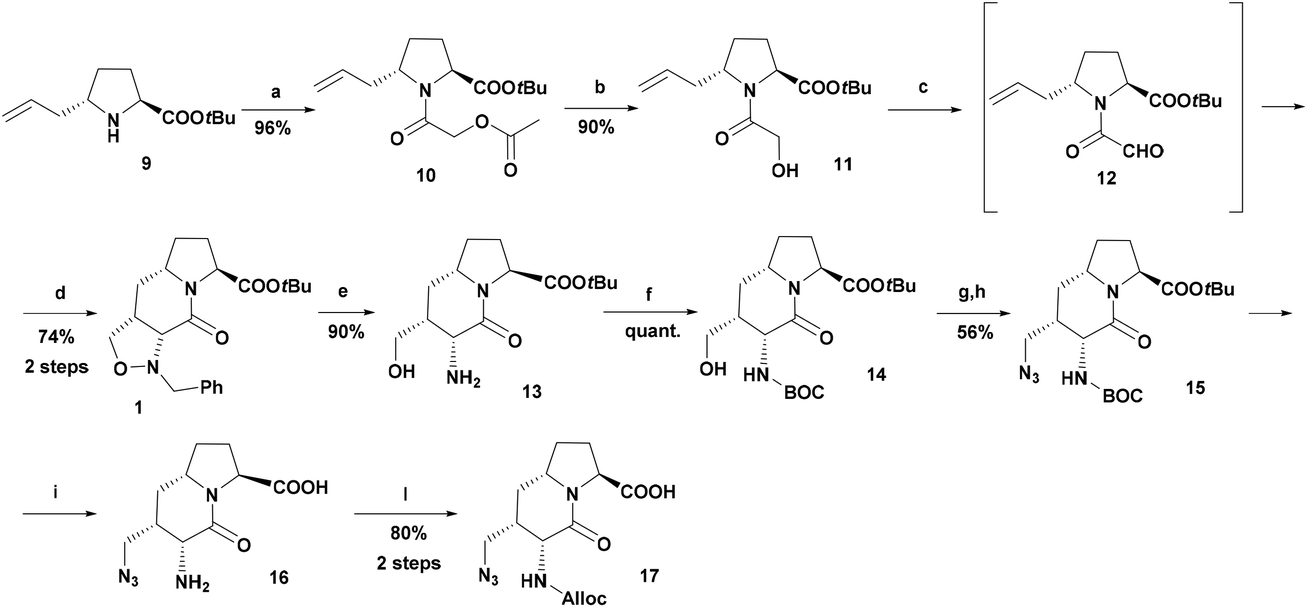 | ||
| Scheme 2 Synthesis of bicyclic scaffold 17. Reagents and conditions (a) AcOCH2COCl, DIPEA, and CH2Cl2; (b) 2 M NH3 in MeOH; (c), Swern oxidation, DMSO, (COCl)2, and CH2Cl2; (d) BnNHOH·HCl, NaHCO3, EtOH, H2O, reflux; (e) H2, Pd/C, MeOH, 70 °C; (f) Boc2O and THF; (g) MsCl, Et3N, and CH2Cl2; (h) NaN3, DMF, 60 °C; (i) TFA; (l) Alloc2O, Et3N, and THF. | ||
Catalytic hydrogenolysis (H2, Pd/C) of the N-benzyl group and the N–O bond led to the amino alcohol 13. The attempted alternative transfer catalytic hydrogenolysis with ammonium formate and Pd/C led to the formation of N-formylated byproducts.
The originally devised strategy was the protection of the amino group in 13 as Fmoc-carbamate but, after mesylation, the treatment with sodium azide led to a concomitant complete cleavage of the Fmoc protecting group, as also reported by Chen.58 An alternative strategy involving the protection of the amino group of 13 as Alloc-carbamate successfully proceeded to the azide stage, but the following deprotection of the tert-butyl ester led to the formation of several by-products. These findings forced us to revise the synthetic strategy to obtain the target compound 17. The amino group in 13 was protected as tert-butyl carbamate, and after mesylation and azidation, the resulting compound 15 was fully deprotected with TFA. The rather unstable amino acid 16 was finally protected with diallyl dicarbonate to give the key building block 17 (Scheme 2).
The azide group in compound 17 was strategically kept as a protected precursor of the amino group and was reduced on the resin right before the final conjugation with sulfo-Cy5.5-COOH, avoiding in this way unnecessary protection/deprotection steps with orthogonal protecting groups.
