
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of haloaromatics from nitroarenes via a sequential one-pot Mo-catalyzed reduction/Sandmeyer reaction†
Raquel
Hernández-Ruiz
 ,
Sara
Gómez-Gil
,
María R.
Pedrosa
,
Samuel
Suárez-Pantiga
and
Roberto
Sanz
*
,
Sara
Gómez-Gil
,
María R.
Pedrosa
,
Samuel
Suárez-Pantiga
and
Roberto
Sanz
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Pza. Misael Bañuelos s/n, 09001 Burgos, Spain. E-mail: rsd@ubu.es
First published on 8th September 2023
Abstract
Herein, we report the direct synthesis of a wide variety of functionalized aromatic bromides, chlorides, iodides, and fluorides from nitroarenes in a sequential one-pot operation. This protocol is based on an air- and moisture-tolerant dioxomolybdenum-catalyzed reduction of nitroaromatics, employing pinacol as a reducing agent, which enables subsequent diazotization and halogenation steps. This methodology represents a step-economical, practical, and alternative procedure for synthesizing haloaromatics directly from nitroaromatics.
Introduction
In the past decades, organic synthesis has reached an extraordinary level of sophistication for the targeted preparation of value-added compounds. However, these syntheses inevitably produce large amounts of waste, closely related to tedious purification and work-up procedures inherent to the traditional step-by-step processes involved. In this context, one-pot processes involving sequential reactions conducted in a single reaction vessel have emerged as valuable alternatives to avoid the need to purify intermediates.1 This approach reduces the amount of chemical waste generated, minimizing separation and purification processes, simplifying practical aspects and ultimately increasing the overall chemical yield. However, one-pot reactions are not straightforward: as no intermediate purification steps are performed, each sequential reaction must proceed in the presence of the previously generated byproducts, unreacted starting materials, or reagents. Consequently, the reaction conditions for a one-pot procedure are very different and a simple combination of optimized steps in a single pot is usually unsuccessful, which constitutes one of the main challenges we face in our approach.Aryl halides are versatile compounds widely used in organic synthesis. On the one hand, aryl iodides, bromides and chlorides constitute pivotal intermediates in cross-coupling or Heck reactions and carbon–heteroatom bond-formation reactions catalyzed by transition-metal complexes.2 The C(sp2)–Hal unit is also a ubiquitous structural motif in many natural products and synthetic drugs.3 In this context, the Sandmeyer reaction represents one of the most general synthetic pathways for their obtention.4 It is a particularly relevant organic transformation that converts arylamines to aryl halides via a diazonium salt intermediate. Ever since Sandmeyer reported in 1884 that benzenediazonium salts undergo copper-mediated decomposition into bromo and chlorobenzenes and benzonitriles,5 multiple reaction variants have been developed, mostly seeking to avoid competing reactions, tedious workups, diazonium salt isolation or excess halogenation.6 Also, almost a century ago, related processes such as the Pschorr, the Gomberg–Bachmann, the Balz–Schiemann, and the Meerwein reactions were reported.7 More recently, new reagent partners have been developed to form C–P, C–S, C–B, C–Sn, C–N, and C–CF3 bonds from diazonium salts, in a clear renaissance of Sandmeyer-type reactions.8 Particularly, one-pot processes starting from anilines have attracted much interest in recent years.8 In addition, Meerwein-type arylations with diazonium salts have also been in the spotlight in the past years with the development of new and improved variants.9 Moreover, aryldiazonium salt intermediates have been recently employed in several new processes involving transition-metal catalyzed transformations.10
However, the classical Sandmeyer reaction for accessing haloaromatics presents unresolved issues of sustainability and practicability as it employs anilines as the nitrogen source, which are usually obtained by a previous reduction reaction of the corresponding nitroaromatic derivatives (Scheme 1a). Therefore, the development of a general, one-pot, and efficient method for the synthesis of aryl halides directly from abundant and readily available nitroarenes, which can be easily accessed through the nitration of arenes, is very attractive and would offer sustainability-related advantages such as saving reagent costs, minimizing tedious purification steps along with waste generation, and suppressing at least one process step. Indeed, there is tremendous interest in synthetic strategies that involve the direct transformation of nitroaromatics into other valuable scaffolds.11 In this field, and based on the selective dioxomolybdenum(VI)-catalyzed reduction of nitroarenes into anilines with pinacol that only releases acetone and water as byproducts (Scheme 1b),12 our group has reported the direct synthesis of nitrogenated polyheterocycles and quinolines from nitroarenes13 with the incorporation of the waste formed in the first reduction step.14
 | ||
| Scheme 1 Previous work and the proposed direct synthesis of haloarenes from nitroaromatics. | ||
In this context, we envisaged that combining the dioxomolybdenum-catalyzed reduction of nitroarenes, using pinacol as a reducing agent, with the well-established Sandmeyer reaction could allow direct access to haloaromatics in a sequential one-pot manner. In particular, the choice of the reductant was anticipated to be decisive in enabling subsequent steps. In this sense, pinacol constitutes an attractive reducing agent, being a renewable glycol typically manufactured by electrolytic dimerization of acetone.15 Equally critical to the success of this proposal would be that the conditions, mainly solvents and reagents, required for the generation of the intermediate diazonium salts are compatible with the initial reduction conditions (Scheme 1c). Herein, we report the direct synthesis of haloaromatics from a wide variety of functionalized nitroarenes by combining the initial Mo-catalyzed chemoselective reduction of the nitro group using pinacol with a subsequent Sandmeyer reaction.
