
Recent advances on non-precious metal-catalysed fluorination, difluoromethylation, trifluoromethylation, and perfluoroalkylation of N-heteroarenes
Purushotam†
,
Atanu
Bera†
and
Debasis
Banerjee
 *
*
Department of Chemistry, Laboratory of Catalysis and Organic Synthesis, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: debasis.banerjee@cy.iitr.ac.in
First published on 12th September 2023
Abstract
This review highlights the recent advances, from 2015 to 2023, on the introduction of organo-fluorine derivatives at the N-heteroarene core. Notable features considering new technologies based on organofluorine compounds such as: (i) approaches based on non-precious metal catalysis (Fe, Co, Mn, Ni, etc.), (ii) the development of new strategies using non-precious metal-catalysts for the introduction of organo-fluorinine derivatives using N-heterocycles with one or more heteroatoms, (iii) newer reagents for fluorination, difluoromethylation, trifluoromethylation, or perfluoroalkylation of N-heteroarenes using different approaches, (iv) mechanistic studies on various catalytic transformations, as and when required, and (v) the synthetic applications of various bio-active organo-fluorine compounds, including post-synthetic drug derivatization, are discussed.
 Purushotam | Purushotam was born in Sirsa, Haryana, in 1998. He obtained his BSc degree in Chemistry (Hons.) from Deshbandhu College, University of Delhi in 2019, followed by an MSc degree in Chemistry from Kirori Mal College, University of Delhi in 2021. He is currently pursuing a PhD at the Department of Chemistry, Indian Institute of Technology, Roorkee, under the supervision of Prof. Debasis Banerjee. His current research work focuses on non-precious 3d-transition metal-catalysed sustainable organic transformations. |
 Atanu Bera | Atanu Bera was born in West Bengal, Kolkata, in 1993. He obtained his BSc degree in Chemistry (Hons.) from St. Paul's Cathedral Mission College (under University of Calcutta) in 2015, followed by an MSc degree in Chemical Science from Pondicherry University in 2017. He is currently pursuing a PhD at the Department of Chemistry, Indian Institute of Technology, Roorkee, under the supervision of Prof. Debasis Banerjee. His current research work focuses on the non-precious 3d-transition metal-catalysed activation of alcohols for sustainable organic transformations. |
 Debasis Banerjee | Debasis Banerjee graduated with an M.Sc. degree in Organic Chemistry from Banaras Hindu University and obtained a PhD in organic chemistry from Indian Institute of Technology Kanpur in 2011 with Prof. M. L. N. Rao. Thereafter, he moved to Leibniz Institute for Catalysis (LIKAT), Germany for a postdoctoral position with Prof. Matthias Beller (2011–14) and subsequently held another postdoctoral position (2014–2015) at Stockholm University, Sweden with Prof. Jan-Erling Bäckvall. In 2015, he accepted the position of Assistant Professor at the Indian Institute of Technology Roorkee (Uttarakhand, India). Currently he is an Associate Professor at the same institute, where he has been since August 2020. He is a recipient of the SERB-Early Career Research Award (2016), DAE-Young Scientist Research Award (YSRA-2016) and winner of the Evonik Call for Research Proposal (ECRP-2016) Award by Evonik Industries GMBH, Germany. He was recently selected for the Thieme Chemistry Journals Award 2020. Since 2021 he has worked as a Guest Editor for Tetrahedron and Tetrahedron Letters on a Special Issue based on Non-Precious Metal-Catalysis for Sustainable Organic Transformations. He has been selected for the Chemical Research Society of India (CRSI) Bronze Medal of 2023. |
1. Introduction
For the past few decades organo-fluorine chemistry has attracted significant attention in the agrochemical and pharmaceutical industries, and in materials science. More than 20% of pharmaceutical and 30% of agrochemical products contain fluorine compounds, which dramatically influence their biological potential, pharmaceutical activities, metabolic stability, and related physical and chemical properties.1–3Fig. 1 shows a few examples of such organo-fluorine compounds, which exhibit diverse medicinal activities, such as anticancer, antibacterial, anti-depressive, anticonvulsant, antipsychotic, and antiemetic properties (Fig. 1).4–6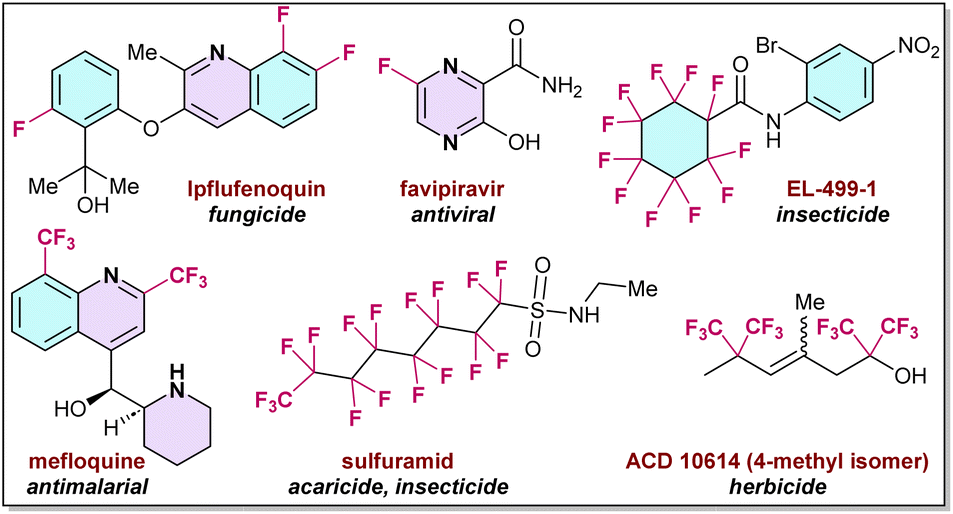 | ||
| Fig. 1 Selected examples of CF3-embaded pharmaceuticals and agrochemicals. | ||
Notably, in recent times, the development of new synthetic processes for fluorine-based molecules have emerged and attracted considerable attention, which has initiated the discovery of more sustainable and cost-efficient organo-fluorine reagents. Interestingly, such fluorine, difluoromethyl, trifluoromethyl, as well as perfluoroalkyl reagents, signify a milestone in fluorination chemistry, and has subsequently led to the design of new procedures to introduce fluorine-based compounds into organic molecules.3,7,8 Therefore, a series of new reagents and compounds are now commercially available, as presented in Fig. 2.9–14
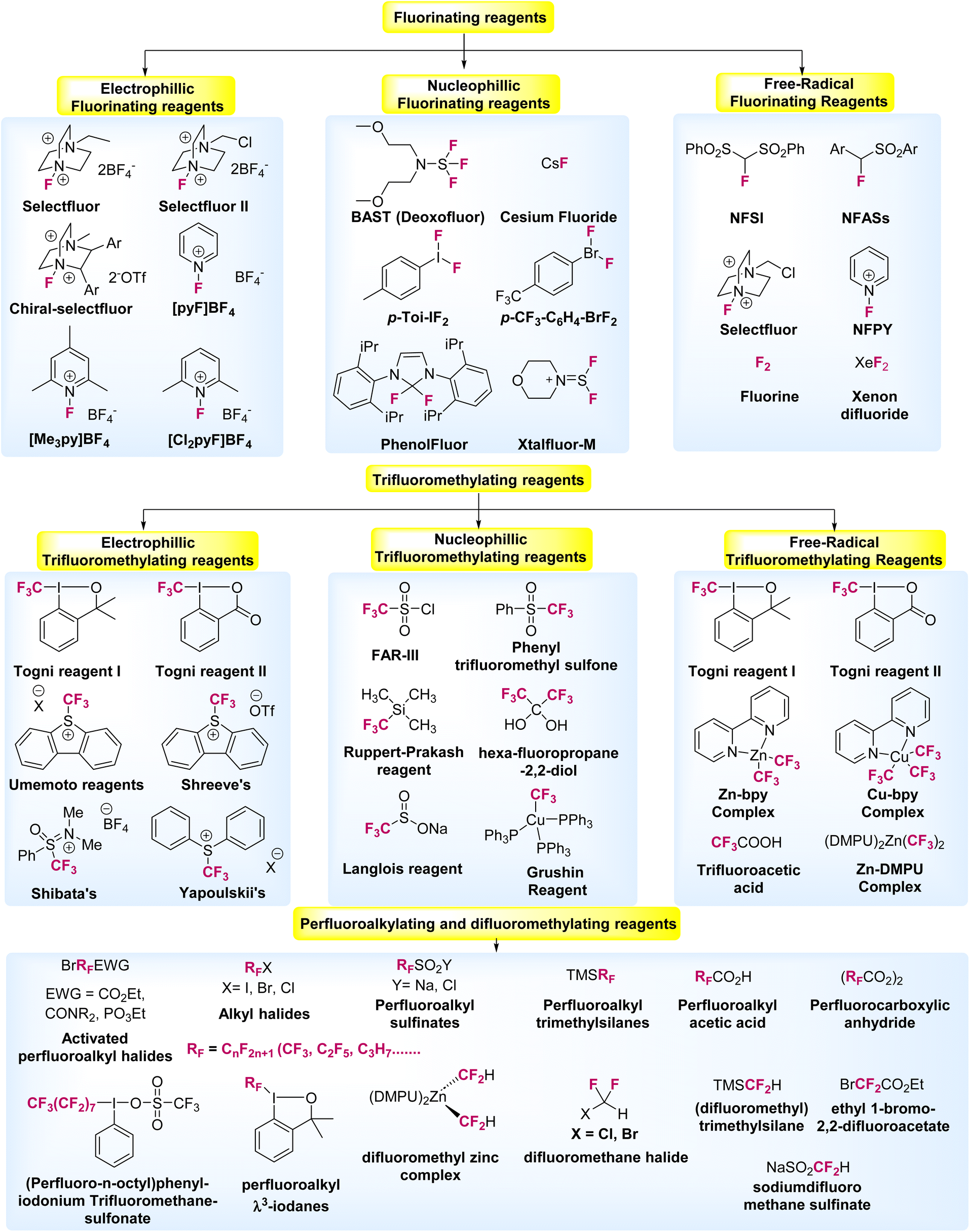 | ||
| Fig. 2 Selected examples of fluorinating and fluoroalkylated reagents developed over the years (ref. 8, 13, 29, 41 and 44). | ||
Notably, rapidly increasing efforts towards this direction have focused on developing efficient strategies, reagents, and catalysts for the incorporation of fluorine, difluoromethyl, trifluoromethyl, perfluoroalkyl groups as well as substituted long chain organofluorine derivatives with strong synthetic and medicinal applications into molecules. In this direction, late-stage modification or functionalization of N-heteroarenes by fine tuning of direct C–H bonds using a suitable fluorinating agent is highly desirable. Importantly, N-heteroarenes bearing trifluoromethyl, perfluoroalkyl or substituted long chain organofluorine derivatives exhibit remarkable medicinal activities and are in great demand (Fig. 2).15,16
Furthermore, precious transition-metal-based catalysts have been extensively studied for the incorporation of organo-fluorine derivatives using N-heteroarenes.17–19 However, due to the toxicity and high costs associated with such noble-metal based catalysis, there has been a recent surge in focusing on more sustainable and environmental benign systems for such applications. Notably, highly abundant and low toxicity 3d-transition metals have proved to be highly beneficial for the desired transformations.20,21
It is noteworthy to mention that, considering the potential importance and upsurge in the demand for organo-fluorine compounds, several reviews on this direction have been reported. For instance, fluorination technologies selectively based on the application of N-fluorobenzenesulfonimide, applications of organo-fluorine compounds in the agrochemical industries or precious metal-based C–H functionalisation using valuable fluorinated building blocks have been well documented.4–7,22–27 In addition, several approaches for metal-catalysed trifluoromethylation,28–41perfluoroalkylation,42–45 and difluoromethylation46–49 have also been reported based on precious or non-precious transition metals as well as under metal-free conditions. However, to the best of our knowledge, to date there has been no such comprehensive review solely focused on the fluorination, difluoromethylation, trifluoromethylation, and perfluoroalkylation of N-heteroarenes using non-precious-metal catalysis.
This review highlights the recent advances made on the functionalisation of N-heteroarenes using a variety of organo-fluorine reagents. Notably, new technologies and important methodologies based on organofluorine compounds developed in the period from 2015 to 2023 were considered for this review. More specifically, we focused on the processes based on non-precious-metal catalysis, employing metals such as Fe, Co, Mn, Ni, etc. Further, we discuss the recently developed fluorination, difluoromethylation, trifluoromethylation, or perfluoroalkylation of N-heterocycles that have one or more heteroatoms. Most importantly, mechanistic studies for various catalytic transformations and synthetic applications, including the post-synthetic drug derivatization of various bio-active organo-fluorine compounds, are discussed.