The RGD peptide synthesis on the solid phase was carried out on a Tribute™ peptide synthesizer by following a standard Fmoc solid-phase strategy.54,55 Commercially available Wang resin 18, with the first amino acid (Fmoc-Asp-OAll) preloaded, was swelled under agitation with DMF and the Fmoc was deprotected with a solution of 20% piperidine in DMF. The following amino acid (Fmoc-Gly-OH) was activated with a combined Oxyma Pure/DIC solution before addition to the solid support. After the completion of the coupling reaction, unreacted amines were capped by treatment with acetic anhydride prior to the Fmoc deprotection of the Gly moiety, performed with a solution of 20% piperidine in DMF. Then, Fmoc-Arg(Pbf)-OH was coupled using the same protocol, followed once more by a capping and a deprotection step. Compound 17 was then coupled to the resin using COMU as the activating agent. The simultaneous deprotection of the allylester and allyloxycarbonyl (Alloc) protecting groups by employing phenylsilane and tetrakis(triphenylphosphine)palladium(0)59 paved the way to intramolecular cyclization, performed through the activation of the free carboxylic function of the aspartic residue by the combination of PyAOP/DIPEA. The reduction of the azido group in 22 was accomplished through a Staudinger reduction60 with triphenylphosphine in water/THF at 60 °C. Finally, the carboxylic group of sulfoCy5.5-COOH was activated with COMU/DIEA before addition to the intermediate 23. Removal of all the protecting groups and cleavage of the final compound from the resin was achieved with trifluoroacetic acid/triisopropylsilane (Scheme 3). The compound DA364 was purified by preparative HPLC, then lyophilized and recovered as a blue powder with an 8% overall yield.
 | ||
| Scheme 3 Solid phase synthesis of DA364. Reagents and conditions (a) 20% piperidine and DMF; (b) FmocGlyOH, DIC, Oxyma Pure, and DMF; (c) Ac2O and DMF; (d) FmocArg(Pbf)OH, DIC, Oxyma Pure, and DMF; (e) 17, COMU, DIPEA, and DMF; (f) PhSiH3, Pd(PPh3)4, and CH2Cl2; (g) PyAOP, DIPEA, and DMF; (h) PPh3 and H2O; (i) sulfo-Cy5.5-COOH, COMU, DIPEA, and DMF; (j) TFA, triisopropylsilane, and H2O. | ||
Experimental
Materials and methods
Reagent-grade chemicals and solvents were obtained from commercial sources and directly used without further purification. Fmoc-Asp(Wang resin)-OAll 18 and Oxyma Pure were purchased from Sigma-Aldrich. Cyanine Cy5.5 activated as succinimidyl ester (sulfo-Cy5.5-NHS) was purchased from VWR International PBI s.r.l. (Italy). Cyanine Cy5.5 carboxylic acid (sulfo-Cy5.5-COOH) was purchased from Fluorochem Ltd (UK) or synthesized on a gram scale by following the procedures reported for the corresponding non-sulfonated derivative.51–53 The 1H and 13C spectra were recorded on Jeol Eclipse ECP300, Bruker Avance Neo 400 and Bruker Avance 600 spectrometers. Reactions were monitored by TLC using MERCK 60 F254 precoated silica gel plates and the products were visualized under UV light. Anion exchange purification was performed on an Äkta Pure system (CYTIVA Europe GmbH) by employing a Capto Q ion exchange chromatography resin (CYTIVA Europe GmbH). Preparative HPLC was performed at room temperature using an Agilent 1200 series instrument equipped with a collection valve on a YMC Triart Phenyl column (YMC, pore size: 120 Å and particle size: 5 μm, 250 × 50 mm) with a linear gradient, from 0 to 95%, of acetonitrile/0.8% ammonium acetate buffer over 25 min at a flow rate of 50 mL min−1 and monitored with a UV detector at 675 nm. Semi-preparative RP-HPLC purification was carried out using an Äkta Pure 25 purification system controlled with UNICORN™ 7.0 (CYTIVA Europe GmbH). The system was equipped with a sample pump P9-S, a fraction collector F9-R, a column valve V9-C, and a UV monitor U9-M (2 mm path length). Purification