Results and discussion
Initially, we centered our efforts on the one-pot bromination of nitroarenes in a two-step fashion. A screening of several reaction conditions was performed using 4-nitrobenzonitrile (1a) as the model starting material (Table 1). The first step, the reduction of the nitroarene, was performed following our previously described methodology using pinacol as the reducing agent in toluene (150 °C, 30 min under microwave irradiation) under dioxomolybdenum catalysis. After that, the mixture was treated with different metallic bromides in the presence of tert-butyl nitrite to promote the bromination of the corresponding in situ-formed diazonium salt. The use of KBr led to decomposition (entry 1), while there was no conversion of the intermediate 4-aminobenzonitrile with CuBr (entry 2). The desired brominated product (2a) was only observed when CuBr2 was employed (entries 3 and 4). However, the formation of an undesired byproduct (likely from a secondary reaction with toluene) was detected. The results did not improve when using α,α,α-trifluorotoluene (entry 5). We then decided to switch to acetonitrile as the solvent, given its wide application in Sandmeyer reactions. Gratifyingly, the Mo-catalyzed reduction of nitroarenes with pinacol also proceeded efficiently in acetonitrile under analogous reaction conditions. Notably, the complementary use of copper(I) bromide and hydrobromic acid allowed the efficient synthesis of 2a in good yields and without further purification (entries 6 and 7). The presence of HBr was crucial for the success of the reaction (entry 8 vs. 7). Interestingly, CuBr could be employed in catalytic amounts, although with a slightly lower yield (entry 9). Moreover, bromination was accomplished even in the absence of CuBr (entries 10 and 11). However, a longer reaction time was required to achieve a high yield (entry 11).|
|
||||
|---|---|---|---|---|
| Entry | Acid (x equiv.) | Bromide | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol), under air. All the reactions were carried out for 1 h. Complete conversion of the starting nitroarene and the intermediate aniline was observed unless otherwise established. b Yield obtained by NMR using 1,3,5-trimethoxybenzene as the internal standard. c Decomposition was observed. d Only 4-aminobenzonitrile was obtained. e 2 equiv. of CuBr2 were used. f Isolated yield of the pure product after extraction. g 10 mol% of CuBr was used. h Reaction time = 4 h. | ||||
| 1 | PTSA (2) | KBr | Toluene | — |
| 2 | — | CuBr | Toluene | —d |
| 3 | — | CuBr2 | Toluene | 56 |
| 4e | — | CuBr2 | Toluene | 66 |
| 5 | — | CuBr2 | PhCF3 | 55 |
| 6 | HBr (2) | CuBr | MeCN | 75 |
| 7 | HBr (1.5) | CuBr | MeCN | 87 (82)f |
| 8 | — | CuBr | MeCN | 14 |
| 9g | HBr (1.5) | CuBr | MeCN | 75 |
| 10 | HBr (1.5) | — | MeCN | 35 |
| 11h | HBr (1.5) | — | MeCN | 68 |

With an optimized set of conditions in hand (Table 1, entry 7), we explored the scope of this new one-pot bromination process of nitroaromatics by employing a wide variety of easily available nitroarenes as depicted in Scheme 2. The ability of the reaction to tolerate sensitive functional groups is exceptional, and in many cases, the corresponding bromides were obtained after simple extraction without the need for further purification. Notable performance is accomplished with nitroarenes bearing electron-withdrawing groups (EWG) such as cyano (2a), ester (2c,o) or amide (2d) affording the corresponding bromides in high yields. Remarkably, despite a reductive process taking place, carbonyl-containing substrates were also selectively brominated (2b,e,m,n), without affecting the carbonyl group. Moreover, aryl substituents remained unaltered (2f), as did an α,β-unsaturated ester (2g). Bromides and chlorides were well tolerated (2h–n), even with nitroarenes