
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible conformational switching of a photo-responsive ortho-azobenzene/2,6-pyridyldicarboxamide heterofoldamer†
Sarah J.
Pike
 *ab,
Richard
Telford
b and
Louise
Male
a
*ab,
Richard
Telford
b and
Louise
Male
a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: s.j.pike@bham.ac.uk
bSchool of Chemistry and Biosciences, Faculty of Life Sciences, University of Bradford, Bradford, West Yorkshire BD7 1DP, UK
First published on 11th August 2023
Abstract
We report on a convenient synthetic route to rapidly access a new photo-responsive ortho-azobenzene/2,6-pyridyldicarboxamide heterofoldamer. The adoption of a stable helical conformation has been established for this scaffold in both the solid state and in solution using single crystal X-ray diffraction and circular dichroism (CD) spectroscopy respectively. Reversible control over the stimuli-driven structural re-ordering of the supramolecular scaffold, from a stable helical conformation under non-irradiative conditions, to a less well-ordered state under irradiative conditions, has been identified. The robust nature of the responsive, conformational, molecular switching behaviour has been determined using UV/Vis, 1H NMR and CD spectroscopy. Minimal loss in the efficiency of the stimuli-driven, structural re-ordering processes of the foldamer scaffold is observed, even upon multiple cyclic treatments with irradiative/non-irradiative conditions.
Introduction
In nature, the ability of biomolecules to demonstrate controlled global conformational changes in response to the presence of external stimuli (e.g., light) is integral for the effective execution of vital functions in our bodies.1,2 Stimuli-responsive conformational switching events, performed by biological systems, often need to be executed in both a reversible and robust manner. The implementation of such efficient operational practices permits function to be achieved with little to no fatigue, thus, ensuring that the system demonstrates limited reduction to its’ operational efficiency over time.Foldamers are synthetic helical oligomers that adopt stable secondary structures in solution.3 In recent years, there has been a significant drive to create stimuli-responsive foldamers that have the ability to, in part, replicate the conformational switching behaviour demonstrated by complex natural systems. Stimuli-responsive foldamers have attracted growing interest due their widespread applications in a number of fields including synthesis, as organocatalysis4 and supramolecular chemistry, as sensors and probes,5 receptors6,7 and for signalling applications.8 The emergent biological properties of foldamers as potential antibiotics has also garnered significant attention.9
Photo-responsive foldamers are attractive scaffolds as they offer a reliable and reversible method by which to control the conformational preferences of these supramolecular systems.10–18 Generally, photo-responsive foldamers are able to demonstrate controlled molecular switching between a stable helical conformation to a random coil or less well-folded state, in the presence and absence of light, respectively.10,11 To achieve photo-driven conformational molecular switching in these supramolecular scaffolds, photo-responsive units (e.g. azobenzene or spiropyran) are incorporated at the side-chain14–16 or in the backbone17,18 of the oligomer. Foldamers based on the latter design strategy are often able to demonstrate higher levels of control over the conformational switching behaviour of the scaffold due to their importance in maintaining the integrity of the global folded structure.19
Many photo-responsive foldamers are created using the heterofoldamer design approach, wherein, the foldamer scaffold is composed of two or more distinct monomer building blocks.16–20 The implementation of this design approach is highly attractive as it offers a convenient method by which structural diversity can be directly incorporated into the scaffold. In 2006, Parquette and co-workers elegantly exploited the heterofoldamer design approach to create a class of photo-responsive foldamers that incorporated alternating sequences of pyridine-2,6-dicarboxamide and meta-(phenylazo)azobenzene units within the oligomer backbone.18a However, despite recent advances in the development of photo-responsive foldamers, existing routes to access these supramolecular systems can be synthetically demanding, for example, in Parquette's original scaffold 12 steps are required to access a two-turn foldamer.18a,21 Additionally, information about the robustness of the light-driven molecular switching behavior of photo-responsive foldamers can also be limited. These drawbacks can both restrict the accessibility of these supramolecular scaffolds and hinder our understanding of their operational practices, which in turn, can limit our ability to optimize the future design of these responsive systems.