was performed at room temperature by eluting on a Luna C18 semi-preparative column (Phenomenex Inc., pore size: 100 Å and particle size: 5 μm, 250 × 10 mm), with a linear gradient, from 2 to 40%, of acetonitrile containing 0.1% TFA/water over 40 min at a flow rate of 5.0 mL min−1. Analytical HPLC of the final product was performed at 40 °C using an Agilent 1200 series instrument on a YMC Triart Phenyl column (YMC, pore size: 120 Å and particle size: 5 μm, 250 × 4.6 mm) with a linear gradient, from 2 to 100%, of acetonitrile/0.1% ammonium acetate buffer over 25 min at a flow rate of 1 mL min−1 and monitored with a UV detector at 675 nm. Solid phase synthesis was performed on an automated Tribute™ Peptide Synthesizer (Gyros Protein Technologies AB, Tucson AZ, USA). Mass spectra were recorded on an Agilent 1100 LC/MSD system composed of an Agilent quaternary pump manager G1311A, an Agilent degasser G1322A, an Agilent autosampler G1313A, a regulated column enclosure with a column switching G1316A and a mass detector G1946D.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 (3.8 g, 18.0 mmol) was dissolved in CH2Cl2 (60 mL) and DIPEA (6.97 g, 54.0 mmol). The solution was cooled to 0 °C and acetoxyacetylchloride (4.91 g, 36.0 mmol) was added dropwise to the reaction. The brown solution was kept at room temperature for 1 h. An aqueous solution of NH4Cl was added and the biphasic mixture was separated. The organic phase was dried over Na2SO4 and evaporated, obtaining a brownish oil (yield = 96%, 5.4 g). 1H NMR (400 MHz, CDCl3, 298 K) δ 5.80–5.69 (m, 1H + 1H′), 5.16–5.04 (m, 2H + 2H′), 4.83 (d, J = 14.4 Hz, 1H′), 4.64 (d, J = 14.3 Hz, 1H′), 4.58 (d, J = 14.4 Hz, 1H), 4.41 (d, J = 14.4 Hz, 1H), 4.39–4.28 (m, 1H + 1H′) 4.07–3.97 (m 1H + 1H′), 2.64 (m, 1H), 2.46 (m, 1H′), 2.39–1.73 (m, 5H + 5H′), 2173 (s, 3H), 2.171 (s, 3H′), 1.48 (s, 9H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3, 298 K) δ 170.9 [C + C′], 170.6 [C′], 170.4 [C], 165.9 [C], 165.7 [C′], 134.8 [CH], 133.6 [CH′], 118.6 [CH2′], 117.5 [CH2], 82.8 [C], 81.4 [C′], 61.9 [CH2], 61.7 [CH2′], 60.3 [CH′], 60.1 [CH], 58.5 [CH] 57.2 [CH′], 39.3 [CH2′], 36.9 [CH2], 29.6 [CH2] 28.5 [CH2′], 27.9 [CH3′], 27.8 [CH3], 26.3 [CH2′], 25.9 [CH2], 20.60 [CH3′], 20.58 [CH3]; HRMS-ESI+ (m/z): [M + H]+ calcd for C16H26NO5, 312.18055, found 312.18050; [M + Na]+ calcd for C16H25NNaO5, 334.16249, found 334.16227; [2M + Na]+ calcd for C32H50N2NaO10, 645.33577, found 645.33575.
57 (3.8 g, 18.0 mmol) was dissolved in CH2Cl2 (60 mL) and DIPEA (6.97 g, 54.0 mmol). The solution was cooled to 0 °C and acetoxyacetylchloride (4.91 g, 36.0 mmol) was added dropwise to the reaction. The brown solution was kept at room temperature for 1 h. An aqueous solution of NH4Cl was added and the biphasic mixture was separated. The organic phase was dried over Na2SO4 and evaporated, obtaining a brownish oil (yield = 96%, 5.4 g). 1H NMR (400 MHz, CDCl3, 298 K) δ 5.80–5.69 (m, 1H + 1H′), 5.16–5.04 (m, 2H + 2H′), 4.83 (d, J = 14.4 Hz, 1H′), 4.64 (d, J = 14.3 Hz, 1H′), 4.58 (d, J = 14.4 Hz, 1H), 4.41 (d, J = 14.4 Hz, 1H), 4.39–4.28 (m, 1H + 1H′) 4.07–3.97 (m 1H + 1H′), 2.64 (m, 1H), 2.46 (m, 1H′), 2.39–1.73 (m, 5H + 5H′), 2173 (s, 3H), 2.171 (s, 3H′), 1.48 (s, 9H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3, 298 K) δ 170.9 [C + C′], 170.6 [C′], 170.4 [C], 165.9 [C], 165.7 [C′], 134.8 [CH], 133.6 [CH′], 118.6 [CH2′], 117.5 [CH2], 82.8 [C], 81.4 [C′], 61.9 [CH2], 61.7 [CH2′], 60.3 [CH′], 60.1 [CH], 58.5 [CH] 57.2 [CH′], 39.3 [CH2′], 36.9 [CH2], 29.6 [CH2] 28.5 [CH2′], 27.9 [CH3′], 27.8 [CH3], 26.3 [CH2′], 25.9 [CH2], 20.60 [CH3′], 20.58 [CH3]; HRMS-ESI+ (m/z): [M + H]+ calcd for C16H26NO5, 312.18055, found 312.18050; [M + Na]+ calcd for C16H25NNaO5, 334.16249, found 334.16227; [2M + Na]+ calcd for C32H50N2NaO10, 645.33577, found 645.33575.