bearing electron-donating functional groups such as alkyl (2i) or methoxy groups at different positions (2j–l), as well as electron-withdrawing carbonyls (2m–n). The presence of an iodide was also compatible with this strategy (2o). At the same time, polyfunctionalized nitroarenes bearing free hydroxyl (2p) and carboxyl (2q) groups were tolerated. Surprisingly, 1-nitronaphthalene (1w) led to a low yield of the corresponding bromide 2w, whereas CF3-functionalized 1ε led to decomposition (Scheme 2). Remarkably, this protocol could be applied to the gram-scale preparation of the selected bromide 2b, which was obtained in 84% yield (877 mg) starting from 4 mmol of 1b (Scheme 2).
 | ||
| Scheme 2 Scope of the direct bromination of nitroarenes 1. Gram-scale synthesis of 2b. | ||
After having established the suitable conditions for the direct bromination of nitroarenes 1, we proceeded to study their one-pot direct chlorination. Starting from analogous reaction conditions (MeCN as the solvent but CuCl as the copper salt), a screening of some Brønsted acids was performed using model starting material 1a.16 Although HCl, in an analogous way to HBr in the bromination reactions, proved to be efficient in promoting this transformation leading to a good yield of the corresponding chloride 3a, we found that HBF4 led to 3a in higher yield and without the need for further purification.
Once the optimized conditions were established, the scope of this transformation was examined (Scheme 3). As expected, the reaction proceeded efficiently in the presence of EWGs such as cyano (3a), carbonyl (3b), ester (3c), amide (3d) and even free carboxyl (3s) groups, without requiring further purification, and resulted in very good yields. Halides are also tolerated as illustrated by the use of polyfunctionalized nitroarenes also bearing methoxy (3k) and carbonyl (3m,r) groups. Moreover, biphenyl- and isophthalate-containing nitro substrates remained unscathed, leading to the corresponding chlorides (3t,u). Unexpectedly some starting nitroarenes such as 1h, l, andv failed to readily undergo the direct chlorination, leading to low yields (10–20%) of the corresponding chlorides (Scheme 3).
 | ||
| Scheme 3 Scope and limitations for the direct chlorination of nitroarenes 1. | ||
At the same time, we studied the one-pot direct iodination of nitroarenes. Given the strong nucleophilicity of the iodide anion, experiments were performed in the absence of copper salts, but using potassium iodide instead (Table 2). Initially, the employment of PTSA as an acid, to promote diazotization, and toluene as the solvent led to the formation of the desired iodide 4a in moderate to good yields (entries 1–3). However, we again observed some unidentified byproducts, likely due to a competitive reaction of the in situ formed diazonium salt with the solvent. Therefore, we switched again to acetonitrile as the solvent and different acids were tested, maintaining KI as the source of iodide (entries 4–8). The best results were obtained when HBF4 was employed (entry 6). As expected, in the absence of an acid intermediate aniline and an unidentified byproduct were observed (entry 9). Finally, it was also checked that the use of CuI did not lead to improved yields compared to the use of KI (entry 10 vs. 6 and 7).16
|
|
||||
|---|---|---|---|---|
| Entry | Acid (x equiv.) | Iodide (x equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol), under air. Complete conversion of the starting nitroarene and the intermediate aniline was observed in all the cases, unless otherwise stated. b Yield obtained by NMR using 1,3,5-trimethoxybenzene as the internal standard. c Isolated yield. d No complete conversion of the amine. | ||||
| 1 | PTSA (1.5) | KI (2.5) | Toluene | 57 |
| 2 | PTSA (2) | KI (2.5) | Toluene | 76 |