Herein, we outline on the development of a short five-step synthetic route to rapidly access a photo-responsive heterofoldamer based on an alternating ortho-azobenzene/2,6-pyridyldicarboxamide motif. The light-responsive heterofoldamer described in this work is related to Parquette's original scaffold18a but the simplified design approach reported, herein, enables a significantly more streamlined synthetic route to be adopted, thus, considerably increasing its synthetic accessibility. The controlled and reversible modulation of conformational preferences of this photo-responsive ortho-azobenzene/2,6-pyridyldicarboxamide heterofoldamer has been established using UV/Vis, circular dichroism (CD) and 1H NMR spectroscopy. Additionally, the robust nature of the molecular, stimuli-induced, conformational switching events demonstrated by this photo-responsive scaffold, has been identified, with low levels of fatigue being observed within the system, even upon multiple cyclic treatments with irradiative and non-irradiative conditions.
Results and discussion
Design and synthesis of photo-responsive foldamers scaffold
Several key design considerations were taken into account when generating photo-responsive foldamer 1 (Fig. 1). Firstly, the foldamer must be able to demonstrate reversible stimuli-responsive, conformational switching between a stable helix in the absence of light to a less stable/partially unfolded structure in the presence of light. This reversible molecular switching event should be robust enough to withstand multiple iterative treatments with external stimuli in order to establish the ability of this scaffold to be repetitively cycled between folded and unfolded states with minimal fatigue to the system. Secondly, the photo-responsive foldamer 1 must be chiral in order to permit the stimuli-induced, global conformational switching behaviour of the scaffold to be directly monitored in solution, using CD spectroscopy. Finally, the foldamer should be readily generated using a short and robust synthetic route to permit rapid synthetic accessibility of the scaffold, which will facilitate facile optimization of the scaffold in the longer-term. | ||
| Fig. 1 Design of synthetically accessible heterofoldamer scaffold composed of alternating ortho-azobenzene (shown in red) and 2,6-pyridyldicarboxamide monomers (shown in blue) as fold-promoting motifs and with the presence of chiral groups at the termini as spectroscopic handles (shown in green) to monitor conformational behaviour of the system in solution. | ||
To accommodate these key design considerations, a simple heterofoldamer design strategy, based on incorporating two well-established fold-promoting motifs of ortho-azobenzene and 2,6-pyridyldicarboxamide monomers, into the backbone of the foldamer was employed (Fig. 1). Crucially, the ortho-azobenzene units in 1 adopt the E-geometrical isomer under non-irradiative conditions which promotes the formation of a helical structure in the foldamers, whilst photo-induced isomerisation of the azobenzene units to the Z-isomer perturbs the helical the scaffold leading to a partially unfolded state upon the application of light.10,11 The helical conformation of 1 is further supported by the inclusion of the central 2,6-pyridyldicarboxamide unit, whose adoption of a syn–syn conformation is a fold-promoting motif commonly used in aromatic oligoamide foldamers and related photo-responsive heterofoldamers (Fig. 1).18,22 Moreover, the well-established reversible and robust nature of the stimuli-induced isomerization of azobenzene units provides the potential for the scaffold to demonstrate the desired repetitive photo-driven conformational switching behaviour with low levels of fatigue.10,11 In 1, (S)-(+)-naproxen groups are incorporated at the termini of the scaffold to bias the system towards the preferential adoption of one helical screw-sense. The positioning of the enantiopure groups at the termini of the scaffold ensures that the conformational preferences of the two fold-promoting motifs, ortho-azobenzene and the 2,6-pyridyldicarboxamide, are not perturbed and, hence, that a stable helical conformation is adopted by the scaffold in the absence of light.
Compound 1 was readily synthesized from 2-aminoacetanilide 2 in a five-step procedure (Scheme 1). Firstly, the treatment of 2 with oxone in a biphasic solvent mixture of dichloromethane/water, led to conversion of the amine group to the corresponding nitroso group, generating 3 as a bright green solid in 71% yield.23,243 underwent a Baeyer–Mills reaction with o-phenylenediamine upon refluxing in toluene, in the presence of acetic acid, to afford 2-acetamido-2′-amino-ortho-azobenzene 4 in 68% yield.23 Treatment of 4 with 2,6-pyridinedicarbonyl chloride, in the presence of pyridine, generated the acetyl terminated oligomer 5 as a bright orange solid in 52% yield. Removal of the acetyl protecting groups in 5 was achieved upon treatment with KOH in an acetone/water/methanol solvent mixture to give the corresponding diamine 6 in 45% yield. The diamine was reacted with an excess of (S)-(+)-naproxen chloride in the presence of pyridine to give 1 in 72% yield.