Alloc2O (150 μL, 0.9 mmol) was added to a solution of compound 16 (190 mg, 0.75 mmol) in dry tetrahydrofuran (12 mL) under a nitrogen atmosphere. The resulting mixture was stirred at 55 °C for 24 h. THF was evaporated, and the crude product was purified by column chromatography (CH2Cl2/MeOH/NH3 8/2/0.2–7/3/0.3) to give compound 17 as an amorphous solid (yield = 80%, 200 mg). 1H NMR (300 MHz, CD3OD, 298 K) selected signals 5.94 (ddt, 1H, J1 = 17.1 Hz, J2 = 10.7 Hz, J3 = 5.5 Hz), 5.31 (dd, 1H, J1 = 17.1 Hz, J2 = 1.2 Hz), 5.18 (d, 1H, J = 10.4 Hz), 4.56 (d, 1H, J = 5.5 Hz), 4.38 (d, 1H, J = 5.8 Hz), 4.34 (bt, 1H), 3.73 (m, 1H), 3.42 (dd, 1H, J1 = 12 Hz, J2 = 6.4 Hz), 3.26 (dd, 1H, J1 = 12.0 Hz, J2 = 7.0 Hz), 2.47 (m, 1H), 2.34 (m, 1H), 2.17–2.01 (m, 2H), 1.84 (m, 1H), 1.53 (m, 1H), 1.42 (m, 1H); 13C NMR (76 MHz, CD3OD, 298 K) δ 166.0 [C], 159.8 [C], 124.7 [CH], 108.2 [CH2], 57.3 [CH2], 50.8 [CH], 50.3 [CH], 44.0 [CH2], 43.0 [CH], 29.2 [CH], 23.8 [CH2], 19.9 [CH2], 19.4 [CH3]; HRMS-ESI− (m/z): [M − H]− calcd for C14H18N5O5, 336.13080, found 336.13079.
Homogeneous synthesis of DA364
Sulfo-Cy5.5-COOH (3.038 g, 2.849 mmol) was dissolved in dry DMF (150 mL) and N-methylmorpholine (1.25 mL, 11.4 mmol) was added. The mixture was stirred for 30 min and then a solution of TBTU (1.83 g, 5.70 mmol) in dry DMF (50 mL) was added. After 30 min, a solution of compound 7 (2.12 g, 2.59 mmol), obtained by quantitative hydrogenation of 6, in dry DMF (120 mL) was added dropwise. After 16 h, the DMF was evaporated under vacuum and the crude product was stirred with a mixture of trifluoroacetic acid/thioanisole/1,2-ethanedithiol/anisole (90/5/3/2 v/v, 200 mL) for 2 h to achieve complete deprotection. The reaction mixture was evaporated, and the crude was dissolved in water (100 mL) and washed with diisopropylether (2 × 50 mL). The aqueous phase was evaporated obtaining a blue solid which was dissolved in 20 mM TRIS (pH 7.5, 2.8 L). This solution was divided into three portions and each portion was loaded on a Capto Q ion exchange chromatography resin (860 mL) previously equilibrated with 20 mM TRIS (pH 7.5). After loading, the column was eluted with 20 mM TRIS (pH 7.5, 4.3 L, 20 mL min−1) eliminating in this way the non-anionic impurities and all the scavenger agents. Compound DA364 was eluted and recovered by adding in the eluent 1 M NaCl up to 80%. Fractions with similar HPLC purity were combined and desalted through passage on SPE C18 cartridges, eluting DA364 with a mixture of water and acetonitrile. The solution was concentrated under vacuum, and the crude was dissolved in 0.8% ammonium acetate buffer and purified by preparative HPLC on a YMC-Triart Phenyl column with a linear gradient, from 0 to 95%, of acetonitrile in 0.8% ammonium acetate buffer to give DA364 as an ammonium salt (yield = 36%, starting from azide 6, 1.04 g; HPLC purity = 99.7% at 675 nm; tR = 14.7 min). 1H NMR (600 MHz, DMSO-d6) δ 12.27 (bs, 1H), 9.02 (t, J = 9.5 Hz, 2H), 8.97 (d, J = 7.9 Hz, 1H), 8.49–8.44 (m, 4H), 8.23 (dd, J = 8.8, 1.5 Hz, 2H), 7.93 (d, J = 9.3 Hz, 1H), 7.78 (dd, J = 11.2, 9.4 Hz, 2H), 7.62 (t, J = 5.8 Hz, 1H), 7.50 (t, J = 5.9 Hz, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.66 (t, J = 12.2 Hz, 1H), 6.39 (dd, J = 14.1, 4.8 Hz, 2H), 4.46 (q, J = 7.4 Hz, 1H), 4.35–4.23 (m, 5H), 4.10 (dd, J = 8.1, 5.0 Hz, 1H), 4.02–3.98 (m, 2H), 3.65 (m, 1H), 3.25 (d, J = 13.0 Hz, 1H), 3.08 (m, 2H), 2.91–2.87 (m, 1H), 2.87 (d, J = 7.8 Hz, 1H), 2.74 (m, 1H), 