2. Fluorination of N-heteroarenes
Pharmaceutical or medicinally important N-heteroarenes often have one or more fluorinated functional groups in their core. There are not many synthetic techniques that can be used to introduce such scaffolds on N-heteroarenes and often demonstrate notable advancements over traditional synthetic approaches. This part of the review gives a detailed explanation of the various fluorination processes available and describes their associated difficulties. Though C–H fluorination is associated with several challenges, the incorporation of a fluorine moiety into inert Csp2–H bonds is a very practical and economical method. Selective C–H bond functionalisations have been performed in the presence of 3d transition metal catalysis because of their extremely inactive nature.2.1 Nickel-catalysed fluorination
In 2017, Li and co-workers established the first Ni-catalysed C-5 fluorination of 8-aminoquinolines with excellent regio-selectivity (Scheme 1a).50 They used NiSO4 as a catalyst and NFSI (N-fluorobenzene sulfonimide) as a fluorine radical source in the presence of DCE as a solvent to achieved the best results. They highlighted the dual role of NFSI, as both a fluorine source and an oxidant. Mechanistic and catalytic control experiments using a potential radical scavenger TEMPO suppressed the reaction drastically. This experimental evidence suggests the formation of free radicals during the progress of the reaction process. As a result of control studies, they proposed a catalytic cycle. In the first step, NiSO4 is co-ordinated with the nitrogen of 8-aminoquinoline amide 1 to the Ni(II)-intermediate 1, which subsequently transforms into intermediate 2. Following oxidation by NFSI, intermediate 2 oxidizes to the Ni(III) species intermediate 3 and simultaneously, generates a fluorine radical. This is followed by single electron transfer (SET), as well as a proton transfer process through intermediates 4–6 to finally release product 2, and the catalytic cycle continues (Scheme 1a).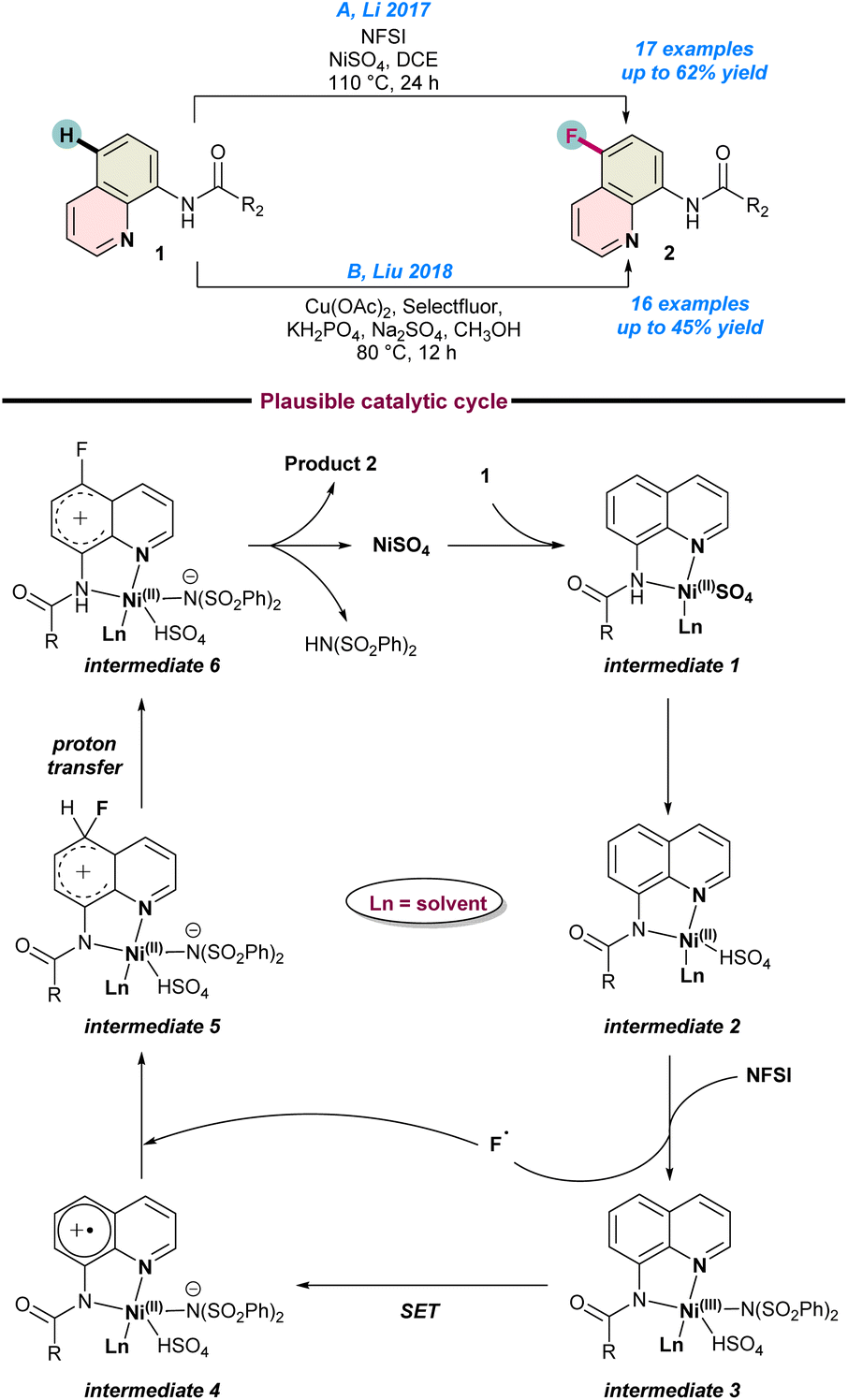 | ||
| Scheme 1 (a) Ni-catalysed fluorination of 8-aminoquinolines (ref. 50), (b) Cu-catalysed fluorination (ref. 53). | ||
2.2 Copper-catalysed fluorination
In 2017, Gouverneur and co-workers disclosed the radio-synthesis of heterocyclic positron emission tomography (PET) using the copper-catalysed oxidative nucleophilic 18F-fluorination of hetero-aryl boronic esters.51 They used potassium 18F-fluoride as the 18F-fluorination source and performed the catalytic reactions in the presence of [Cu(OTf)2py4] in combination with DMF as a solvent at 110 °C. A high Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate ratio (up to 1.5:1), was needed and the radiochemical conversion was enhanced for electron-deficient hetero-arenes when DMA was used as a solvent. Following this strategy, a series of medicinally important 18F-labeled N-heteroarenes were synthesized (Scheme 2A). In the same year, Sanford and co-workers also reported the (MeCN)4CuOTf catalysed radio-fluorination of electron-rich N-heteroarenes using [18F]KF as a fluorine source (Scheme 2b).52
:substrate ratio (up to 1.5:1), was needed and the radiochemical conversion was enhanced for electron-deficient hetero-arenes when DMA was used as a solvent. Following this strategy, a series of medicinally important 18F-labeled N-heteroarenes were synthesized (Scheme 2A). In the same year, Sanford and co-workers also reported the (MeCN)4CuOTf catalysed radio-fluorination of electron-rich N-heteroarenes using [18F]KF as a fluorine source (Scheme 2b).52
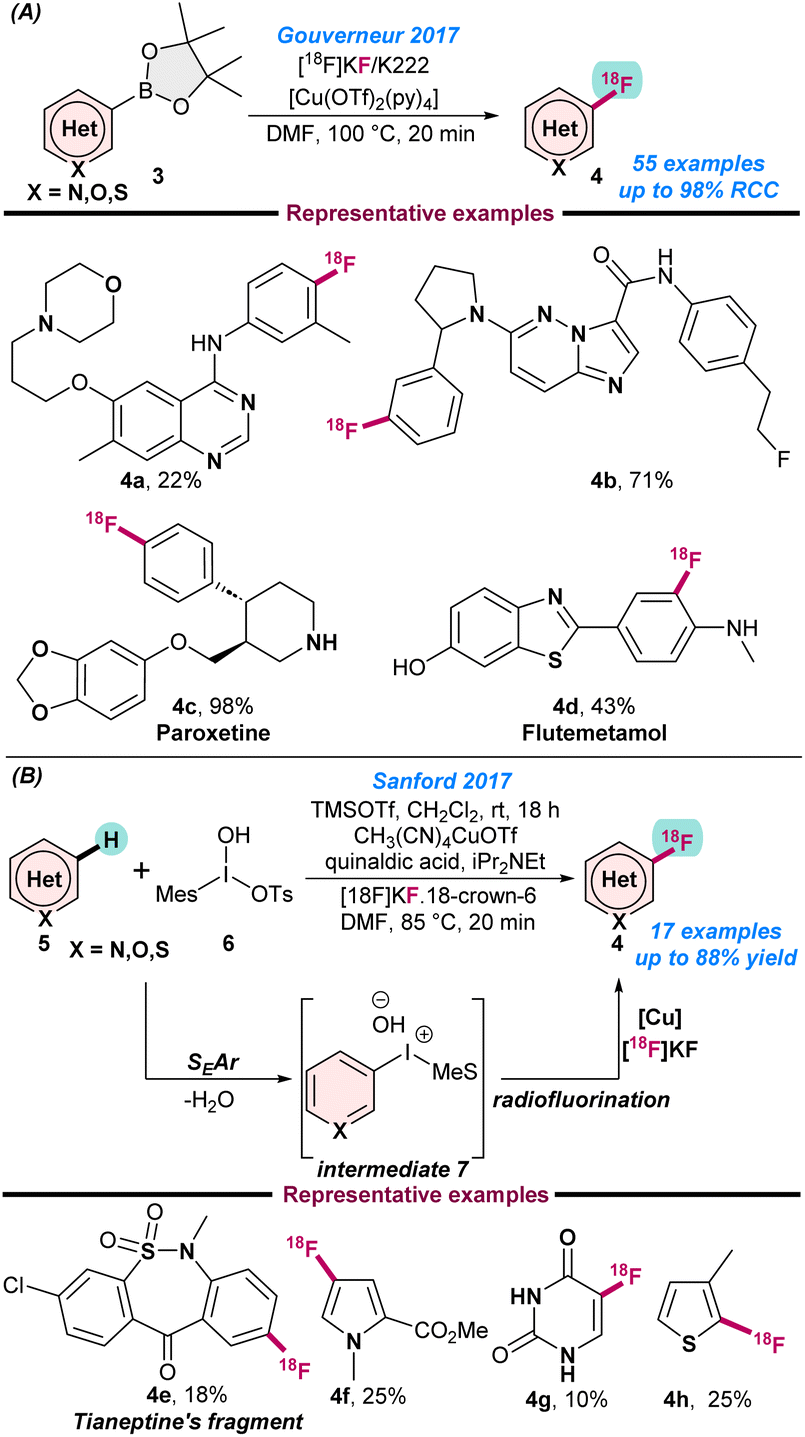 | ||
| Scheme 2 (a) Cu-catalysed radiosynthesis of heterocyclic PET radioligands (ref. 51). (b) Nucleophilic radio fluorination of heteroarenes (ref. 52). | ||
Additionally, the presence of a Lewis or Brønsted acid activator is the key for successful transformations, as the activator facilitates the SEAr-step. Mechanistic investigations show that in the first step electrophilic addition of 5 takes place and results in the formation of intermediate 7, which further undergoes nucleophilic radio-fluorination in the presence of a Cu catalyst (Scheme 2b). This technique highlighted few key advantages, such as that the electrophilic addition of 5 is highly site selective following the SEAr process and the in situ generated intermediate readily participates in nucleophilic radio-fluorination for successful transformations. The catalytic protocol is tolerant to a variety of functional groups and can be used for a broad scope of N-heteroarenes.