| 3 | PTSA (2) | KI (2) | Toluene | 71 |
| 4 | HI (1.5) | KI (2.5) | MeCN | 40 |
| 5 | HBr (1.5) | KI (2.5) | MeCN | 63 |
| 6 | HBF4 (1.5) | KI (2.5) | MeCN | 92 (89)c |
| 7 | HBF4 (1.5) | KI (1.5) | MeCN | 83 |
| 8 | PTSA (1.5) | KI (2.5) | MeCN | 43 |
| 9 | — | KI (2.5) | MeCN | 28d |
| 10 | HBF4 (1.5) | CuI (1.5) | MeCN | 83 |

After the optimization of the reaction, we proceeded to study the scope of this new one-pot iodination process by employing a wide variety of readily available nitroarenes (Scheme 4). Once again, this methodology shows a remarkable ability to tolerate sensitive functional groups. The presence of EWGs such as cyano (4a), carbonyl (4b), ester (4c), amide (4d) and free carboxyl (4s) groups not only is tolerated but also leads to the formation of the corresponding iodides in excellent yields. Moreover, both aryl substituents (4f) and α,β-unsaturated carbonyls (4g) remain unaltered. Alkyl (4i) and methoxy (4k–l) EDGs were also tested and they remained unscathed. Different polyfunctionalized substrates bearing carbonyl groups were converted (4n,r,v), thus demonstrating the exceptional ability of the process to tolerate this potentially reducible group. In addition, the method is compatible with polycyclic nitroarenes (4w). The tolerance towards halides is widely demonstrated throughout the explored scope by the presence of bromine (4h,r), chlorine (4i,k,l,n,y) and fluorine (4v) atoms in either simpler or more sophisticated polyfunctionalized substrates. In addition, simple 4-nitroanisole proved to be a useful substrate for this process affording 4-iodoanisole (4x), although in a slightly lower yield.17
 | ||
| Scheme 4 Scope of the direct iodination of nitroarenes 1. | ||
Surprisingly, when 4-(alkynyl)-1-nitrobenzenes 1z,α were employed as substrates, an additional and regioselective hydration of the triple bond occurred, likely due to the concomitant generation of water during the Mo-catalyzed reduction process. The resulting intermediate anilines were efficiently converted into brominated aryl ketones 2z,α, chlorinated aryl ketones 3z,α, and iodinated aryl ketones 4z,α (Scheme 5). The almost complete regioselectivity of the water addition to the triple bond could be rationalized by the electron-donating nature of the amino group that polarizes the alkyne moiety. This effect was demonstrated in the reaction of a (3-alkynyl)-1-nitrobenzene such as 1β, which under the reported bromination conditions gave rise to the 3-alkynylbromobenzene derivative 2β, thus confirming the compatibility of the alkyne moiety with this process. Interestingly, this is another example in which the byproduct generated in the first step of a sequence (H2O from the reduction of the nitroarene with pinacol) is advantageously employed, in this case as a reagent, for the downstream hydration of the triple bond. The development of such strategies that internally reuse the waste generated in the first step of a sequence as a (co)-catalyst or reagent for subsequent steps is a highly interesting topic in modern organic synthesis.18
 | ||
| Scheme 5 Sequential reduction/hydration/Sandmeyer reaction of 4-(alkynyl)-1-nitrobenzenes 1z,α. | ||
On the other hand, it is worth mentioning the ever-increasing demand for fluorinated compounds, given their importance in medicinal and agrochemical chemistry as well as in materials science.19 Consequently, there is continuous interest in developing efficient methods for preparing fluorine-containing organic molecules.20 In this field, the Balz–Schiemann reaction21 is one of the most conventional methods for synthesizing aryl fluorides.22 This reaction based on the decomposition of aryl diazonium salts under heating in the presence of a fluoride source was demonstrated to be more challenging due to the lower reactivity of the fluoride.