2.48 (dd, J = 16.7, 6.7 Hz, 4H), 2.29 (m, 1H), 2.13–2.08 (m, 1H), 2.05–1.94 (m, 15H), 1.78 (quint, J = 7.4 Hz, 2H), 1.62 (m, 1H), 1.52 (m, 2H), 1.42 (m, 2H), 1.36 (t, J = 7.2 Hz, 3H), 1.32 (d, J = 7.0 Hz, 2H), 1.21 (m, 1H), 0.96 (m, 1H); 13C NMR (151 MHz, DMSO-d6) δ 174.16 [C], 173.79 [C], 173.27 [C], 172.62 [C], 172.49 [C], 171.42 [C], 171.29 [C], 169.54 [C], 169.16 [C], 157.13 [C], 153.75 [CH], 153.60 [CH], 146.14 [C], 145.79 [C], 145.76 [C], 145.69 [C], 140.51 [C], 139.81 [C], 134.10 [C], 133.87 [C], 130.21 [CH], 129.99 [CH], 128.03 [C], 128.00 [C], 127.34 [C], 127.16 [C], 125.96 [CH], 122.02 [CH], 119.65 [CH], 119.44 [CH], 118.73 [C], 116.74 [C], 112.32 [CH], 111.73 [CH], 103.34 [CH], 102.98 [CH], 62.28 [CH], 55.15 [CH], 53.27 [CH], 51.32[CH], 51.22[CH], 51.16 [C], 50.59 [C], 44.04 [CH2], 43.48 [CH2], 40.89 [CH2], 40.44 [CH2], 39.29 [CH2], 36.32 [CH], 35.37 [CH2], 34.11 [CH2], 33.17 [CH2], 33.09 [CH2], 29.90 [CH2], 27.96 [CH2], 27.52 [CH3], 27.51 [CH3], 27.46 [CH2], 27.42 [CH3], 25.90 [CH2], 25.81 [CH2], 25.44 [CH2], 12.92 [CH3].; HRMS-ESI+ (m/z): [M + 2H]2+ calcd 718.71925, found 718.71883; [M + H + Na]2+ calcd 729.71026, found 729.71032; [M + H]+ calcd for C63H78N11O20S4 1436.43074, found 1436.43152; [M + Na]+ calcd for C63H77N11NaO20S4 1458.41269, found 1458.40595; HRMS-ESI− (m/z): [M − 2H]2− calcd 716.70635, found 716.70593; [M − H]− calcd for C66H76N11O20S4 1434.41509, found 1434.42144.Solid phase synthesis of 21
Fmoc-Asp(Wang resin)-OAll (Novabiochem®) was used in the whole study (1.0 g, loading capacity: 0.469 mmol g−1 and active capacity: 0.469 mmol). All steps were performed in DMF with a reactor of 40 mL. The resin was covered with 10 mL of DMF and swelled under agitation (vortex and nitrogen bubbling) for ten minutes. This operation was repeated three more times. The first step consisted of the Fmoc deprotection with 10 mL of a piperidine/DMF 1:4 (v/v) solution for 30 s. Then, the resin was washed three times with DMF. Next, Fmoc-Gly-OH (1394.3 mg, 4.69 mmol, 10 eq.) was activated with Oxyma Pure (667.0 mg, 4.69 mmol) and DIC (6.61 mL, 4.69 mmol) in DMF (8 mL) solution for 10 min before being added on the resin. The coupling reaction lasted for 4 h. Capping with acetic anhydride (6.64 mL, 70.4 mmol, 150 eq.) solution in DMF (8 mL) was performed for 5 min. The resin was filtered again and washed with DMF. Then Fmoc deprotection was performed with 10 mL of a piperidine/DMF 1:4 (v/v) solution for 30 s. After that, Fmoc-Arg(Pbf)-OH (3042.9 mg, 4.69 mmol) was coupled by following the same procedure described above. At the end of the reaction, the medium was filtered and washed with DMF. At the end of the synthesis, the resin 20 was washed extensively with DMF and methanol and dried under nitrogen flow for 15 min.
Resin 20 (953 mg) was Fmoc deprotected with 10 mL of a piperidine/DMF 1:4 (v/v) solution for 30 s, then washed and swelled in DMF (8 mL × 10 min × 4). In a round bottom flask, 17 (96.5 mg, 286 μmol, 98 eq.) was solubilized in DMF and the carboxylic function was activated with COMU (119.9 mg, 280 μmol) and DIPEA (149.5 μL, 858 μmol) before being added to deprotected resin 20. The mixture was agitated by vortexing under a nitrogen atmosphere for 20 h. The resin was filtered and washed by stirring (30 s) with DMF (3 mL). Then, DMF was filtered, and the washing procedure was repeated four times. At the end, the resin 21 was washed extensively with DMF and methanol and dried under nitrogen flow for 15 min.