In 2018, Liu and co-workers reported Cu-catalysed regio-selective C–H bond fluorination at the C-5 position of 8-aminoquinolines (Scheme 1b).53 Selectfluor was used as the electrophilic fluorinating reagent and 20 mol% Cu(OAc)2 in MeOH gave the best result, where KH2PO4 and Na2SO4 were used as an additive. However, the major limitation of this method was the low product yield despite the application of higher catalyst loading. The catalytic process followed a similar reaction pathway as presented in Scheme 1a. In 2021, Ritter and co-workers demonstrated the Cu(II)-catalysed photo-induced LMCT (ligand–metal charge transfer)-enabled de-carboxylative fluorination of heteroaryl carboxylic acids (Scheme 3).54 Application of two different Cu salts, Cu(OTf)2 and Cu(MeCN)4BF4 in MeCN was the key for successful transformations. It was observed that unprecedented photo-induced LMCT facilitated a milder approach and generated an oxygen-centred radical through the cleavage of metal–oxygen bonds. Notably, Cu(OTf)2 was responsible for the decarboxylation process and Cu(MeCN)4BF4 suppressed the formation of the unreactive photo-decarboxylative side product. Mechanistic studies revealed that initially the carboxylic acid coupled with the Cu(II)-species and transformed into photoactive Cu(II)aryl carboxylate intermediate 8, which subsequently resulted in aryl-carboxyl radical intermediate 9 and a Cu(I)-species being generated (Scheme 3). Next, decarboxylation of intermediate 9 to hetero aryl-radical successively resulted in the desired product by the reductive elimination of intermediate 12 (Scheme 3).
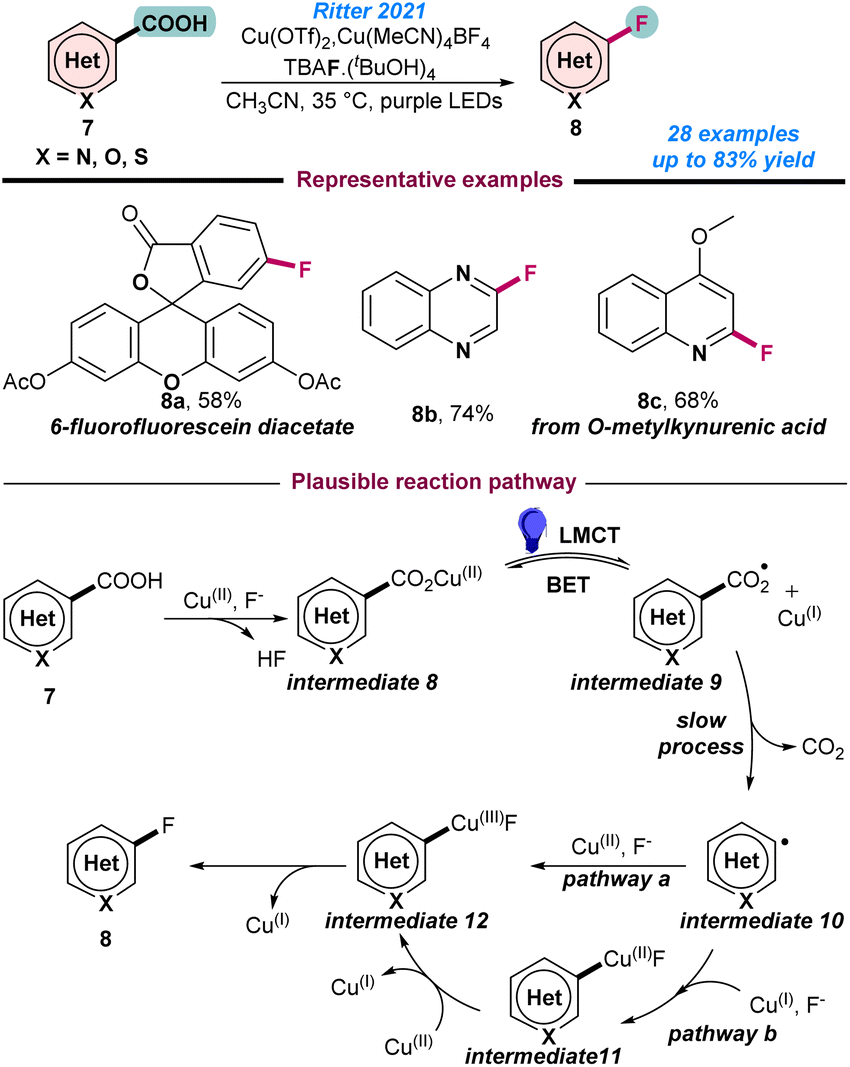 | ||
| Scheme 3 Cu-catalysed photoinduced decarboxylative fluorination of aryl acids (ref. 54). | ||
3. Difluoromethylation of N-heteroarenes
Considering their potential importance, the introduction of difluoromethyl functionalities on N-heterocycles has attracted significant attention. This part of the review summarizes the various difluoromethylation processes and associated challenges of using 3d-metal based catalysts for this process, including their applications.3.1 Fe-catalysed difluoromethylation
In 2020, Zhao and co-workers reported an interesting para-selective C–H difluoromethylation of N-heteroarenes using BrCF2CO2Et (10) as a difluoroalkylating agent in combination with an Fe(TPP)Cl catalyst (Scheme 4).55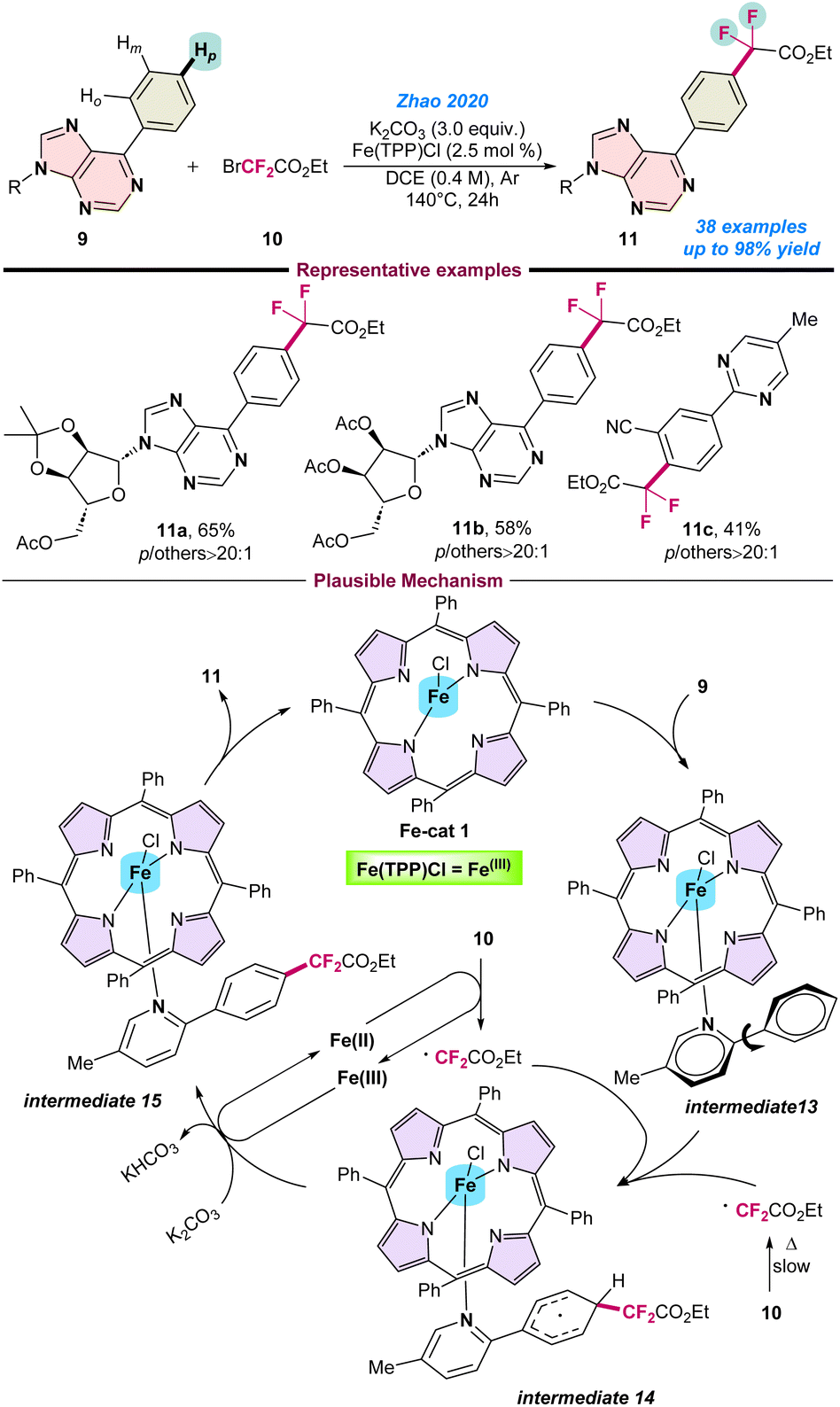 | ||
| Scheme 4 Fe-catalysed para-selective C–H difluoroalkylation of N-heteroarenes (ref. 55). | ||
Notably, the application of an earth-abundant Fe-catalyst for such para-site selective C–H difluoromethylation is quite challenging. The key to success for such reactions is the application of suitably designed bulky ligands. Ru, Fe, Co, Cu, Ni, and Mn-based metal complexes have been used for such transformations with porphyrin-based ligands, however, the Fe–porphyrin complex gave the desired product with high para selectivity. The catalytic protocol was compatible with various heteroaromatic compounds and gave the desired product in moderate to good yield. Here, the role of Fe is to activate the benzene ring adjacent to the N-atom of the heteroaromatic compounds. A series of control experiments were performed to understand the reaction pathway. It is proposed that initially Fe(TPP)Cl is coordinated with the substrate 9, leading to the formation of intermediate-13. Subsequently, BrCF2CO2Et (10) transforms into a ˙CF2CO2Et radical species, and reacts at the para position of intermediate 13. Finally, the desired product 11 is obtained through intermediate 14 followed by sequential deprotonation and aromatization, with the iron complex regenerated to continue the catalytic cycle (Scheme 4).
3.2 Ni-catalysed difluoromethylation
In 2016, Zhang and co-workers reported an excellent catalytic system based on a Ni catalyst for cross-coupling between (hetero)arylborons (12) and 1-bromo-1,1-difluoroalkanes (13) for a series of difluoroalkyl compounds 14 (Scheme 5a).56 Conceptually, a combination of a bidentate and a monodentate ligand system were utilized to modulate the steric and electronic properties at the Ni centre. For instance, 4,4′-ditBu-bpy (L) and DMAP were used as bidentate and monodentate ligands, respectively, to facilitate the activation of various 1-bromo-1,1-difluoroalkanes moieties. The notable features of the reactions are, the use of commercially available low-cost catalytic systems and the late-stage difluoroalkylation of biologically active molecules (Scheme 5a). Following a similar conceptual technology, the cross-coupling reaction between (hetero)arylboronic acids (15) with BrCF2H (16) or (hetero)aryl chloride (18) and ClCF2H (19) in the presence of a suitable Ni-catalyst was also studied (Schemes 5b and c).58,59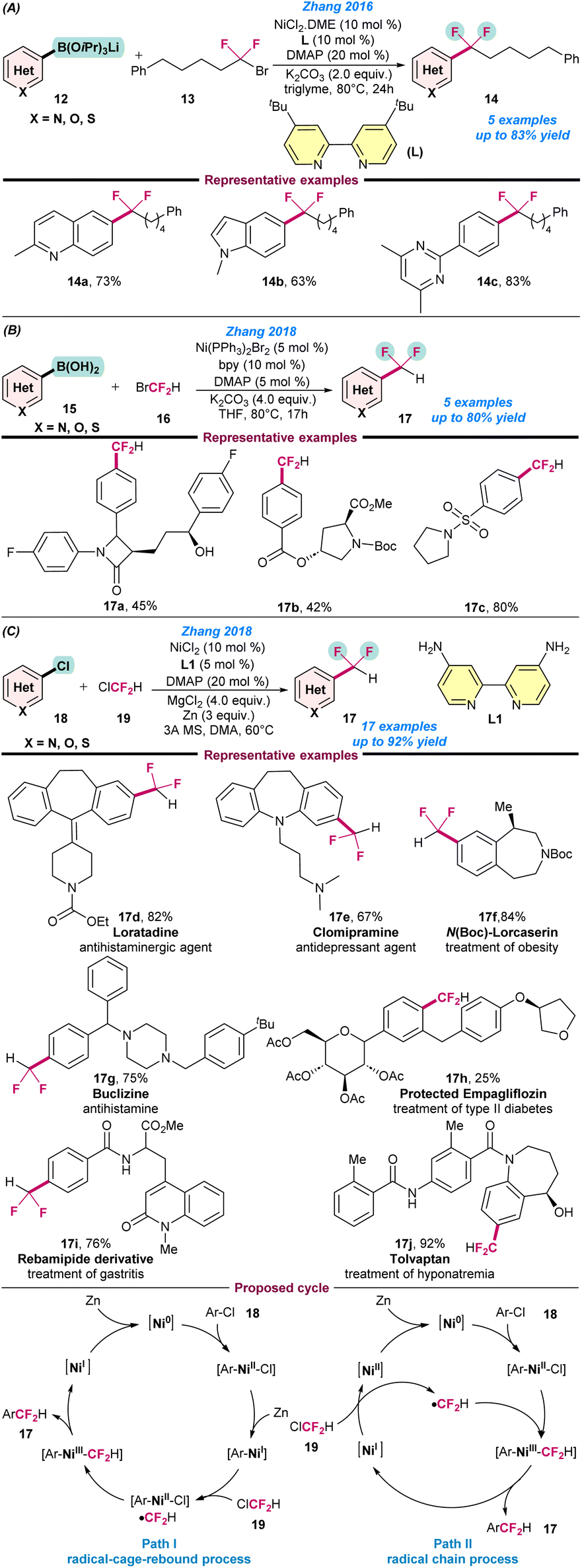 | ||
| Scheme 5 . Ni-catalysed (a) difluoroalkylation of (hetero)arylborons (ref. 56), (b) difluoromethylation of arylboronic acids using bromodifluoromethane (ref. 58), and (c) difluoromethylation of (hetero)aryl chloride with chlorodifluoromethane (ref. 59). | ||
Based on those experiments, two possible catalytic cycles were proposed: a radical-cage-rebound process or a radical chain mechanism. In the radical-cage-rebound process, the first step is the oxidative addition of an aryl chloride to Ni(0) to produce a Ni(II) species [Ar–Ni(II)–Cl], which is then reduced by Zn to generate an [Ar–Ni(I)] species. In the next step, a second oxidative addition of ClCF2H with Ni(I) species produces [Ar–Ni(II)–Cl]. Besides this, ˙CF2H is generated via a SET process, which then readily recombines with [Ar–Ni(II)–Cl] to produce [Ar–Ni(III)–CF2H]. The formation of [Ar–Ni(III)–CF2H] from [Ar–Ni(I)] proceeds via a radical-cage-rebound process. Finally, the reductive elimination of [Ar–Ni(III)–CF2H] gives the desired difluoromethylated arenes and a Ni(I) species is generated, which is then reduced to Ni(0) in the presence of Zn. However, in the radical chain mechanism, ClCF2H (19) directly reacts with Ni(I) to generate ˙CF2H, and results in the formation of the intermediate species [Ar–Ni(III)–CF2H], which undergoes reductive elimination to the desired difluoromethylated product, as presented in Scheme 5c.