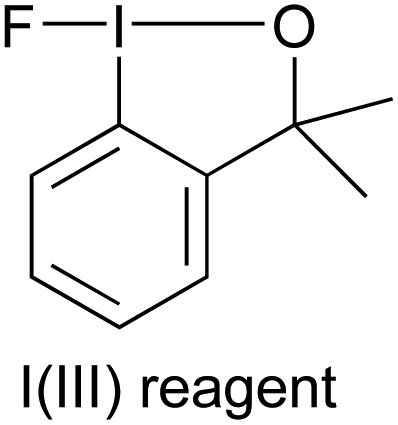
Hence, we decided to tackle the last and most challenging step of this work, which would be the one-pot fluorination of nitroarenes. After trying some approaches for the one-pot diazotization–fluorination sequence,16 we focused our attention on a previously communicated procedure reported by Hu and co-workers.23 We thought that their hypervalent iodine(III)-catalyzed Balz–Schiemann fluorination24 could be combined with our reduction of nitroarenes in a one-pot operation. Therefore, we then tried to use 1-fluoro-3,3-dimethylbenziodoxole [I(III)] in substoichiometric amounts as an additive to promote the fluorination reaction (Table 3). Following the reported method, initially, BF3·OEt2 was used as an acid for the diazotization with tBuONO in α,α,α-trifluorotoluene as the solvent at 0 °C for 15 min. After that, a substoichiometric amount of the commercially available iodine(III) reagent 1-fluoro-3,3-dimethylbenziodoxole was added and the reaction was heated overnight at 100 °C, leading to the expected fluoride 5f but with a low yield. In addition, the intermediate 2-aminobiphenyl (∼22%) was also obtained (entry 1). We then decided to change the acid to HBF4 trying to achieve a complete conversion of the intermediate aniline. A moderate isolated yield was afforded at 100 °C (entry 2). Gratifyingly, lowering the reaction temperature and time led to improved results (entries 3 and 4). The best results were achieved at 60 °C in only 3 h (entry 5). It was finally checked that further decreasing the temperature affected the obtained yield negatively (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Acid | Temp (°C) | t (h) | Yieldb (%) |
| a Reaction conditions: 1f (1 mmol), under air. Complete conversion of the starting nitroarene and the intermediate aniline was observed in all the cases, unless otherwise stated. b Isolated yield referred to 2-nitrobiphenyl 1f. c ∼22% of 2-aminobiphenyl was obtained. | ||||
| 1 | BF3·OEt2 | 100 | 19 | 30c |
| 2 | HBF4 | 100 | 19 | 39 |
| 3 | HBF4 | 80 | 19 | 49 |
| 4 | HBF4 | 80 | 3 | 58 |
| 5 | HBF4 | 60 | 3 | 64 |
| 6 | HBF4 | 40 | 3 | 56 |
|
||||

With an optimized set of conditions in hand, we explored the scope of this one-pot fluorination process starting from easily available nitroarenes as depicted in Scheme 6. As observed in previous transformations with other halides, the carbonyl groups of benzophenones (5b,v) and acetophenones (5δ) are not reduced. Moreover, aryl substituents at different positions of the ring remain unaltered (5f,t), including a more sterically demanding example featuring two tert-butyl groups (5γ). α,β-Unsaturated carbonyls are also tolerated (5g). In addition, the method is compatible with polycyclic nitroarenes, delivering the corresponding fluoride in good yield (5w). However, even though the best yield was obtained when heating at 60 °C for the model substrate 1f, some nitroarenes required a higher temperature to maximize the yield. Although moderate to good yields were obtained for a selection of nitroarenes, some of the substrates that efficiently underwent the bromination, chlorination, or iodination reactions failed in this case, leading to decomposition under the standard conditions.25
 | ||
| Scheme 6 Scope and limitations of the direct fluorination of nitroarenes 1. | ||
Conclusions
In summary, we have demonstrated that the dioxomolybdenum-catalyzed reduction is compatible with subsequent diazotization followed by Sandmeyer or Balz–Schiemann reactions enabling a one-pot synthesis of haloaromatics from nitroarenes. Moreover, the developed method proved to be an efficient, step-economical, practical, scalable, chemoselective, and air and moisture-tolerant procedure for the synthesis of haloaromatics directly from nitroarenes in a one-pot procedure. As these results demonstrate, a wide variety of aryl bromides, chlorides, iodides, and even more challenging fluorides can be synthesized through this nitro reduction–diazotization–halogenation sequence, circumventing intermediate workup operations and purification steps. Consequently, not only are the practical aspects of more traditional synthetic routes simplified but also a minimization of both waste generation and time is realized.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033 (PID2020-115789GB-C21) and the Junta de Castilla y León and FEDER (BU049P20) for financial support. R. H.-R. and S. G.-G. thank the Ministerio de Educación for FPU predoctoral contracts. S. S.-P. thanks the Ministerio de Ciencia e Innovación and the “NextGenerationEU”/PRTR EU for a Ramón y Cajal contract (RYC2021-031533-I).References
- (a) Y. Hayasi, Chem. Sci., 2016, 7, 866–880 RSC; (b) Y. Hayasi, Acc. Chem. Res., 2021, 54, 1385–1398 CrossRef PubMed.