Solid phase synthesis of 22
The resin 21 was swelled in dichloromethane (10 mL) and then, under an argon atmosphere, phenylsilane (853 μL, 6.91 mmol, 25 eq.) was added. After 3 min, a solution of Pd(PPh3)4 (127 mg, 110 μmol) in dichloromethane (4 mL) was added. After 40 min, the resin was washed subsequently with dichloromethane (4 × 4 mL × 30 s), with a solution of dioxane/H2O 9:1 (v/v) (4 mL × 1 min), DMF (4 mL × 2 min) and again with dichloromethane (2 × 4 mL × 2 min). The whole process, including the washing steps, was repeated a second time with a fresh solution of Pd(PPh3)4. The resin (861 mg) was swelled in DMF (10 mL), a PyAOP solution (201.8 mg, 387 μmol, 1.5 eq.) in DMF was added to the resin, and then DIPEA (135 μL, 774 μmol, 3 eq.) was added. The cyclization was performed under agitation with vortexing and nitrogen bubbling. After 20 h, the solution was filtered and DMF (3 mL) was added. The medium was stirred for 30 s and then DMF was filtered. This washing step was repeated four additional times.
Solid phase synthesis of 23
The resin 22 (100 mg) was swelled in THF (10 mL), and PPh3 (78.7 mg, 30 μmol) and H2O (27.0 μL, 150 μmol) were added. The mixture was agitated at 60 °C for 20 min. Upon completion of the reaction, the resin was washed with 1% Et3N in dichloromethane (3 × 5 mL × 2 min) and with Et2O (2 × 5 mL × 2 min). The expected resin 23 was then used without further purification.On bead fluorophore final coupling
The resin 23 (82 mg) was swelled in DMF (4 × 1 mL × 10 min). Sulfo-Cy5.5-COOH was activated with COMU (10.4 mg, 24.3 μmol, 0.98 eq.) and DIPEA (12.9 μL, 49.6 μmol, 2 eq.) in DMF for 3 min before being added to the resin. After 20 h of reaction, the resin was filtered and washed thoroughly with DMF (4 × 3 mL).Peptide cleavage
The resin was dried beforehand under vacuum for 30 min. Then, a freshly prepared scavenger cocktail solution (10 mL) of trifluoroacetic acid/triisopropylsilane/water 95:2.5:2.5 (v/v/v) was added to the resin. The mixture was stirred at room temperature for 3 h. The reaction mixture was filtered, and the blue solution was diluted with excess diethyl ether. The resulting blue crude solid was washed twice with diethyl ether (2 × 10 mL) and then centrifuged. The supernatant was removed each time, and the solid was finally dried on a rotary evaporator in order to remove the last traces of solvent.
DA364 purification
The crude DA364 obtained was purified by semi-preparative RP-HPLC by elution at room temperature on a Luna C18 semi-preparative LC column (Phenomenex Inc., pore size: 100 Å and particle size: 5 μm, 250 × 10 mm), with a linear gradient, from 2 to 40%, of acetonitrile containing 0.1% TFA/water over 40 min at a flow rate of 5.0 mL min−1. The collected fractions were analysed by analytical HPLC-UV and mass spectrometry. Fractions of interest were combined and acetonitrile was evaporated. The resulting solution was dynamically frozen with a mechanical stirrer in a 2-propanol bath at −35 °C. The product was then lyophilized for 12 h and was recovered as a blue powder (54 mg, 8% yield).Conclusions
In summary, we successfully improved the overall yield of DA364 by 50% by performing the conjugation of sulfo-Cy5.5-COOH on the protected peptidomimetic 7. This simplified the chromatographic purification by reducing time and amounts of solvents. With the synthesis of the new key building block 17, we easily obtained DA364 with an automated Fmoc SPPS strategy in an 8% overall yield and demonstrated the possibility of performing the cyclization and the conjugation reactions on the resin.These results allow the synthesis of larger amounts of DA364 and reduction of the time and cost of the preparation of a single batch. Thus, we were able to obtain enough material to support a preclinical study, in which DA364 was employed as a fluorescent probe in the guided surgical resection of tumours in dogs.
Author contributions
Conceptualization: L. L. and G. B. G.; supervision: L. L., F. B., F. M., G. B. G., and A. C.; synthesis and analysis: R. M., F. B., L. O., L. P., R. C., A. C., and C. C.; HPLC methods and purification: F. C., F. F., F. M., R. C., and A. C.; writing – original draft: L. L., G. B. G., and R. M.; writing – review and editing: all authors.Conflicts of interest
There are no conflicts to declare.References
- R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580–589, DOI:10.1038/nature06917.
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897–965, DOI:10.1152/physrev.00049.2010.
- G. Pirovano, S. Robert, S. Kossatz and T. J. Reiner, Nucl. Med., 2020, 61, 1419–1427, DOI:10.2967/jnumed.119.238279.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634, DOI:10.1016/j.cbpa.2003.08.007.
- S. H. Yun and S. J. J. Kwok, Nat. Biomed. Eng., 2017, 1, 0008, DOI:10.1038/s41551-016-0008.