In 2016, Vicic and co-workers disclosed the first Ni-catalysed difluoromethylation of (hetero)aryl iodide (20) using a difluoromethyl zinc reagent (21) (Scheme 6a).57 They synthesized a series of pyridine-based difluoromethylated zinc reagents in the laboratory and utilized them for the catalytic transformations. It was observed that the ligand and solvent play a crucial role in achieve higher product yield of 17. However, the combination of dppf as a ligand and DMSO as a solvent dramatically changed the product yields and selectivity (Scheme 6a). In the same year, Wang and co-workers also developed the difluoromethylation of an aryl boronic acid (15) using Ni(OTf)2 as a catalyst (Scheme 6b).60
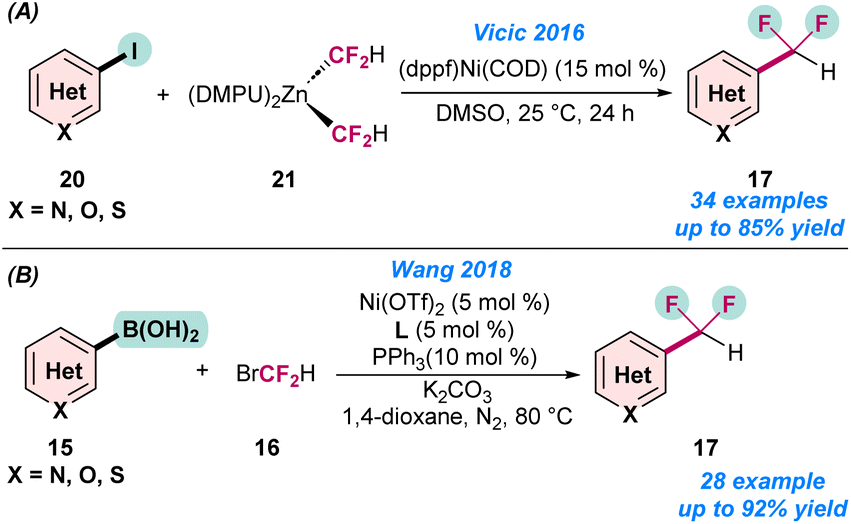 | ||
| Scheme 6 Ni-catalysed (a) difluoromethylation of aryl iodide using a difluoromethyl zinc reagent (ref. 57). (b) Difluoromethylation of aryl boronic acids (ref. 60). | ||
In 2018, MacMillan and co-workers reported an elegant strategy for the difluoromethylation of (hetero)aryl bromides (22) with bromodifluoromethane (16) under a dual Ni/Ir metallophotoredox platform (Scheme 7).61 A combination of an Ir-based photocatalyst, NiBr2·dtbubpy as a pre-catalyst, commercially available (TMS)3SiH, 2,6-lutidine as a base in DME, and a blue LED as a photon source are the key to the successful transformations. Late-stage functionalisation of several important drug molecules, such as celecoxib, sulfadimethoxine and indometacine analogues is noteworthy. Initial mechanistic studies revealed that, in the first step, Ir(III)(1) photocatalyst proceeds via an excited state *Ir(III)(2) complex under visible light. Next, the *Ir(III)(2) complex oxidizes the bromide anion to a bromine radical (Br˙), which further engages the hydrogen atom transfer with (TMS)3SiH, resulting in the generation of a nucleophilic silyl radical. Thereafter, the silyl radical abstracts the bromine from bromodifluoromethane (16) and affords the key difluoromethyl radical intermediate species. Simultaneously, the (dtbbpy)Ni(0) species undergoes facile oxidative addition with aryl bromide (22) to produce Ni(II)–aryl species intermediate 16, which is transformed into the Ar–Ni(III)–CF2H intermediate 17 after trapping the difluoromethyl radical. Finally, intermediate 17 undergoes reductive elimination to afford the desired difluoromethylated product (17) along with the Ni(I)-species intermediate 18, which is further transformed into a low valent Ni(0) species along with the ground state photocatalyst Ir(III)(1), as presented in Scheme 7.
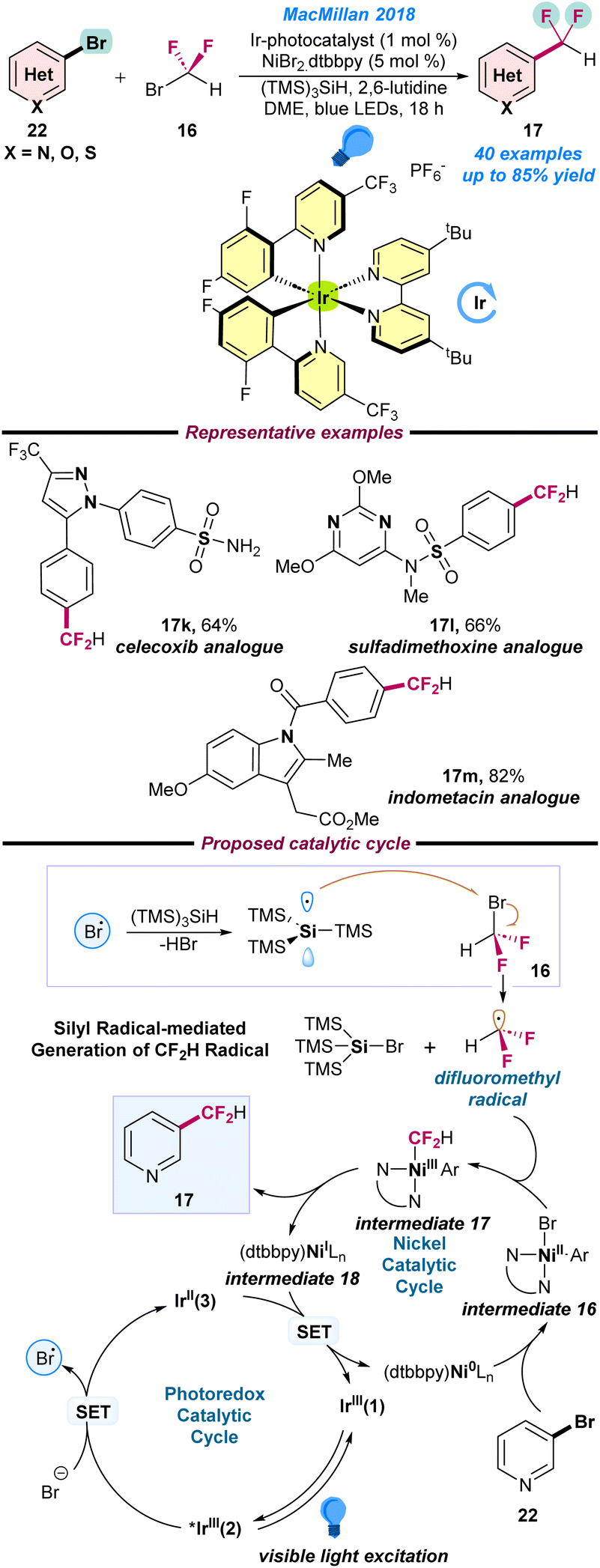 | ||
| Scheme 7 Ni/Ir-based metallophotoredox catalyzed difluoromethylation of an aryl bromide (ref. 61). | ||
In 2021, Pan and co-workers disclosed the Ni-catalysed cross-coupling strategy of difluoromethylation using aryl iodide (20) under electrochemical conditions (Scheme 8).62 A combination of NiBr2.diglyme and 4,7-diphenyl-1,10-phenanthroline (L2) along with sodium difluoromethanesulfinate (23) as a source of CF2H radicals was used in an undivided cell (graphite cathode/anode, 3.5 V cell voltage system) for 16 h to achieve the best product yield. It was observed that tetra-butylammonium tetrafluoroborate (TBABF4) played a key role and assisted the electron transfer reaction. Two possible catalytic pathways were postulated for the electrochemical fluoro-alkylation based on the control experiments. In path a, the oxidative addition of Ar–I to Ni(I) generates intermediate 19, which then undergoes cathodic reduction to Ni(II) intermediate 20. In path b, initially the cathodic reduction of Ni(I) species to Ni(0) is observed, followed by oxidative addition of Ar–I to form Ar–Ni(II)–I intermediate 20. Simultaneously, sodium difluoromethanesulfinate provides the difluoroalkyl radical through anodic oxidation. Next, intermediate 20 traps the CF2H radical to give intermediate 21, which undergoes reductive elimination to fluoroalkylated product 17 and regenerates the Ni(I)-catalyst, as depicted in Scheme 8.