- For selected reviews, see: (a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed; (b) P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721–5745 CrossRef CAS; (c) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed; (d) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (e) J.-Q. Xie, R.-X. Liang and Y.-X. Jia, Chin. J. Chem., 2021, 39, 710–728 CrossRef CAS.
- See, for instance: (a) J. Latham, E. Bradenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 CrossRef CAS PubMed; (b) N. K. Shinada, A. G. de Brevern and P. Schmidtke, J. Med. Chem., 2019, 62, 9341–9356 CrossRef CAS PubMed; (c) K. Mu, Z. Zhu, A. Abula, C. Peng, W. Zhu and Z. Xu, J. Med. Chem., 2022, 65, 4424–4435 CrossRef CAS PubMed.
- For selected reviews, see: (a) H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS PubMed; (b) W. A. Cowdrey and D. S. Davies, Q. Rev., Chem. Soc., 1952, 6, 358–379 RSC; (c) F. Csende, Mini-Rev. Org. Chem., 2015, 12, 5618–5627 Search PubMed; (d) R. Akhtar, A. F. Zahoor, N. Rasool, M. Ahmad and K. G. Ali, Mol. Divers., 2022, 26, 1837–1873 CrossRef CAS PubMed.
- T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 2650–2653 CrossRef.
- See, for instance: (a) M. P. Doyle, B. Siegfried and J. F. Dellaria, J. Org. Chem., 1977, 42, 2426–2431 CrossRef CAS; (b) E. A. Krasnokutskaya, N. I. Semenischeva, V. V. Filimonov and P. Knochel, Synthesis, 2007, 81–84 CrossRef CAS; (c) M. E. Trusova, E. A. Krasnokutskaya, P. S. Postnikov, Y. Choi, C.-W. Chi and V. D. Filimonov, Synthesis, 2011, 2154–2158 CAS; (d) D. A. Leas, Y. Dong, J. L. Vennerstrom and D. E. Stack, Org. Lett., 2017, 19, 2518–2521 CrossRef CAS PubMed; (e) M. D. Bochare and M. S. Degani, ACS Sustainable Chem. Eng., 2017, 5, 3716–3720 CrossRef CAS; (f) S. Mukhopadhyay and S. Batra, Chem. – Eur. J., 2018, 24, 14622–14626 CrossRef CAS PubMed; (g) N. Sivendran, F. Belitz, D. S. Prendes, A. M. Martínez, R. Schmid and L. J. Gooßen, Chem. – Eur. J., 2022, 28, e202103669 CrossRef CAS PubMed.
- (a) R. Pschorr, Ber. Dtsch. Chem. Ges., 1896, 29, 496–501 CrossRef CAS; (b) M. Gomberg and W. E. Bachmann, J. Am. Chem. Soc., 1924, 46, 2339–2343 CrossRef CAS; (c) H. Meerwin, E. Büchner and K. van Emster, J. Prakt. Chem., 1939, 152, 237–266 CrossRef.
- C–P bonds: (a) S. Wang, D. Qiu, F. Mo, Y. Zhang and J. Wang, J. Org. Chem., 2016, 81, 11603–11611 CrossRef CAS PubMed; (b) M. Estruch-Blasco, D. Felipe-Blanco, I. Bosque and J. C. González-Gómez, J. Org. Chem., 2020, 85, 14473–14485 CrossRef CAS PubMed. C–S bonds: (c) S. Chen, S. Cao, C. Liu, B. Wang, X. Ren, H. Huang, Z. Peng and X. Wang, Org. Lett., 2021, 23, 7428–7433 CrossRef CAS PubMed; (d) T. Zhong, M.-K. Pang, Z.-D. Chen, B. Zhang, J. Weng and G. Lu, Org. Lett., 2020, 22, 3072–3078 CrossRef CAS PubMed. C–S bonds: (e) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS PubMed; (f) D. Qiu, Y. Zhang and J. Wang, Org. Chem. Front., 2014, 1, 422–425 RSC. C–S bonds: (g) D. Qiu, H. Meng, L. Jin, S. Wang, S. Tang, X. Wang, F. Mo, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 11581–11584 CrossRef CAS PubMed. C–S bonds: (h) M. Cui, R. Wang, Q. Yang and C. Kuang, J. Org. Chem., 2022, 87, 9654–9662 CrossRef CAS PubMed. C–CF3 bonds: (i) J.-J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z.-J. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436–8439 CrossRef CAS PubMed; (j) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330–10333 CrossRef CAS PubMed; (k) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 7972–7975 CrossRef CAS PubMed; (l) B. Bayarmagnai, C. Matheis, E. Risto and L. J. Gooßen, Adv. Synth. Catal., 2014, 356, 2343–2348 CrossRef CAS. C–OCF3 bonds: (m) Y.-M. Yang, J.-F. Yao, W. Yan, Z. Luo and Z.-Y. Tang, Org. Lett., 2019, 21, 8003–8007 CrossRef CAS PubMed. For a review, see: (n) F. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496–506 CrossRef CAS PubMed.