 | ||
| Scheme 1 (i) Oxone, CH2Cl2/H2O, rt, 1 h, 71%; (ii) o-phenylenediamine, toluene, acetic acid, 60 °C, 18 h, 68%; (iii) 2,6-dipyridinecarbonyl chloride, pyridine, CH2Cl2, 4 Å MS, 48 h, 52%; (iv) KOH, acetone/water/methanol, 70 °C, 18 h, 45%; (v) (S)-(+)-naproxen, pyridine, CH2Cl2, rt, 48 h, 72%. | ||
Conformational analysis and molecular switching behaviour of foldamer
The ability of compound 1 to adopt a stable helical conformation was first investigated in the solid state using crystal X-ray diffraction analysis.25 Single crystals of 1 suitable for X-ray diffraction, were obtained through the slow diffusion of di-isopropyl ether into a chloroform solution (Fig. 2). In the unit cell, there are two crystallographically-independent molecules of 1 and three chloroform molecules, of which two of the solvent molecules are disordered. Solid state analysis of 1, identified that both molecules adopt a stable folded conformation displaying P-helicity, and each molecule consists of a one-turn helix with a pitch of 3.4 Å and possesses a central cavity running along the a axis (Fig. 2). | ||
| Fig. 2 X-ray structure of 1 (a) viewed from the a axis showing the central cavity within the foldamers, (b) showing a side-on view, highlighting the crystal packing arrangement in the solid-state with columnar stacks of helices. C atoms are shown in grey, O in red, N in light blue. Three molecules of chloroform and H atoms have been removed for clarity. | ||
The importance of the two structural fold-promoting design features, of E-geometries of the azobenzene units and syn–syn conformation of 2,6-pyridyldicarboxamide unit in the backbone of the heterofoldamer scaffold of 1 is observed in the solid state. The adoption of the latter fold-promoting motif,20,25 results in the pyridyl amide protons being directed in towards the central cavity of the helix, leading to the formation of an intramolecular bifurcated hydrogen-bonding interaction26 involving the pyridyl N atom and the NH's of the adjacent amide bonds as anticipated (2.706(7)–2.742(7) Å, see Fig. S27 and S28†).27 In the crystal lattice of 1, the helices that aligned in columnar stacks orientated along the a axis (Fig. 2b). This crystal packing arrangement is supported by a diverse array of intermolecular non-covalent interactions formed between molecules within the same and adjacent columnar stacks including, C–H⋯O hydrogen bonding interactions28 involving aromatic and aliphatic protons and the O atoms of the carbonyl groups in neighbouring molecules, off-set π–π stacking interactions29 and C–H⋯π interactions30 (see ESI, Fig. S38–49†). Additionally, there are three intermolecular Cl3C–H⋯O hydrogen-bonding interactions31 involving chloroform solvent molecules and O atoms on the carbonyl group of amide bonds in 1 (see ESI, Fig. S50–52†).
Due to the presence of the azobenzene units in the backbone of the scaffold, 1 exhibits a UV/Vis absorption spectrum that displays a strong π → π* band at 412 nm which overlaps with the n → π* band at 464 nm (Fig. 3). The presence of the pyridyl carboxamide and the naproxen carboxamide groups at the ortho positions of the azobenzene units in 1 causes a larger red shift of the π → π* band compared to unsubstituted E-azobenzene, where it typically occurs at 320 nm. Accordingly, the photo- and thermally-driven configurational E/Z isomerization of the azobenzene units in 1 was studied using UV/Vis spectroscopy. Irradiation of a 4.8 × 10−5 M acetonitrile solution of 1 with 365 nm light induces photo-driven E → Z isomerization of the azo bonds in 1 as demonstrated by the observed decrease in the 412 nm and 464 absorption bands and an observed increase in the 530 nm absorption band corresponding to the Z isomer (Fig. 3).32 A photostationary state (PSS) is reached within 10 min of irradiation and thermal Z → E isomerization of the azo bonds in 1 proceeded over 220 min at ambient temperature, whereupon the reversibility of the photo-switching event was established through complete restoration of the initial UV/Vis signal obtained before irradiation (see Fig. 3).