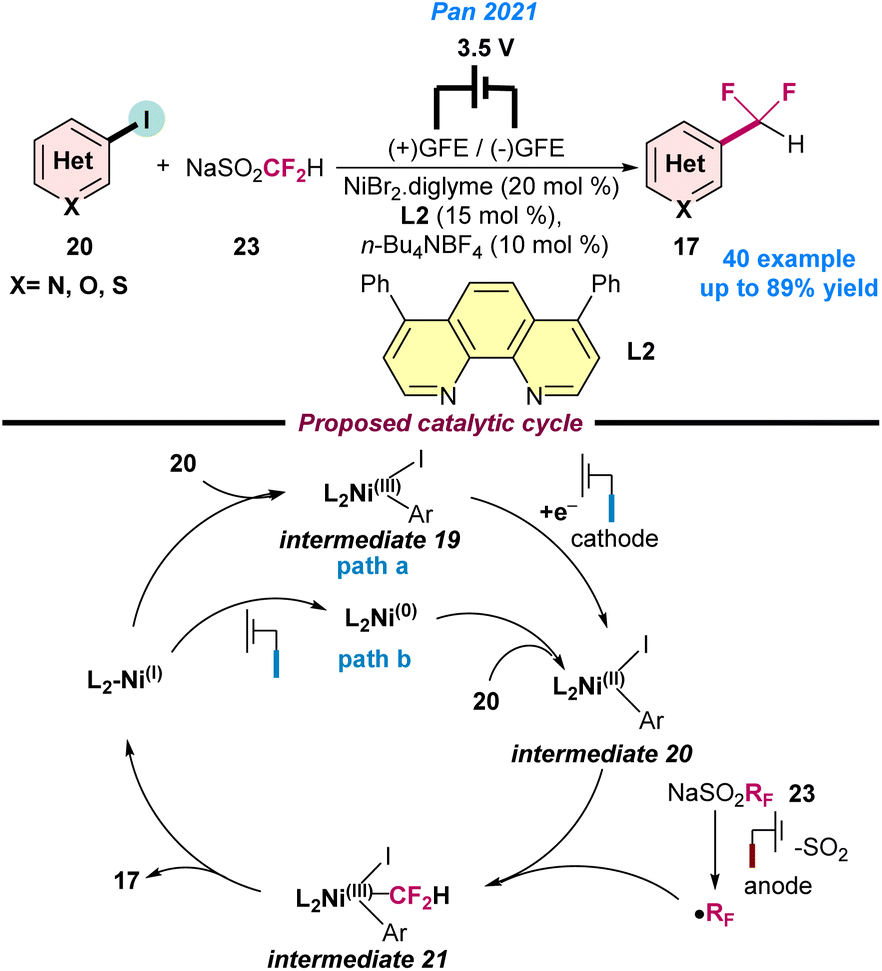 | ||
| Scheme 8 Electrochemical Ni-catalysed oxidative fluoroalkylation of aryl iodide and the difluoroalkylating reagent (ref. 62). | ||
3.3 Cu-catalysed difluoromethylation
In 2016, Hu and co-workers reported the Cu-catalysed (phenylsulfonyl)difluoromethylation of arylboronic acids (15) using a PhSO2CF2HCu-complex prepared from PhSO2CF2H (Scheme 9a).63 In a mixture of CuCl (0.2 mmol) and t-BuONa (2 equiv.) in DMF at −20 °C, PhSO2CF2H (1.6 equiv.) was added and kept under an argon atmosphere 30 min to obtain the PhSO2CF2HCu-complex. Cu(OAc)2·H2O and AgNO3 were used as additives in the reaction. Various functional groups such as formyl, nitrile, sulfonyl, halides, nitro, and different N-, O-, and S-containing heterocycles were easily tolerated to afford the desired PhSO2CF2 substituted products in moderate to good yield (Scheme 9a). In the same year, Mikami and co-workers reported the direct difluoromethylation of (hetero)aryl iodides (20) using a Cu catalyst (Scheme 9b).64 They used (difluoromethyl)zinc (21) as a fluorine source, which was prepared using diethylzinc and difluoroiodomethane in DMF, with a mixture of products with (DMF)2Zn(CF2H)I and (DMF)2Zn(CF2H)2 in a 18:82 ratio obtained. Thereafter, the ligand exchange to (DMPU)2Zn(CF2H)2, followed by the addition of CuI resulted in the formation of Cu(III) intermediate 22 and afforded the desired products (Scheme 9b).
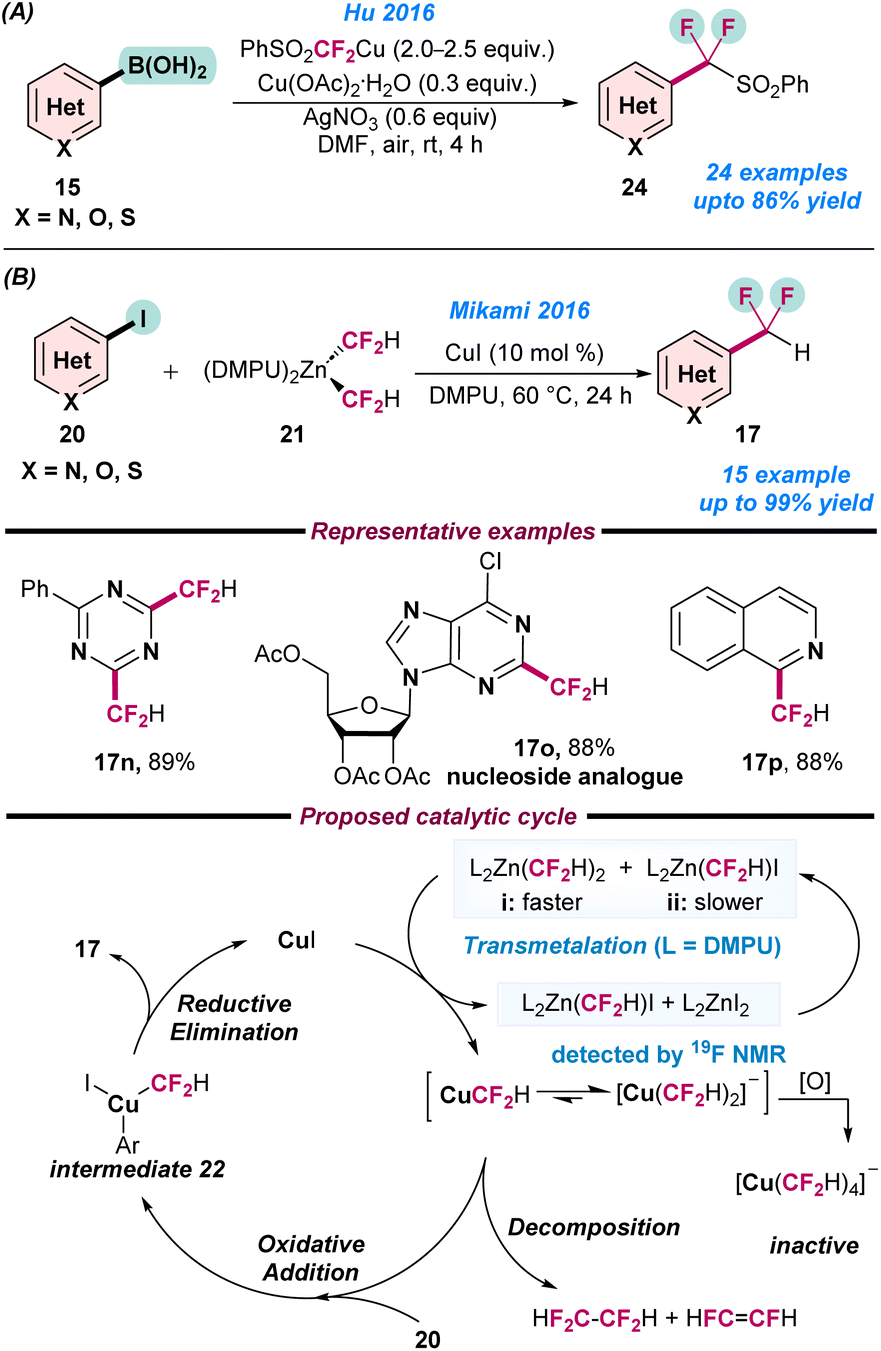 | ||
| Scheme 9 Cu-catalysed (a) aerobic (phenylsulfonyl)difluoromethylation of arylboronic acid (ref. 63) and (b) difluoromethylation of aryl iodides using a (difluoromethyl)zinc reagent (ref. 64). | ||
In 2018, Qing and co-workers demonstrated a new strategy for the Cu- catalysed oxidative difluoromethylation of heteroarenes (25) with TMSCF2H (26) using t-BuOK as a base with 9,10-phenanthrenequinone (PQ) as an oxidant (Scheme 10a).65 When the reaction was performed without PQ, a trace amount of product was obtained, which indicates that the oxidant played an important role in successful transformations. Heteroarenes including oxazole, thiazole, imidazole, 1,3,4-oxadiazole, benzoxazole, benzothiazole, benzothiophene, pyridine, thiophene, and thiazolo[5,4-c]pyridine was easily coupled with TMSCF2H (26) in moderate to good yield. Based on control experiments, it was proposed that the reaction begins with the generation of CuCF2H and Cu(CF2H)2−via the treatment of TMS(CF2H) (26) with CuCN and t-BuOK. Next, t-BuOK abstracts a proton from the heteroarenes (25) and affords intermediate 23 followed by reductive elimination to give the desired product in the presence of PQ (Scheme 10a).
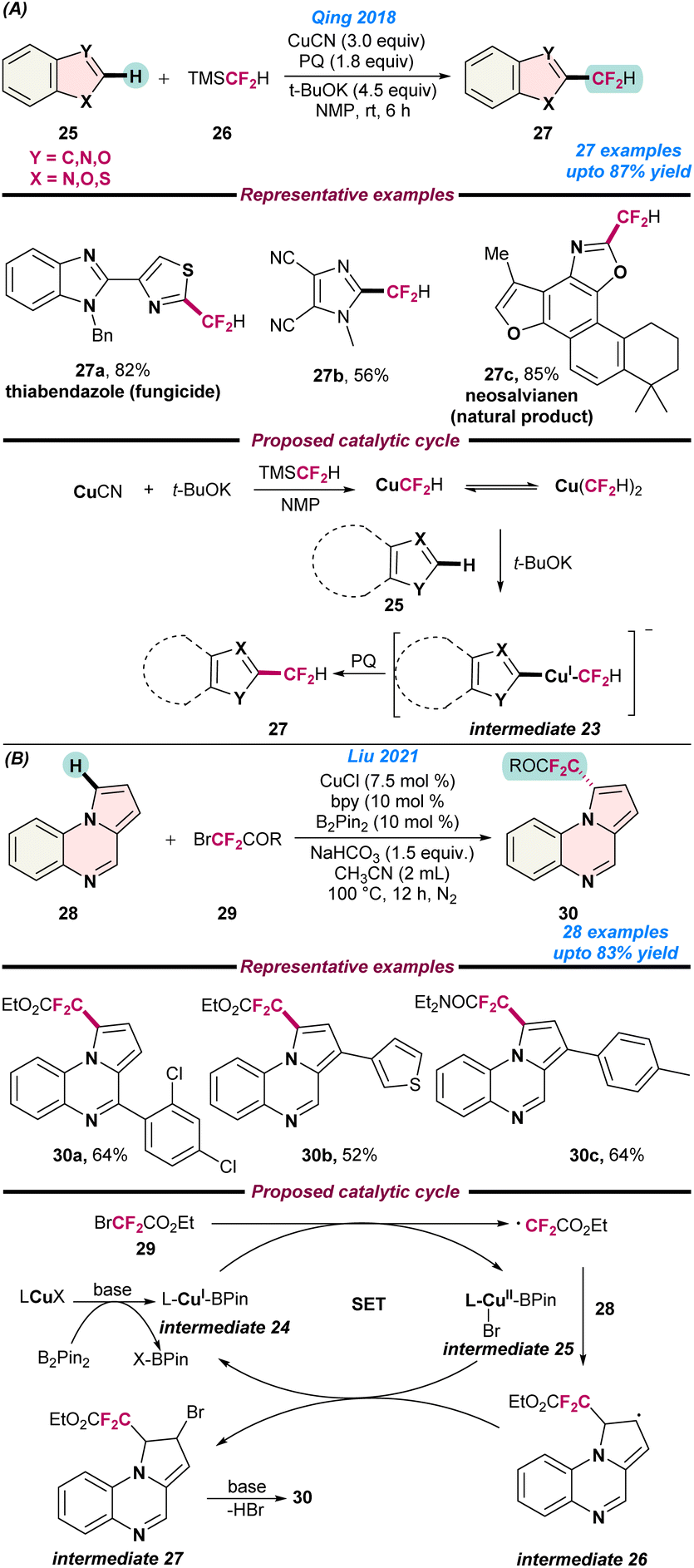 | ||
| Scheme 10 Cu-catalysed (a) regiosective C–H oxidative difluoromethylation of heteroarenes (ref. 65) and (b) the direct difluoromethylation of pyrrolo[1,2-a]quinoxalines (ref. 66). | ||
In 2021, Liu and co-workers reported the direct difluoromethylation of pyrrolo[1,2-a]quinoxalines (28) with BrCF2CO2Et or BrCF2CONEt2 (29) using a Cu catalyst (Scheme 10b).66 B2Pin2 played a vital role during the formation of the metal–ligand complex. The reaction followed radical pathways, as no desired product was observed in the presence of 3.0 equiv. of TEMPO. Based on control experiments, it was proposed that initially L-CuX and B2Pin react in the presence of base and generate the L-CuI-Bpin intermediate 24, which eventually reacts with 2-bromo-2,2-difluoroacetate (29) to give intermediate 25 and a difluoroacetate radical via a single-electron transfer pathway. Subsequently, heteroarene 28 traps the difluoroacetate radical to form radical intermediate 26, resulting in the formation of the desired product via intermediate 27 on treatment with HBr (Scheme 10b).