- See, for instance: (a) C. Molinaro, J. Mowat, F. Gosselin, P. D. O′Shea, J.-F. Marcoux, R. Angelaud and I. W. Davies, J. Org. Chem., 2007, 72, 1856–1858 CrossRef CAS PubMed; (b) F. P. Crisóstomo, T. Martín and R. Carrillo, Angew. Chem., Int. Ed., 2014, 53, 2181–2185 CrossRef PubMed; (c) S. Kindt, K. Wicht and M. R. Heinrich, Org. Lett., 2015, 17, 6122–6125 CrossRef CAS PubMed; (d) S. Kindt, K. Wicht and M. R. Heinrich, Angew. Chem., Int. Ed., 2016, 55, 8744–8747 CrossRef CAS PubMed; (e) D. Felipe-Blanco and J. C. González-Gómez, Adv. Synth. Catal., 2018, 360, 2773–2778 CrossRef CAS.
- For selected reviews, see: (a) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed; (b) F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC; (c) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed.
- For selected recent references, see: (a) B. Feng, Y. Yang and J. You, Chem. Commun., 2020, 56, 790–793 RSC; (b) G. Li, L. Yang, J.-J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS PubMed; (c) Z. Bao, J. Zhou, C. Mou, Z. Jin, S.-C. Ren and Y. R. Chi, Org. Lett., 2022, 24, 8907–8913 CrossRef CAS PubMed; (d) S. Wang, T. Li, C. Gu, J. Han, C.-G. Zhao, C. Zhu, H. Tan and J. Xie, Nat. Commun., 2022, 13, 2432 CrossRef CAS PubMed; (e) G. Li, M. N. Lavagnino, S. Z. Ali, S. Hu and A. T. Radosevich, J. Am. Chem. Soc., 2023, 145, 41–46 CrossRef CAS PubMed; (f) J. Yao, L. Yu, W. Duan and C.-J. Li, Org. Chem. Front., 2023, 10, 524–530 RSC; (g) J. Bai, S. Li, R. Zhu, Y. Li and W. Li, J. Org. Chem., 2023, 88, 3714–3723 CrossRef CAS PubMed; (h) L. Duff, H. Meakin, A. Richardson, A. J. Greener, G. W. A. Smith, I. Ocaña, V. Chechik and M. J. James, Chem. – Eur. J., 2023, 29, e202203807 CrossRef CAS PubMed. For a review, see: (i) D. Zou, W. Wang, Y. Hu and T. Jia, Org. Biomol. Chem., 2023, 21, 2254–2271 RSC.
- (a) N. García, P. García-García, M. A. Fernández-Rodríguez, R. Rubio, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Adv. Synth. Catal., 2012, 354, 321–327 CrossRef. For a related reduction of heteroaromatic N-oxides, see: (b) R. Rubio-Presa, M. A. Fernández-Rodríguez, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Adv. Synth. Catal., 2017, 359, 1752–1757 CrossRef CAS.