- E. A. Owens, S. Lee, J. M. Choi, M. Henary and H. S. Choi, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 828–838, DOI:10.1002/wnan.1337.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518, DOI:10.1038/nrclinonc.2013.123.
- S. L. Gibbs, Quant. Imaging Med. Surg., 2012, 2, 177–187, DOI:10.3978/j.issn.2223-4292.2012.09.04.
- B. P. Joshi and T. D. Wang, Cancers, 2010, 2, 1251–1287, DOI:10.3390/cancers2021251.
- A. Haque, Md. S. H. Faizi, J. A. Rather and M. S. Khan, Bioorg. Med. Chem., 2017, 25, 2017–2034, DOI:10.1016/j.bmc.2017.02.061.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010, DOI:10.1038/s41551-016-0010.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138, DOI:10.1016/j.biomaterials.2011.06.024.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640, DOI:10.1021/cr900263j.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 60–74, DOI:10.1016/j.cbpa.2009.10.022.
- L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385–9397, 10.1039/d0ob01962c.
- H. A. Shindy, Dyes Pigm., 2017, 145, 505–513, DOI:10.1016/j.dyepig.2017.06.029.
- C. Shi, J. B. Wu and D. Pan, J. Biomed. Opt., 2016, 21, 050901, DOI:10.1117/1.JBO.21.5.050901.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2001, DOI:10.1021/cr990402t.
- J. T. Alander, I. Kaartinen, A. Laakso, T. Pätilä, T. Spillmann, V. V. Tuchin, M. Venermo and P. Välisuo, Int. J. Biomed. Imaging, 2012, 940485, DOI:10.1155/2012/940585.
- M. V. Marshall, J. C. Rasmussen, I. Tan, M. B. Aldrich, K. E. Adams, X. Wang, C. E. Fife, E. A. Maus, L. A. Smith and E. M. Sevick-Muraca, Open Surg. Oncol. J., 2010, 2, 12–25, DOI:10.2174/1876504101002010012.
- M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805, DOI:10.1016/j.tet.2011.10.069.
- T. L. Dost, M. T. Gressel and M. Henary, Anal. Chem. Insights, 2017, 12, 1–6, DOI:10.1177/1177390117711938.
- D. S. Pisoni, L. Todeschini, C. A. Borges, C. L. Petzhold, F. S. Rodembusch and L. F. Campo, J. Org. Chem., 2014, 79, 5511–5520, DOI:10.1021/jo500657s.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–154, DOI:10.1038/nbt.2468.
- G. Chapman, M. Henary and G. Patonay, Anal. Chem. Insights, 2011, 6, 29–36, DOI:10.4137/ACI.S6568.
- R. K. Das, A. Samanta, H.-H. Ha and Y.-T. Chang, RSC Adv., 2011, 1, 573–575, 10.1039/c1ra00498k.
- L. L. Jiang, B.-L. Li, F.-T. Lv, L.-F. Dou and L.-C. Wang, Tetrahedron, 2009, 65, 5257–5264, DOI:10.1016/j.tet.2009.04.086.
- M. V. Kvach, A. V. Ustinov, I. A. Stepanova, A. D. Malakhov, M. V. Skorobogatyi, V. V. Shmanai and V. A. Korshun, Eur. J. Org. Chem., 2008, 2107–2117, DOI:10.1002/ejoc.200701190.
- C. Bouteiller, G. Clavé, A. Bernardin, B. Chipon, M. Massonneau, P.-Y. Renard and A. Romieu, Bioconjugate Chem., 2007, 18, 1303–1317, DOI:10.1021/bc0700281.
- Y. Lin, R. Weissleder and C.-H. Tung, Bioconjugate Chem., 2002, 12, 605–610, DOI:10.1021/bc0155723.
- B. P. Joshi and T. D. Wang, Contrast Media Mol. Imaging, 2018, 2015237, DOI:10.1155/2018/2015237.
- M. Staderini, A. Megia-Fernandez, K. Dhaliwal and M. Bradley, Bioorg. Med. Chem., 2018, 26, 2816–2826, DOI:10.1016/j.bmc.2017.09.039.
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271, 10.1039/c6cs00908e.
- O. J. Mezu-Ndubuisi and A. Maheshwari, Pediatr. Res., 2021, 89, 1619–1626, DOI:10.1038/s41390-020-01177-9.
- M. Bachmann, S. Kukkurainen, V. P. Hytönen and B. Wehrle-Haller, Physiol. Rev., 2019, 99, 1655–1699, DOI:10.1152/physrev.00036.2018.
- S. J. Shattil, C. Kim and M. H. Ginsberg, Nat. Rev. Mol. Cell Biol., 2010, 11, 288–300, DOI:10.1038/nrm2871.
- Y. Ye and X. Chen, Theranostics, 2011, 1, 102–126, DOI:10.7150/thno/v01p0102.