4. Trifluoromethylation of heteroarenes
Over the past few decades, notable advancement in the development of trifluoromethylation reagents has been documented, as presented in Fig. 2. Such trifluoromethylation technologies comprise transition-metal catalysed electrophilic, nucleophilic, and radical-assisted processes. A large number of successful methodologies relying on the direct introduction of Csp2-trifluoromethyl bonds into N-heteroarenes have been intensively investigated over the past few decades. Herein, we summarise a series of different trifluoromethylation processes of N-heteroarenes based on commercially available reagents using non-precious metal-based catalysts.4.1 Nickel-catalysed trifluoromethylation
In 2016, Wang and co-workers disclosed the nickel-catalysed trifluoromethylation of electron-rich N-heteroarenes, imidazopyridines, indoles and thiophenes. Application of a catalytic amount of NiCl2(dppp) in combination with CF3I as the trifluoromethylating agent facilitated the introduction of -CF3 on an imidazo[1,2-a] pyridine core (31) (Scheme 11).67 A plausible reaction mechanism established single-electron transfer from the in situ generated Ni(I)-species to CF3I, and a CF3-radical was generated. Thereafter, reaction of CF3-radical with imidazo[1,2-a] pyridine 31, resulted in the formation of intermediate 28, which is subsequently oxidised to intermediate 29 in the presence of a Ni(II) species. Finally, base-mediated deprotonation afforded the desired product 32 from intermediate 29. The key advantages in this process are the applications of relatively inexpensive iodotrifluoromethane (CF3I) as the trifluoromethylating reagent and also the direct tri-fluoromethylation of medicinally important melatonin and zolmitriptan (Scheme 11).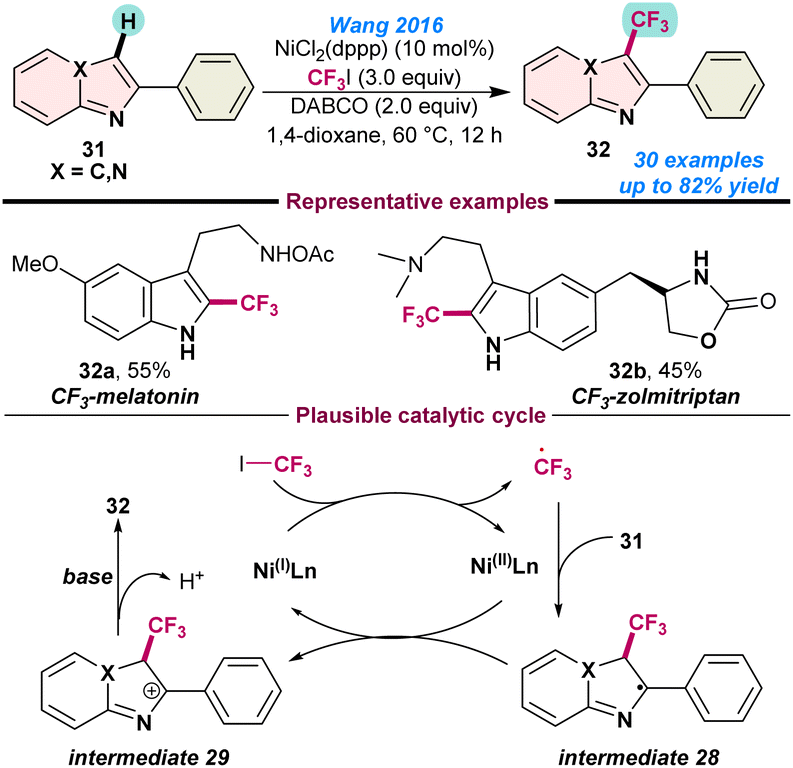 | ||
| Scheme 11 Ni-catalysed trifluoromethylation of N-heteroarenes (ref. 67). | ||
In 2019, Sanford and co-workers for the first time demonstrated the synthesis and applications of the NiIV-catalysed C–H trifluoromethylation of electron-rich arenes and heteroarenes. A series of experimental studies were conducted, including a computational mechanistic report supporting the radical chain process and evidence of the involvement of NiIV, NiIII, and NiII intermediates species (Scheme 12).68 Initially, the NiIV–CF3 (Ni-cat. 1) complex was independently prepared and used to perform the C–H trifluoromethylation of heteroarenes. The NiIV-complex was synthesized from the NiII precursor after 2e− oxidation. They envisioned two possible catalytic pathways, where in the first step Ni-cat-1 directly transforms the +CF3 group to an organic substrate and generates the cationic organic species intermediate 30 along with the NiII product, resulting in the generation of the desired trifluoro-methylated product after deprotonation. In the case of the second pathway, a ˙CF3 radical was generated from Ni-cat-1 and attached to the organic substrate to form radical intermediate 31 along with the NiIII species. Finally, this intermediate 31 underwent sequential oxidation and deprotonation to the desired product. Again, radical quenching experiments with 1.0 equiv. of TEMPO diminished the C–H trifluoromethylation product yield and a TEMPO-CF3 species was detected, which directly supported the involvement of a ˙CF3-radical intermediate (Scheme 12).
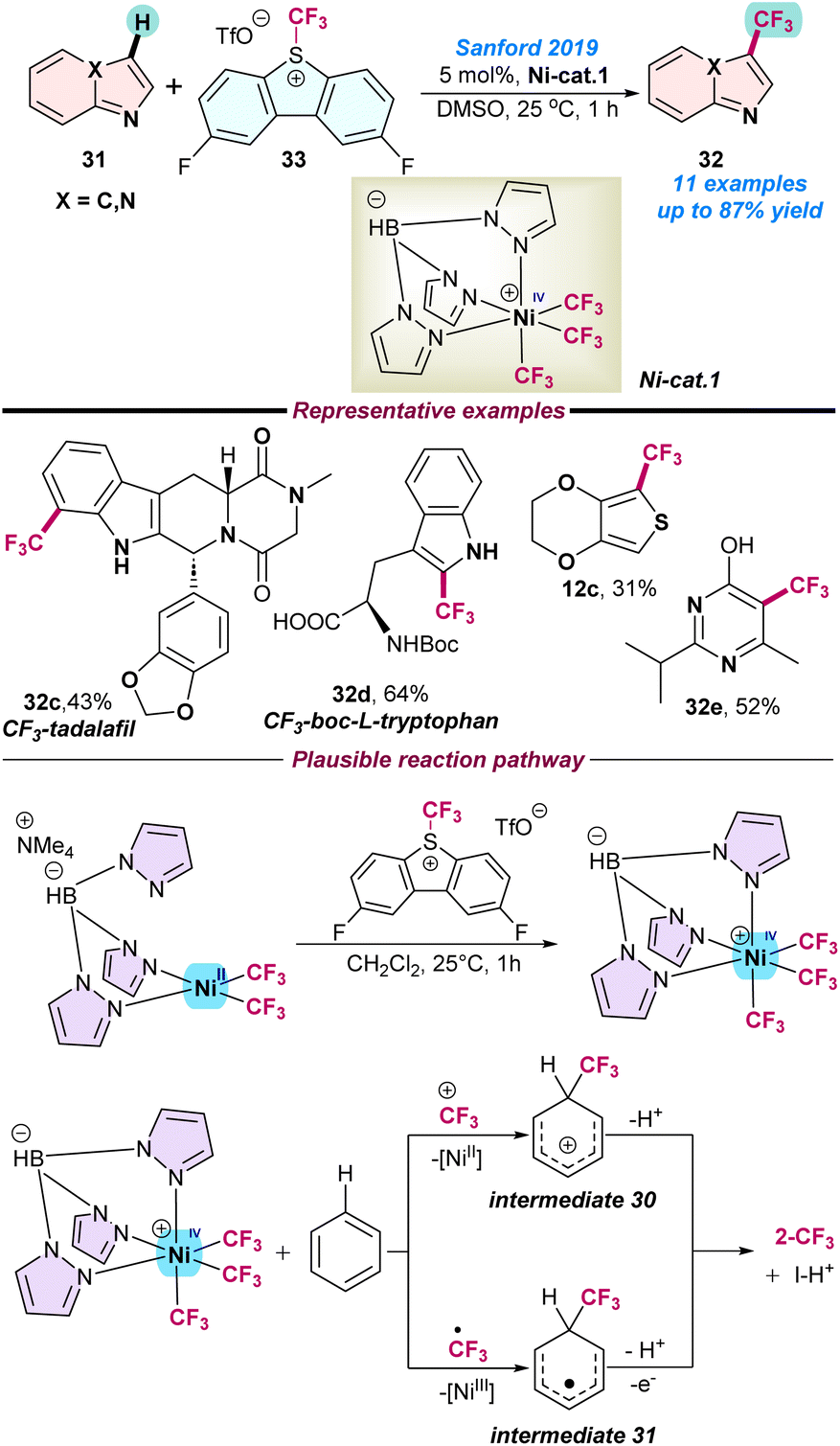 | ||
| Scheme 12 Ni(IV)-catalysed tri-fluoromethylation of electron-rich N-heteroarenes (ref. 68). | ||
Recently, in 2021, Khusnutdinova and co-workers used a series of robust naphthyridine-based air-stable high-valent NiIII-complexes for photoinduced (blue LED 465 nm) tri-fluoromethylation of the CSP2–H bond of (hetero)arenes (Scheme 13).69 Three bench-stable NiIII–CF3 complexes were used in the presence of air as an oxidant and facilitated the catalytic C–H tri-fluoromethylation either using a radical or an electrophilic CF3 source. The key advantages of this reaction are that it does not require any complicated set up and can be performed under mild conditions. Initial control and mechanistic studies supported that the reaction proceeds via a radical pathway. The protocol could be used for the late-stage trifluoromethylation of natural products; however, NiIII–CF3 complexes have limited applications in the case of trifluoromethylations of aliphatic substrates (Scheme 13).