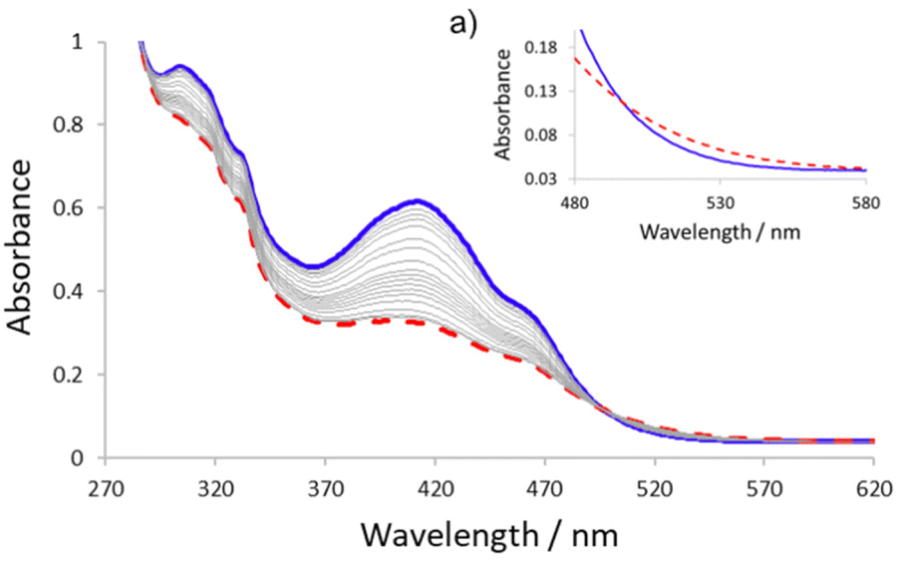 | ||
| Fig. 3 UV/Vis spectra of 1 before irradiation (shown in blue line) and at the photostationary state (PSS) (shown in the dashed red line) after irradiation of a 4.8 × 10−5 M acetonitrile solution with 365 nm for 10 min. Grey lines show thermal relaxation of 1 observed over 220 min. (a) Inset highlighting the region between 480–600 nm. | ||
To gain a better understanding of the photo-driven isomerization processes in 1, 1H NMR spectroscopy was used to follow the stimuli-driven molecular switching event. Irradiation of a 1.1 mM CD3CN sample of 1 with 365 nm light, for 45 min, afforded a PSS (Fig. 4b). Analysis of the 1H NMR spectrum of the PSS showed that, due to the presence of two azo bonds in the scaffold, three isomers (E/E, Z/Z and E/Z) of 1 were generated under irradiative conditions in 28%, 30% and 42% respectively.33 The symmetrical E/E and Z/Z-isomers of 1 display a single resonance for the NH amide proton of the carboxamide unit, Hc, at δ 11.20 ppm and δ 10.39 ppm respectively, wherein the Z-isomer displays significant upfield shifting of δ 0.81 ppm compared to the E-isomer (Fig. 4b).18a Whilst the asymmetrical E/Z isomer of 1 observed in the PSS, displays two resonances corresponding to the NH amide of the carboxamide unit, Hc, at δ 11.33 ppm (in the E-form) and at δ 10.52 ppm (in the Z-form) (Fig. 4b). Thermal Z → E isomerization of 1 occurred at ambient temperature over 285 min as determined by restoration of the original 1H NMR spectrum obtained before exposure to irradiative conditions (Fig. 4a–h).34 The Z isomer exhibited a half-life for the thermally-promoted Z → E isomerization of ∼ 100 min at 298 K (see ESI†).