- M. Schottelius, B. Laufer, H. Kessler and H.-J. Wester, Acc. Chem. Res., 2009, 42, 969–980, DOI:10.1021/ar800243b.
- A. J. Beer and M. Schwaiger, Cancer Metastasis Rev., 2008, 27, 631–644, DOI:10.1007/s10555-008-9158-3.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H.-J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 39805, DOI:10.1038/srep39805.
- L. Manzoni, L. Belvisi, D. Arosio, M. Civera, M. Pilkington-Miksa, D. Potenza, A. Caprini, E. M. V. Araldi, E. Monferini, M. Mancino, F. Podestà and C. Scolastico, ChemMedChem, 2009, 7, 615–632, DOI:10.1002/cmdc.200800422.
- D. Arosio, L. Manzoni, E. M. V. Araldi, A. Caprini, E. Monferini and C. Scolastico, Bioconjugate Chem., 2009, 20, 1611–1617, DOI:10.1021/bc900155j.
- D. Arosio, L. Manzoni, E. M. V. Araldi and C. Scolastico, Bioconjugate Chem., 2011, 22, 664–672, DOI:10.1021/bc100448r.
- L. Manzoni, L. Belvisi, D. Arosio, M. P. Bartolomeo, A. Bianchi, C. Brioschi, F. Buonsanti, C. Cabella, C. Casagrande, M. Civera, M. De Matteo, L. Fugazza, L. Lattuada, F. Maisano, L. Miragoli, C. Neira, M. Pilkington-Miksa and C. Scolastico, ChemMedChem, 2012, 7, 1084–1093, DOI:10.1002/cmdc.201200043.
- L. Conti, S. Lanzardo, M. Iezzi, M. Montone, E. Bolli, C. Brioschi, A. Maiocchi, G. Forni and F. Cavallo, Contrast Media Mol. Imaging, 2013, 8, 350–360, DOI:10.1002/cmmi.1529.
- S. Lanzardo, L. Conti, C. Brioschi, M. P. Bartolomeo, D. Arosio, L. Belvisi, L. Manzoni, A. Maiocchi, F. Maisano and G. Forni, Contrast Media Mol. Imaging, 2011, 6, 449–458, DOI:10.1002/cmmi.444.
- S. Favril, C. Brioschi, K. Vanderperren, E. Abma, E. Stock, N. Devriendt, I. Polis, H. De Cock, A. Cordaro, L. Miragoli, P. Oliva, G. Valbusa, C. Alleaume, I. Tardy, A. Maiocchi, F. Tedoldi, F. Blasi and H. de Rooster, Oncotarget, 2020, 11, 2310–2326, DOI:10.18632/oncotarget.27633.
- https://www.sigmaaldrich.com/IT/en/search/pa15602?focus=products&page=1&perPage=30&sort=relevance&term=PA15602&type=product (July 8th, 2023).
- L. Manzoni, D. Arosio, L. Belvisi, A. Bracci, M. Colombo, D. Invernizzi and C. Scolastico, J. Org. Chem., 2005, 70, 4124–4132, DOI:10.1021/jo0500683.
- https://www.chemscene.com/210892-23-2.html (July 8th, 2023).
- S. Xu, Q. Wang, Q. Zhang, L. Zhang, L. Zuo, J.-D. Jiang and H.-Y. Hu, Chem. Commun., 2017, 53, 11177–11180, 10.1039/c7cc07050k.
- Y. Lin, R. Weissleder and C.-H. Tung, Bioconjugate Chem., 2002, 13, 605–610, DOI:10.1021/bc0155723.
- S. R. Mujumdar, R. B. Mujumdar, C. M. Grant and A. S. Waggoner, Bioconjugate Chem., 1996, 7, 356–362, DOI:10.1021/bc960021b.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis, Oxford University Press, 2000 Search PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27, DOI:10.1002/psc.2836.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524, DOI:10.1038/NCHEM.1062.
- P. W. R. Harris, M. A. Brimble and P. D. Gluckman, Org. Lett., 2003, 5, 1847–1850, DOI:10.1021/ol034370e.
- C. C. Chen, B. Rajagopal, X. Y. Liu, K. L. Chen, Y. C. Tyan, F. Lin and P. C. Lin, Amino Acids, 2014, 46, 367–374, DOI:10.1007/s00726-013-1625-7.
- F. Guibé, Tetrahedron, 1998, 54, 2967–3042, DOI:10.1016/S0040-4020(97)10383-0.
- D. C. Lenstra, P. E. Lenting and J. Mecinović, Green Chem., 2018, 20, 4418–4422, 10.1039/c8gc02136h.
Footnotes |
| † This article is dedicated to the memory of the late Dr Leonardo Manzoni (CNR-ISTM, Milan, Italy). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01206a |
| This journal is © The Royal Society of Chemistry 2023 |