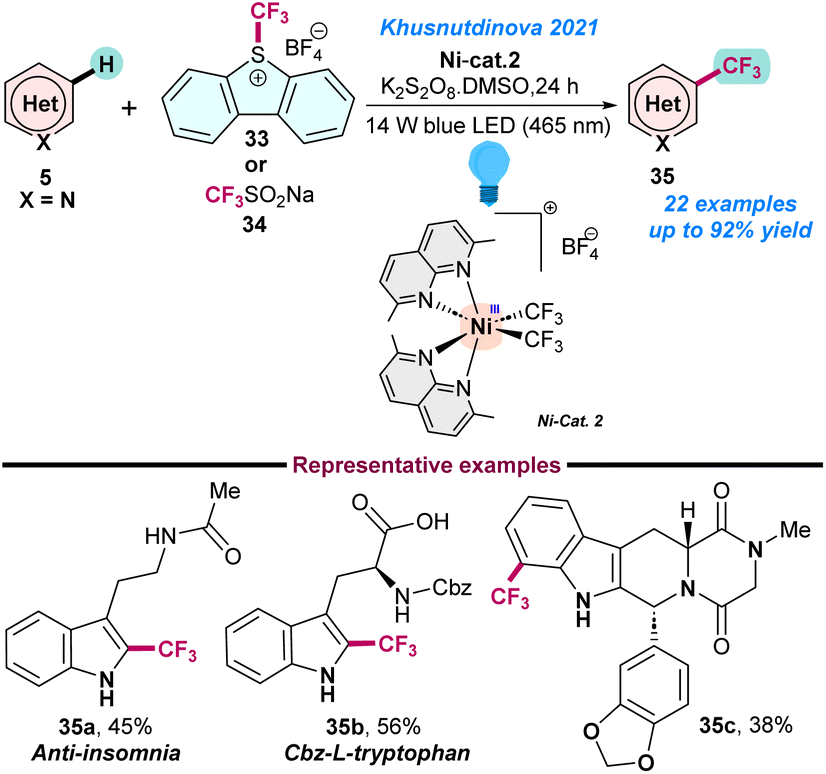 | ||
| Scheme 13 Trifluoromethylation of (hetero)arenes using high-valent nickel complexes (ref. 69). | ||
4.2 Manganese-catalysed trifluoromethylation
Zou and co-workers (2016), developed the Mn-catalysed Csp2–H trifluoromethylation of pyrimidinones and pyridinones (36) using a combination of Mn(OAc)3 and CF3SO2Na (34) and afforded the regioselective formation of 5-trifluoromethyl pyrimidinones and 3-trifluoromethyl pyridinones (37) in moderate to good yields.70 They observed that attack by the trifluoromethyl radical had a strong influence on the steric hindrance of the substrates and therefore influenced the product yield. Initial mechanistic investigations revealed that the CF3 radical was generated from CF3SO2Na in the presence of Mn3+ and formed radical intermediate 32 from 36. Next, intermediate 32 was readily oxidised to the cationic intermediate 33, followed by de-protonation to afford the desired product 37 (Scheme 14).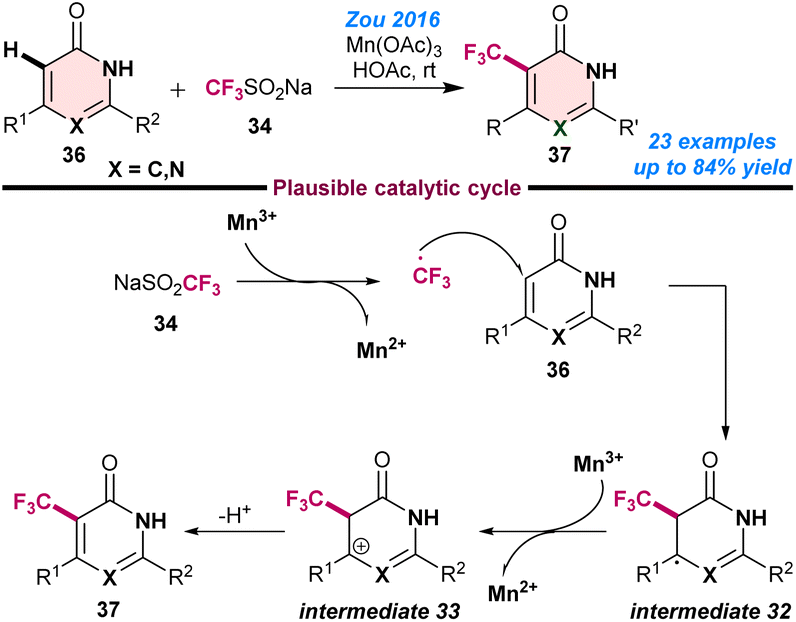 | ||
| Scheme 14 Mn-catalysed Csp2–H trifluoromethylation of pyrimidinones and pyridinones (ref. 70). | ||
4.3 Copper-catalysed tri-fluoromethylation
In 2015, Tang and co-workers reported the Cu(OAc)2 catalysed regioselective C–H trifluoromethylation of imidazoheteroarene (31) using a combination of Langlois reagent as a CF3 source and tBuOOH as the terminal oxidant. The reaction was performed using a recyclable mixed medium of and ionic liquid ([Bmim]BF4) and water at room temperature (Scheme 15a).71 The protocol was tolerant to various substituents on the imidazopyridine ring and afforded moderate to good product yields. Reaction mechanism studies revealed that initially a tert-butoxyl radical was generated from TBHP, which then reacted with the Langlois reagent and facilitated the formation of the CF3 radical. Thereafter, CF3-radical participated with N-heteroarene 31 and the desired product 32 was obtained via the radical intermediates 34 and 35.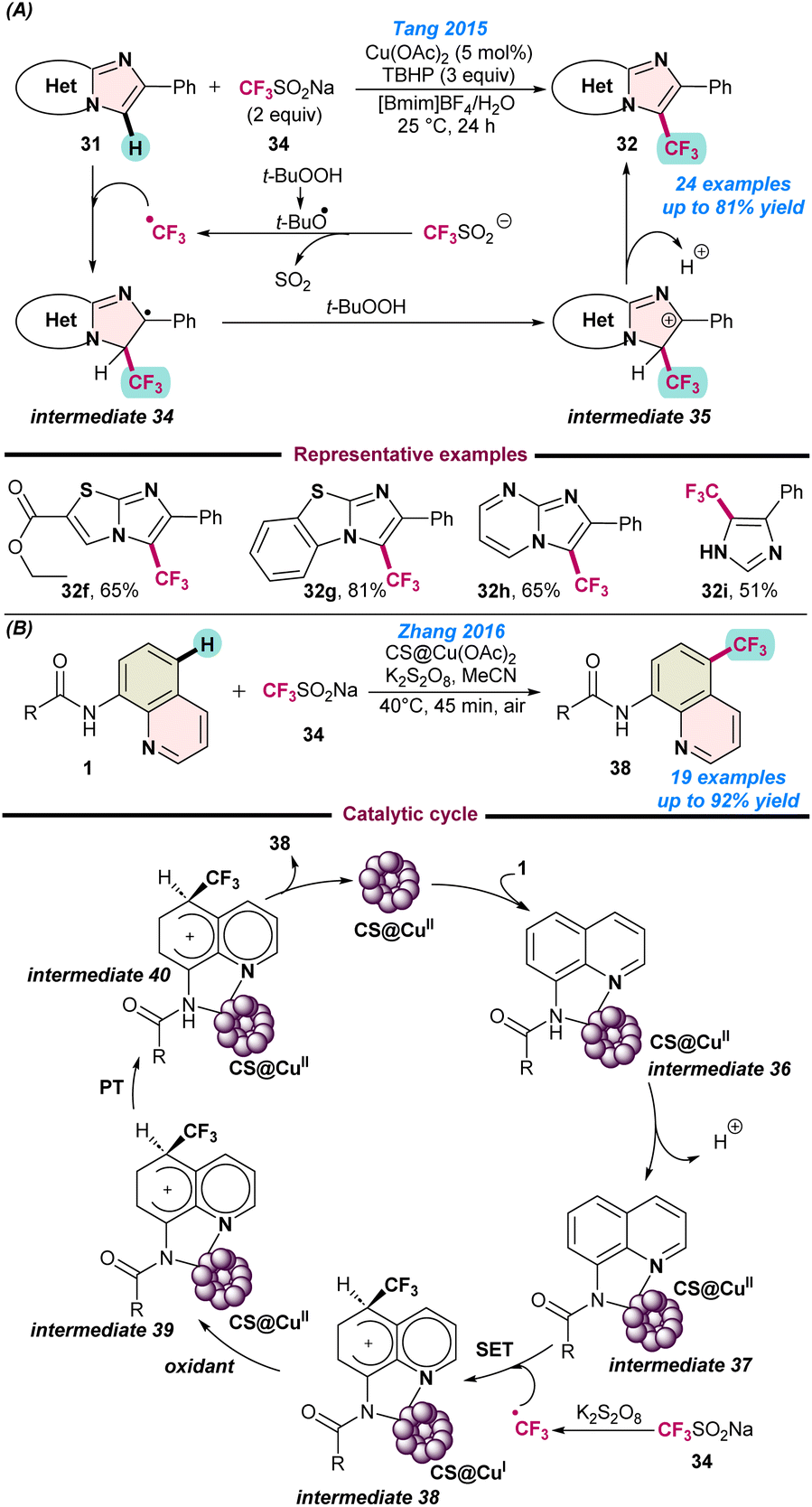 | ||
| Scheme 15 (a) Cu-catalysed regioselective C–H trifluoromethylation of imidazoheteroarene (ref. 71). (b) Cu-chitosan based heterogeneous catalyst for trifluoromethylation of aminoquinoline (ref. 72). | ||
In the next year (2016), Zhang and co-workers demonstrated a user-friendly chitosan-based heterogeneous copper-catalyst for the remote trifluoromethylation of aminoquinoline derivatives (Scheme 15b).72 They also used the low-cost and stable Langlois reagent (CF3SO2Na) as the CF3 source. As is evident from Scheme 15a, independent radical quenching experiments using TEMPO and hydroquinone (HQ) did not provide any desired product and a radical coupling adduct was detected. Based on the experimental evidence, a catalytic route was proposed, as depicted in Scheme 15b, which follows similar pathways to those shown in Scheme 15a.
In 2019, Shi and co-workers demonstrated the copper-catalysed trifluoromethylation of indole derivatives 39 through functionalization of a Csp2–H bond (Scheme 16a).73 Among the different copper-salts used, a CuSO4 catalyst was found to be more suitable than CuI, and the addition of a small amount of KF as a base along with tBuOOH dramatically improved the product yield. The catalytic process efficiently tolerated a broad range of functional groups on the indole derivatives. Thereafter, the same group also expanded their studies on the trifluoromethylation of N-Boc protected indoles 41 under milder conditions without the application of external ligands or additives (Scheme 16a).74 Both catalytic protocols used the Langlois reagent as a potential CF3 source for successful transformations.
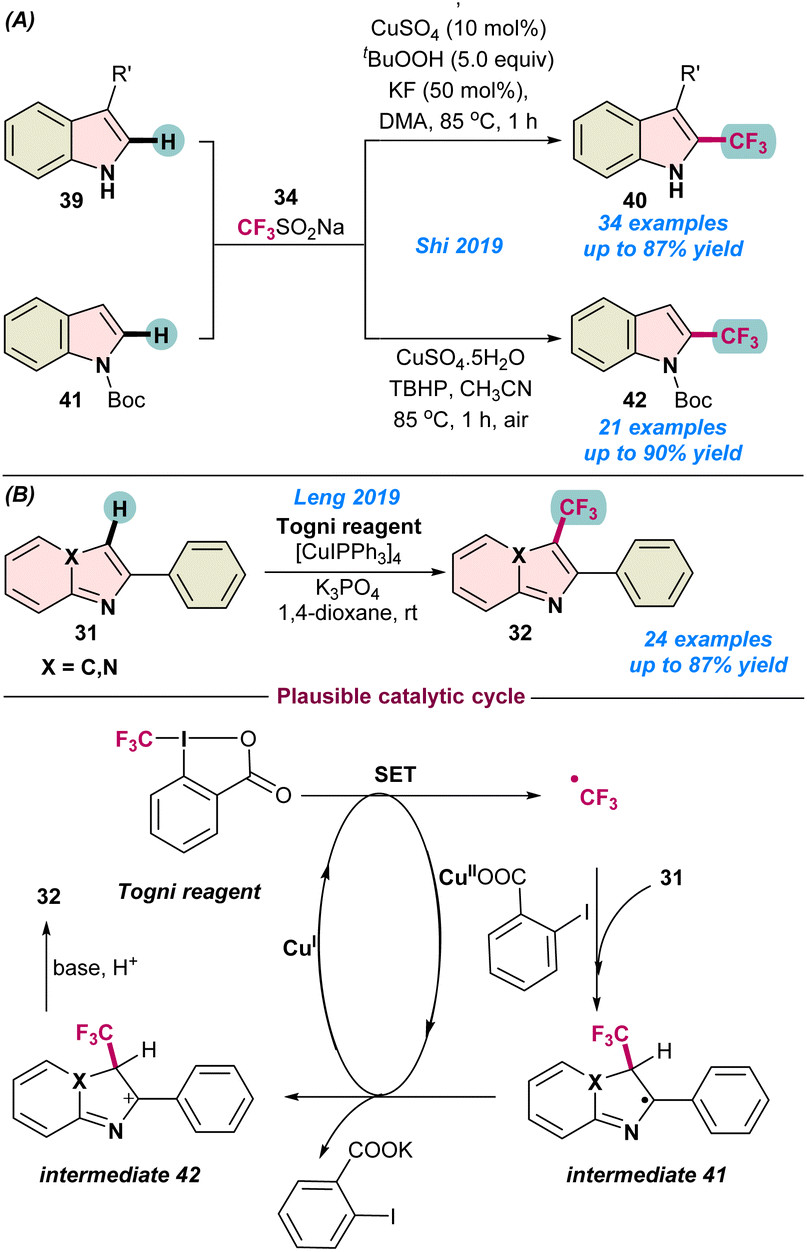 | ||
| Scheme 16 (a) Cu-catalysed oxidative trifluoromethylation of indoles (ref. 73 and 74). (b) Trifluoromethylations of imidazoheterocycles with the Togni reagent (ref. 75). | ||
In the same year (2019), Leng and co-workers demonstrated the direct Csp2–H trifluoromethylation of imidazo-heterocycles 31 using an efficient [Cu(I)IPPh3]4 pre-catalyst in combination with Togni's reagent as a CF3 source (Scheme 16b).75 The process facilitated the transformations in the presence of an inorganic base, K3PO4, and no external oxidant was required. A wide range of functional groups were tolerant to the process and the reaction followed similar catalytic pathways to those shown in Scheme 11.