 | ||
| Fig. 4 Partial 1H NMR spectra (500 MHz, 298 K, CD3CN) of 1.1 mM solution of 1 showing the photo-driven E → Z isomerisation of the azobenzene units within the scaffold: (a) before irradiation, (b) PSS obtained after 45 min irradiation of the sample at 365 nm, (c) 45 min after initial irradiation of the sample, (d) 105 min after initial irradiation of the sample, (e) 135 min after initial irradiation of the sample, (f) 195 min after initial irradiation of the sample, (g) 225 min after initial irradiation, (h) 285 min after initial irradiation, (i) PSS obtained after fourth treatment of the sample with irradiative conditions at 365 min for 45 min (with 285 min thermal relaxation time allowed between each of treatments with irradiative conditions), (j) 280 min after fourth treatment with irradiative conditions. The NH amide proton of the carboxamide unit in 1, HC, is highlighted with the assignments corresponding to the lettering shown in Scheme 1. | ||
To obtain further information about the stimuli-induced changes on the global helical conformational switching behaviour of 1, CD spectroscopy was employed to monitor the system. The CD spectrum of a 6.7 × 10−5 M acetonitrile solution of 1 displays a large minima centred around 472 nm which is in the absorption region associated with the azobenzene units of the scaffold and a maxima centred at 394 nm which is in the absorption region associated with the azobenzene and oligoamide units of the scaffold.18c Accordingly, the observed CD signal indicates the presence of stable secondary helical conformation in solution in 1 in the absence of irradiative conditions (Fig. 5, blue line).18c The signs of the Cotton effects for the peaks at 472 and 394 nm (negative and positive respectively) are indicative of the preferential adoption of P-helicity by 1 in solution and aligns well with conformational preferences observed in the solid state.34 It is the presence of the terminal chiral (S)-(+)-naproxen groups in 1, that shift the equilibrium between left- and right-handed helical conformations, giving the rise to the preferential adoption of one helical screw-sense over the other and, thus leading to the observed CD signals associated with its secondary structure. Irradiation of a 6.7 × 10−5 M acetonitrile solution of 1 with 365 nm, for 10 min, afforded a PSS wherein a significant reduction in the magnitude of the Cotton effects in the negative peak at 472 nm (from −57.8 mdeg to −14.4 mdeg) and the positive peak at 394 nm (from 25.6 mdeg to 1.6 mdeg) (see Fig. 5, dashed red line) is observed. The observed photo-induced changes to these peaks in the CD spectrum of compound 1, indicates a conformational change is seen within the scaffold under irradiative conditions. This photo-induced change could be caused by depopulation of the helical conformation of the scaffold, due to the controlled local photo-induced E → Z isomerization of the azobenzene linkages in the backbone of the system (i.e., helix-coil transition), aggregation of the foldamer in this solvent and/or it could be caused by photo-induced re-orientation of the terminal naproxen group causing a change in the helical twist sense bias of the system (i.e., through chirality transfer from the naproxen-capping group to the foldamer chain).35 Whilst the latter case could be at least, in part, responsible for the observed photo-induced changes in the CD signal, controlled photo-induced conformational changes, in related azobenzene-based foldamers, are often attributed to structural helix-coil transitions.18c,36 Moreover, these literature reports combined with the pronounced conformational switching behaviour observed in 1 by 1H NMR spectroscopy (Fig. 4 and ESI), wherein significant shifting is observed for many of the signals of 1 upon irradiation which indicates that a large conformational change is induced across the whole of the foldamer, suggests that the significant changes observed by CD spectroscopy are likely to be at least, in part, caused by light-driven conformational switching from a globally stable well-folded helix to a partially unfolded scaffold. Thermal Z → E isomerization of the azo bonds in 1 occurs at ambient temperature to completely restore the initial CD signal, after 230 min, indicating regeneration of the original well-folded helical conformation of 1 under non-irradiative conditions (see Fig. 5).