- (a) R. Rubio-Presa, M. R. Pedrosa, M. A. Fernández-Rodríguez, F. J. Arnáiz and R. Sanz, Org. Lett., 2017, 19, 5470–5473 CrossRef CAS PubMed; (b) R. Rubio-Presa, S. Suárez-Pantiga, M. R. Pedrosa and R. Sanz, Adv. Synth. Catal., 2018, 360, 2216–2220 CrossRef CAS; (c) R. Hernández-Ruiz, R. Rubio-Presa, S. Suárez-Pantiga, M. R. Pedrosa, M. A. Fernández-Rodríguez, M. J. Tapia and R. Sanz, Chem. – Eur. J., 2021, 27, 13613–13623 CrossRef PubMed.
- For reviews about dioxomolybdenum(VI)-catalyzed reactions, see: (a) R. Hernández-Ruiz and R. Sanz, Synthesis, 2018, 4019–4036 Search PubMed; (b) S. Suárez-Pantiga and R. Sanz, Org. Biomol. Chem., 2021, 19, 10472–10492 RSC.
- (a) O. C. Slotterbeck, Trans. Electrochem. Soc., 1947, 92, 377–390 CrossRef; (b) F. D. Popp and H. P. Schultz, Chem. Rev., 1962, 62, 19–41 CrossRef CAS; (c) H. Nohe, F. Beck, D. Wetner and E.-J. Schier, U.S. Patent, 3984294A1, 1976 Search PubMed.
- See the ESI† for further details.
- The corresponding bromination and chlorination reactions of 4-nitroanisole (1x) failed, likely due to the incompatibility with the conditions used for the Sandmeyer reaction, as the initial reduction of the nitro group with pinacol is clean after 40 min.
- For pioneering work, see: (a) T. Konoshita, S. Okada, S.-R. Park, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2003, 42, 4680–4684 CrossRef PubMed. See, also: (b) J. Zhou and X.-P. Zeng, in Multicatalyst System in Asymmetric Catalysis, Ed. J. Zhou, John Wiley & Sons, New York, 2014, Chapter 9 CrossRef.
- (a) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochisnky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (c) R. Ragni, A. Punzi, F. Babudri and G. M. Farinola, Eur. J. Org. Chem., 2018, 3500–3519 CrossRef CAS; (d) N. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) Y. Li, Y. Wu, G.-S. Li and X.-S. Wang, Adv. Synth. Catal., 2014, 356, 1412–1418 CrossRef CAS; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed; (d) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed; (e) S. Caron, Org. Process Res. Dev., 2020, 24, 470–480 CrossRef CAS.
- G. Balz and G. Schiemann, Chem. Ber., 1927, 60, 1186–1190 CrossRef.
- For selected reports, see: (a) D. J. Milner, Synth. Commun., 1992, 22, 73–82 CrossRef CAS; (b) K. K. Laali and V. J. Gettwert, J. Fluorine Chem., 2001, 107, 31–34 CrossRef CAS; (c) L. Garel and L. Saint-Jalmes, Tetrahedron Lett., 2006, 47, 5705–5708 CrossRef CAS; (d) J. Heredia-Moya and K. L. Kirk, J. Fluorine Chem., 2007, 128, 674–678 CrossRef CAS PubMed; (e) Z.-q. Yu, Y.-w. Lv, C.-m. Yu and W.-k. Su, Tetrahedron Lett., 2013, 54, 1261–1263 CrossRef CAS; (f) N. H. Park, T. J. Senter and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 11907–11911 CrossRef CAS PubMed; (g) T. M. El Dine, O. Sadek, E. Gras and D. M. Perrin, Chem. – Eur. J., 2018, 24, 14933–14937 CrossRef PubMed; (h) L. Yang and C.-P. Zhang, ACS Omega, 2021, 6, 21595–21603 CrossRef CAS PubMed; (i) R. M. Pérez-García, G. Grøvnnevik and P. J. Riss, Org. Lett., 2021, 23, 1011–1015 CrossRef PubMed.
- B. Xing, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2018, 57, 9896–9900 CrossRef CAS PubMed.
- For other fluorination reactions using fluoro-benziodoxole hypervalent iodine(III) reagents, see also: (a) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897–12901 CrossRef CAS PubMed; (b) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, Chem. Commun., 2013, 49, 9263–9265 RSC.
- The reason for the limited scope of the fluorination reaction is not clear at this moment, but competitive hydroxylation processes, due to the presence of water in the reaction medium, were observed with some substrates. In addition, classical protocols for the Balz–Schiemann reaction typically require isolation and drying of the aryl diazonium salt to avoid competitive reaction pathways.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01187a |
| This journal is © The Royal Society of Chemistry 2023 |