 | ||
| Fig. 5 CD spectra of 1 before irradiation (shown in blue line) and at the photostationary state (PSS) (shown in the dashed red line) after irradiation of a 6.7 × 10−5 M acetonitrile solution with 365 nm for 10 min. Grey lines show thermal relaxation of 1 observed over 230 min. (a) Inset shows light- and thermally-driven cycles of the E–Z isomerisation of the azo bonds of 1. Blue points correspond to 1 in the absence of application of irradiative conditions and red points correspond to the PSS obtained at 472 nm after irradiation of a 6.7 × 10−5 M acetonitrile solution of 1 with 365 nm for 10 min over four cycles. | ||
Robust and reversible conformational switching of the foldamer
The ability of compound 1 to demonstrate reversible photo- and thermally-driven cycling of the molecular conformational switching event, through repeated E/Z isomerizations of the integral azobenzene units in the scaffold, was assessed using UV/Vis, 1H NMR and CD spectroscopy. The 4.8 × 10−5 M UV/Vis, 1.1 mM NMR and 6.7 × 10−5 M CD samples were treated with four repeated cycles of exposure to 365 nm light for 10 min (for UV/Vis and CD samples) and 45 min (for NMR sample) followed by thermal relaxation of 220 min (for the UV/Vis sample), 230 min (for the CD sample) and 285 min (for NMR sample). Spectroscopic analysis of the PSS obtained after each treatment of 1 with exposure to 365 nm light showed that the comparable levels of Z-isomer content were observed even after four cycles (45–51% of Z-isomer by NMR spectroscopy, see Fig. 4 and ESI†) whilst the absorbance of the UV/Vis signals ranged from 0.317 to 0.329 (at 412 nm) in the PSS upon four cycles (see ESI†), and the magnitude of the Cotton effects for the characteristic peaks at 472 and 394 nm ranged from −14.4 mdeg to −13.7 mdeg (at 472 nm) and from 0.2 mdeg to 1.6 mdeg (at 394 nm) upon four cycles (see inset Fig. 5a and ESI†). Accordingly, all three spectroscopic techniques identify that minimal fatigue is observed in the PSS after four cyclic treatments with irradiative/non-irradiative conditions, thus, indicating the robust nature of photo-induced chiral modulation of the local and global conformational preferences of 1. Subsequent thermal Z → E relaxation of the azo bonds in the PSS of 1 was observed after each cycle by 1H NMR, CD and UV/Vis spectroscopy (see Fig. 4, 5 and ESI†) indicating full restoration of the original E/E isomer of the heterofoldamer in all cases.Conclusions
We have developed a simple and short synthetic route to rapidly access a photo-responsive ortho-azobenzene/2,6-pyridyldicarboxamide heterofoldamer scaffold. The conformational stability of the foldamer has been determined in solid state and solution using single-crystal X-ray diffraction and CD spectroscopy respectively. We have established the stimuli-responsive molecular conformational switching behaviour demonstrated by the ortho-azobenzene/2,6-pyridyldicarboxamide heterofoldamer, using UV/Vis, 1H NMR and CD spectroscopy. Additionally, it has been established that the stimuli-induced conformational switching behaviour of the foldamer is robust, with the system demonstrating little fatigue in these molecular switching events, even upon treatment with four cycles of irradiative/non-irradiative conditions. We anticipate that the rapid synthetic accessibility of this heterofoldamer scaffold, combined with its robust, reversible stimuli-responsive behaviour, will serve to open up opportunities for this foldamer class towards important future smart applications, with particular relevance, to the smart sensing of small molecules or catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the UK National Crystallography Service (University of Southampton) for having acquired the crystal data for 1. We thank Dr Richard Grainger (University of Birmingham) and Prof. Robin Perutz (University of York) for access to photochemistry equipment. S. J. P. gratefully acknowledges the Royal Society of Chemistry Research Grant [RF19-4013], Royal Society Research Grant [RGS/R1/231242], Leverhulme Trust Research Project Grant [RPG-2022-118] and the UKRI Future Leaders Fellowship [MR/S035486/2] for financial support. S. J. P. acknowledges the University of Birmingham for support through the Birmingham Fellowship Scheme and the Talent Stabilisation Fund. S. J. P. is a Birmingham Fellow and UKRI Future Leaders Fellow.References
- H. Ishikawa, K. Kwak, J. K. Chung, S. Kim and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8619 CrossRef CAS PubMed.
- J.-H. Ha and S. N. Loh, Chem. – Eur. J., 2012, 18, 7984 CrossRef CAS PubMed.
- (a) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (b) S. H. Gellmann, Acc. Chem. Res., 1998, 31, 173 CrossRef; (c) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed; (d) T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687 RSC; (e) B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852 RSC.
- (a) Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellmann, Science, 2019, 366, 1528 CrossRef CAS PubMed; (b) D. Bécart, V. Diemer, S. Salaün, M. Oiarbide, Y. R. Nelli, B. Kauffmann and G. Guichard, J. Am. Chem. Soc., 2017, 36, 12524 CrossRef PubMed; (c) C. Mayer, M. M. Müller, S. H. Gellmann and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978 CrossRef CAS PubMed; (d) G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13679 CrossRef CAS PubMed.
- (a) Y. Zhao and Z. Zhog, J. Am. Chem. Soc., 2006, 128, 9988 CrossRef CAS PubMed; (b) H.-G. Jeon, H. B. Jang, P. Kang, Y. R. Choi, J. Kim, J. H. Lee, M.-G. Choi and K.-S. Jeong, Org. Lett., 2016, 18, 4404 CrossRef CAS PubMed; (c) K. M. Bąk, K. Masłowska and M. J. Chmielewski, Org. Biomol. Chem., 2017, 15, 5968 RSC; (d) F. G. A. Lister, N. Eccles, S. J. Pike, R. A. Brown, G. F. S. Whitehead, J. Raftery, S. J. Webb and J. Clayden, Chem. Sci., 2018, 9, 6860 RSC; (e) S. J. Pike, M. De Poli, W. Zawodny, J. Raftery, S. J. Webb and J. Clayden, Org. Biomol. Chem., 2013, 11, 3168 RSC; (f) M. G. Lizio, V. Andrushchenko, S. J. Pike, A. D. Peters, G. F. S. Whitehead and I. J. Vitórica-YrezábalS. T. Mutter, J. Clayden, P. Bouř, E. W. Blanch and S. J. Webb, Chem. – Eur. J., 2018, 24, 9399 CrossRef PubMed.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119 CrossRef CAS PubMed.
- A. Tanatani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325 CrossRef CAS PubMed.
- (a) F. G. A. Lister, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420 CrossRef CAS; (b) M. D. Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575 CrossRef PubMed; (c) J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680 CrossRef CAS PubMed.
- A. D. Peters, S. Borsley, F. della Sala, D. F. Cairns-Gibson, M. Leonidou, J. Clayden, G. F. S. Whitehead, I. Vitórica-Yrezábel, E. Takano, J. Burthem, S. L. Cockcroft and S. J. Webb, Chem. Sci., 2020, 11, 7023 RSC.
- Z. Yu and S. Hecht, Chem. Commun., 2016, 52, 6639 RSC.
- (a) S. Pramanik, B. Kauffmann, S. Hecht, Y. Ferrand and I. Huc, Chem. Commun., 2021, 57, 93 RSC; (b) A. Khan and S. Hecht, Chem. – Eur. J., 2006, 12, 4764 CrossRef CAS PubMed; (c) J. Atcher, P. Mateus, B. Kauffmann, F. Rosu, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2021, 60, 2574 CrossRef CAS PubMed.
- (a) Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630 CrossRef CAS PubMed; (b) Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838 CrossRef CAS PubMed; (c) S. Lee, Y. Hua and A. H. Flood, J. Org. Chem., 2014, 79, 8383 CrossRef CAS PubMed.
- (a) M. Pollastrini, G. Marafon, J. Clayden and A. Moretto, Chem. Commun., 2021, 57, 2269 RSC; (b) G. Marafon, M. Crisma, A. Masato, N. Plotegher, L. Bubacco and A. Moretto, Angew. Chem., Int. Ed., 2021, 60, 5173 CrossRef CAS PubMed; (c) D. Mazzier, M. Crisma, M. De Poli, G. Marafon, C. Peggion, J. Clayden and A. Moretto, J. Am. Chem. Soc., 2016, 138, 8007 CrossRef CAS PubMed.
- A. Ueno, K. Takahashi, J. Anzai and T. Osa, J. Am. Chem. Soc., 1981, 103, 6410 CrossRef CAS.
- H. Sogawa, M. Shiotsuki, H. Matsuoka and F. Sanda, Macromolecules, 2011, 44, 3338 CrossRef CAS.
- F. Ciardelli, D. Fabbri, O. Pieroni and A. Fissi, J. Am. Chem. Soc., 1989, 111, 3470 CrossRef CAS.
- Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 1640 CrossRef CAS PubMed.
- (a) C. Tie, J. C. Gallucci and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 1162 CrossRef CAS PubMed; (b) C. J. Gabriel and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 13708 CrossRef CAS PubMed; (c) E. D. King, P. Tao, T. T. Sanan, C. M. Hadad and J. R. Parquette, Org. Lett., 2008, 10, 1671 CrossRef CAS PubMed.
- (a) A. Roy, P. Prabhakaran, P. K. Baruah and G. J. Sanjayan, Chem. Commun., 2011, 47, 11593 RSC; (b) V. V. E. Ramesh, S. S. Kale, A. S. Kotmale, R. L. Gawade, V. G. Puranik, P. R. Rajamohanan and G. J. Sanjayan, Org. Lett., 2013, 15, 1504 CrossRef CAS PubMed; (c) P. K. Baruah, N. K. Sreedevi, B. Majumdar, R. Pasricha, P. Poddar, R. Gonnade, S. Ravindranathan and G. J. Sanjayan, Chem. Commun., 2008, 712 RSC.
- (a) C. M. Grison, S. Robin and D. J. Aitken, Chem. Commun., 2016, 52, 7802 RSC; (b) H. Wu, Q. Qiao, P. Teng, Y. Hu, D. Antoniadis, X. Zuo and J. Cai, Org. Lett., 2015, 17, 3524 CrossRef CAS PubMed.
- The 12 step synthesis outlined by Parquette and co-workers which were undertaken in ref. 18a to access a two turn pyridine-2,6-dicarboxamide and meta-(phenylazo)azobenzene heterofoldamer includes the synthetic steps required to access 2-nitrosoacetanilide which is employed twice within the synthetic route.
- (a) I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS; (b) H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316 RSC.
- S. Bellotto, R. Reuter, C. Heinis and H. A. Wegner, J. Org. Chem., 2011, 76, 9826 CrossRef CAS PubMed.
- S. J. Pike, A. Heliot and C. C. Seaton, CrystEngComm, 2020, 22, 5040 RSC.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1997, 119, 10587 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 9925 CrossRef CAS.
- The folded helical conformation of the ortho-azobenzene/2,6-pyridyldicarboxamide scaffold in 1 could potentially be further supported by a series of short intramolecular contacts between the pyridyl carboxamide NH's and the adjacent azo nitrogen atoms (2.215–2.338(Å) and, additionally, between the NH's in the terminal groups and the adjacent azo nitrogen atoms (2.210–2.258 Å), see ref. 18a.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576 CrossRef CAS PubMed.
- (a) M. Egli, V. Tereshko, G. N. Mushudov, R. Sunushvilli, X. Liu and F. D. Lewis, J. Am. Chem. Soc., 2003, 125, 10842 CrossRef CAS PubMed; (b) C. A. Hunter, J. Singh and J. M. Thornton, J. Mol. Biol., 1991, 218, 837 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- F. H. Allen, P. A. Wood and P. T. A. Galek, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 379 CrossRef CAS PubMed.
- These observed changes in the UV/Vis spectrum upon irradiation are characteristic of E/Z-photoisomerisation of azobenzene (see ref. 17 and 18) with the Z-isomer reportedly showing an increase in intensity the n → π* transition, see: L. Vetráková, V. Ladányi, J. Al Anshori, P. Dvořák, J. Wirz and D. Heger, Photochem. Photobiol. Sci., 2017, 16, 1749 CrossRef PubMed.
- This level of photo-induced E → Z isomerisation of the azo bonds in 1 is comparable to those observed in pyridinecarboxamide/m-(phenylazo)azobenzene heterofoldamers (see ref. 18a) and for some of the internal azo bonds in oligo(azobenzene) foldamers developed in Hecht and co-workers in ref. 17.
- The observed sign of the peaks at 472 and 394 nm in the CD spectrum of 1 indicates the adoption of P-helicity in solution when compared to the profile of the CD spectra of related, chiral 2,6-pyridinedicarboxamide/m-(phenylazo)azobenzene foldamers in ref. 18c.
- To ascertain if these ortho-azobenzene/2,6-pyridyldicarboxamide foldamers display any self-aggregation in acetonitrile, as has been observed in other azobenzene-based foldamers see: Z. Yu and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 313 CrossRef CAS . A series of 1H NMR spectroscopic dilution experiments of 1 in CD3CN were also carried out between 1.7 mM and 0.3 mM using 1H NMR spectroscopy. Over this concentration range there was no shifting of any of the resonances indicating that aggregation was not observed for this foldamer under the studied reaction conditions, see ESI for further details.†.
- Z. Yu and S. Hecht, Chem. – Eur. J., 2012, 18, 10519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterisation of new compounds as well as crystallographic data for compounds 1 and 4. CCDC 2121319 (1) and 2130347 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01137b |
| This journal is © The Royal Society of Chemistry 2023 |