In 2021, Reddy and co-workers disclosed the first Cu(I)-catalysed oxidative trifluoromethylation of imidazo[1,5-a]N-heteroarenes 43 using the Langlois reagent as the CF3 source, and TBPB as an oxidant (Scheme 17a).76 The catalytic protocol afforded regio-selective synthesis of medicinally important fused trifluoromethylated imidazoles in moderate to good yields. However, the amount of catalyst used for this transformation was slightly higher and the mechanism followed radical pathways, as no desired product 44 was observed when the standard reaction was performed with 3.0 equiv. of TEMPO and BHT. As proposed, initially in situ generated CF3 radical reacted with 43 and a Cu(II)CO2Ph species to organo-copper intermediate 43, and the desired product was obtained after reductive elimination from Cu(III) species intermediate 44, with the continuation of the catalytic cycle (Scheme 17a).76
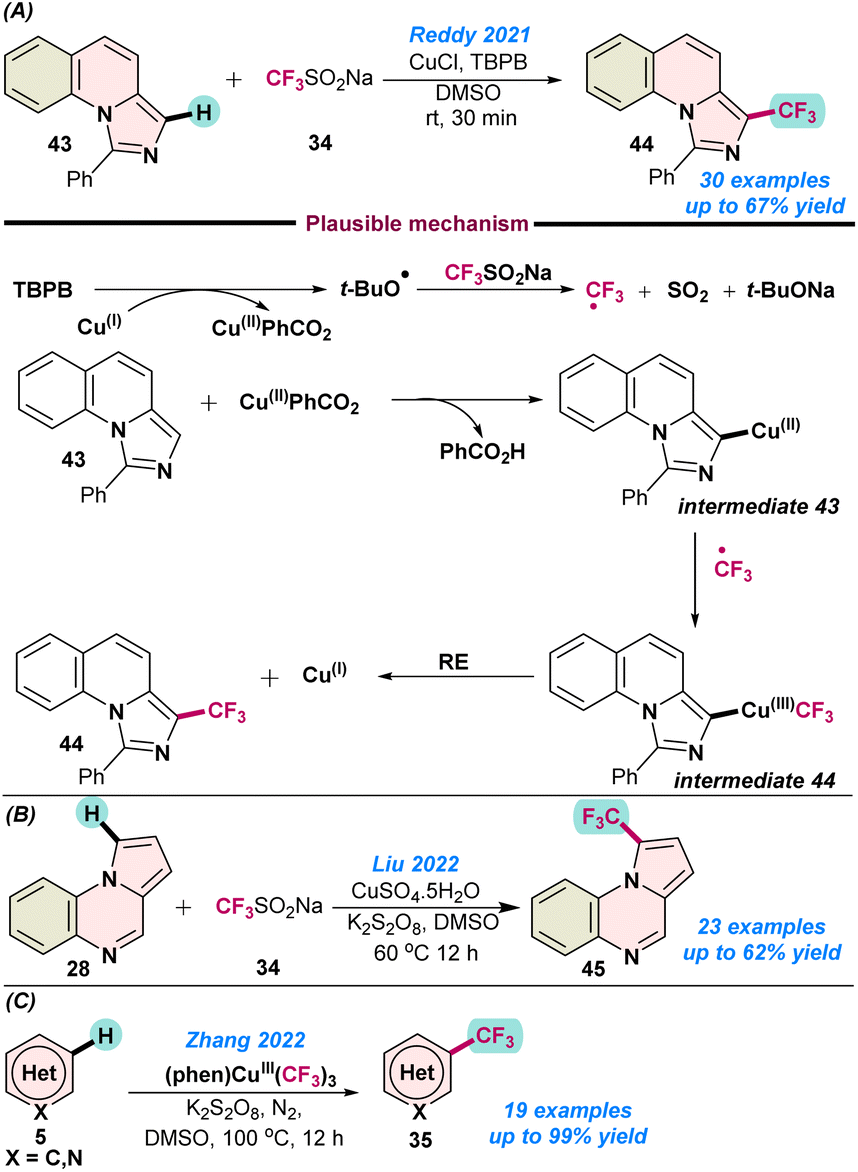 | ||
| Scheme 17 Cu-catalysed (a) trifluoromethylation of imidazo[1,5-a]N-heteroarenes (ref. 76), (b) trifluoromethylation of pyrolo{1,2-a} quinoxalines, (ref. 77) and (c) trifluoromethylation of N-hetero(arenes) (ref. 78). | ||
In 2022, Liu and co-workers demonstrated the Cu(II)SO4·5H2O catalysed direct Csp2-H trifluoromethylation of pyrolo[1,2-a] quinoxalines 28 in the presence of K2S2O8 as an oxidant in combination with the Langlois reagent (Scheme 17b).77 A series of trifluoromethylated pyrolo[1,2-a] quinoxaline derivatives were synthesized in moderate to good yields. The high-scale synthesis of 45 indicated the practical utility of the protocol.
In the same year, Zhang and co-workers established the first C–H trifluoromethylation of N-heteroarenes (5) using a high-valent Cu(III)–CF3 complex (Scheme 17c).78 It was observed that the (phen)CuIII(CF3)3 complex served as the source of the CF3 radical. Partial increase in the product yield was observed upon the addition of co-oxidant K2S2O8 in DMSO as a solvent. However, when DMF was used as a solvent, the addition of a co-oxidant did not have much influence on the product yields and indicated the potential role played by the solvent in product conversion. A series of control experiments suggested the homolytic cleavage of Cu(III)–CF3 and the involvement of the generated CF3-radical, which were confirmed by time-dependent electron paramagnetic resonance (EPR) spectroscopy analysis (Scheme 17c).78
Later in that year (2022), Shen and co-workers, described a multi-component reaction involving indoles 39, quinoxaline-2(1H)-ones 46 and the Langlois reagent in the presence of CuF2 as a catalyst for the synthesis of 3-[2-(trifluoromethyl)1H-indole-3-yl] quinoxaline-2(1H)-ones 47 (Scheme 18).79 A series of control experiments were performed to detect the key intermediate 48 along with the radical trapping process. Based on these experimental findings, a plausible catalytic cycle was proposed, as shown in Scheme 18.
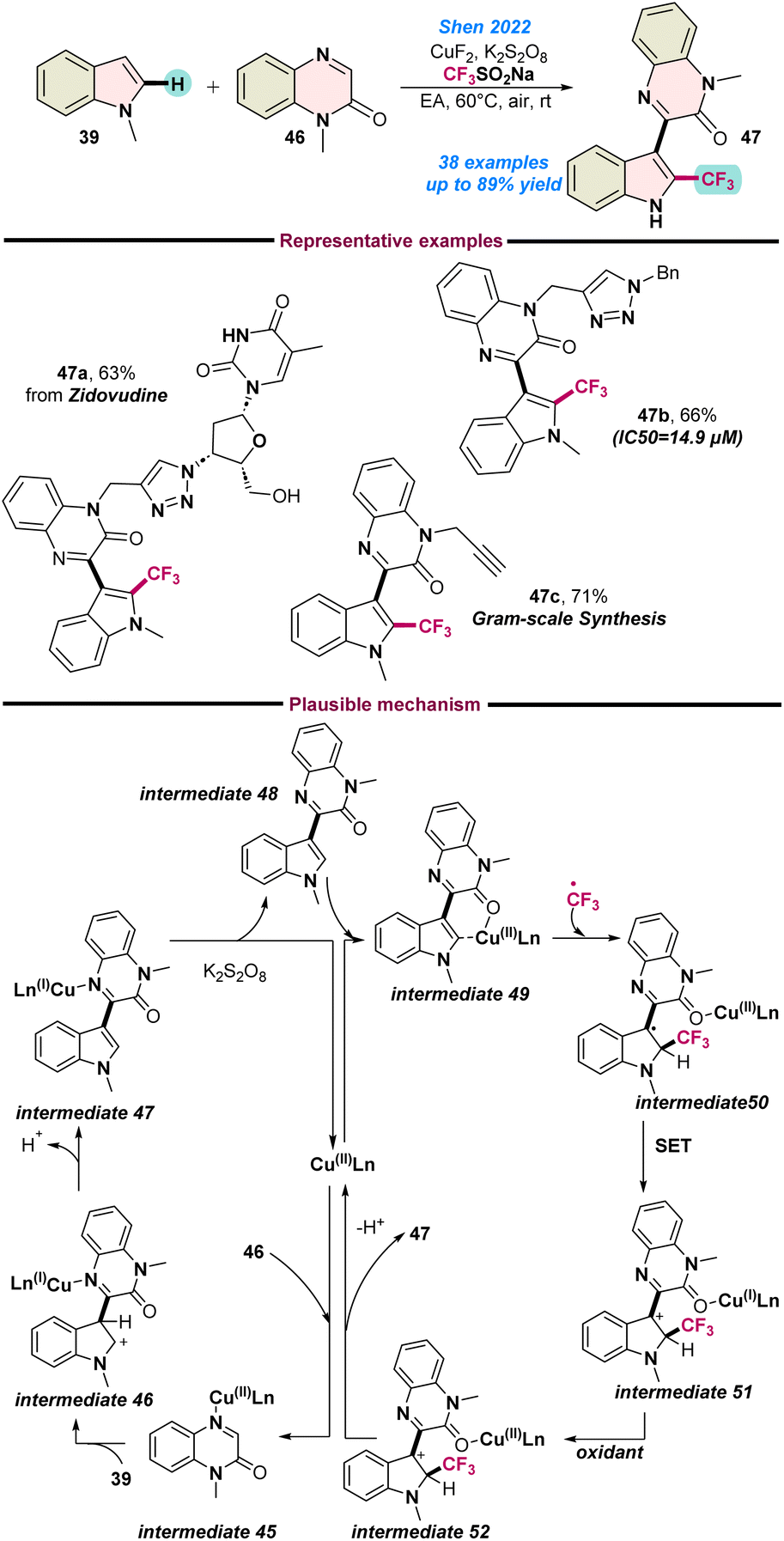 | ||
| Scheme 18 Cu(II)-catalysed multicomponent synthesis of fluorinated indole-quinoxalin-2(1H)-ones (ref. 79). | ||
Initially, the Cu(II) species weakly co-ordinates with the nitrogen of quinoxaline-2(1H)-ones 46 and generates an organo-Cu(II) species intermediate 45, which readily couples with indole 39 to afford intermediate 46. Subsequently, intermediate 46 converts to intermediate 49 following an oxidative deprotonation process. In the meantime, the generated CF3-radical attaches to the indole ring of intermediate 49 and transforms into intermediate 50. Thereafter, single-electron transfer occurs from intermediate 50, followed by oxidation and deprotonation that result in the formation of the target product 47, as shown in Scheme 18.
5. Perfluoroalkylation of heteroarenes
Perfluoroalkyl compounds (PFA) are rarely found in naturally occurring substances, however, their introduction into biologically active molecules enables significant alteration of the physical or chemical properties of these molecules. More specifically, perfluoroalkylated N-heteroarenes are widely used as lubricants, surfactants, and often as flame retardants due to their notable chemical stability. In this part of the review, we broadly discuss the various recently developed perfluoroalkylation reactions for the direct introduction of various perfluoroalkyl groups on an N-heteroarene core.5.1 Iron- and nickel-catalysed perfluoroalkylation
In 2015, Budnikova and co-workers reported the [(bpy)Fe(II)] catalysed perfluoroalkylation of 1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione 48 with C6F13I. To improve the product yields, the reactions were performed under electrochemical conditions at room temperature. Notably, when the electrode potential of the electrolysis was −1.0 V vs. Ag/AgCl in DMF, they observed a high yield of perfluoroalkylated products. The electrochemical transformation proceeded through radical pathways, as was evident from electron spin resonance (ESR) spectroscopy and cyclic voltammetry (CV) studies (Scheme 19a).80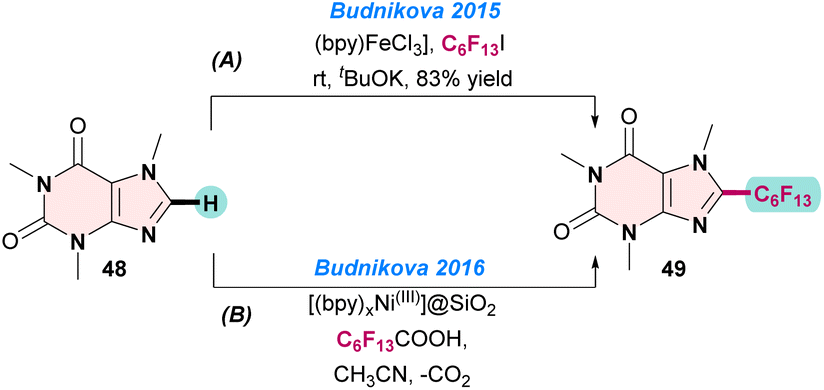
In the next year (2016), the same group prepared recyclable Ni(III)-doped silica nanoparticles ([(bpy)xNiIII]@SiO2) for perfluoroalkylations of heteroarenes (48). The catalyst was prepared electrochemically by doping a [(bpy)3NiII] salt on silica nanoparticles and oxidizing it to [(bpy)3NiIII]. The coupling reaction between 48 and perfluoroheptanoic acid gave the desired product 49 in an almost quantitative yield (Scheme 19b).81
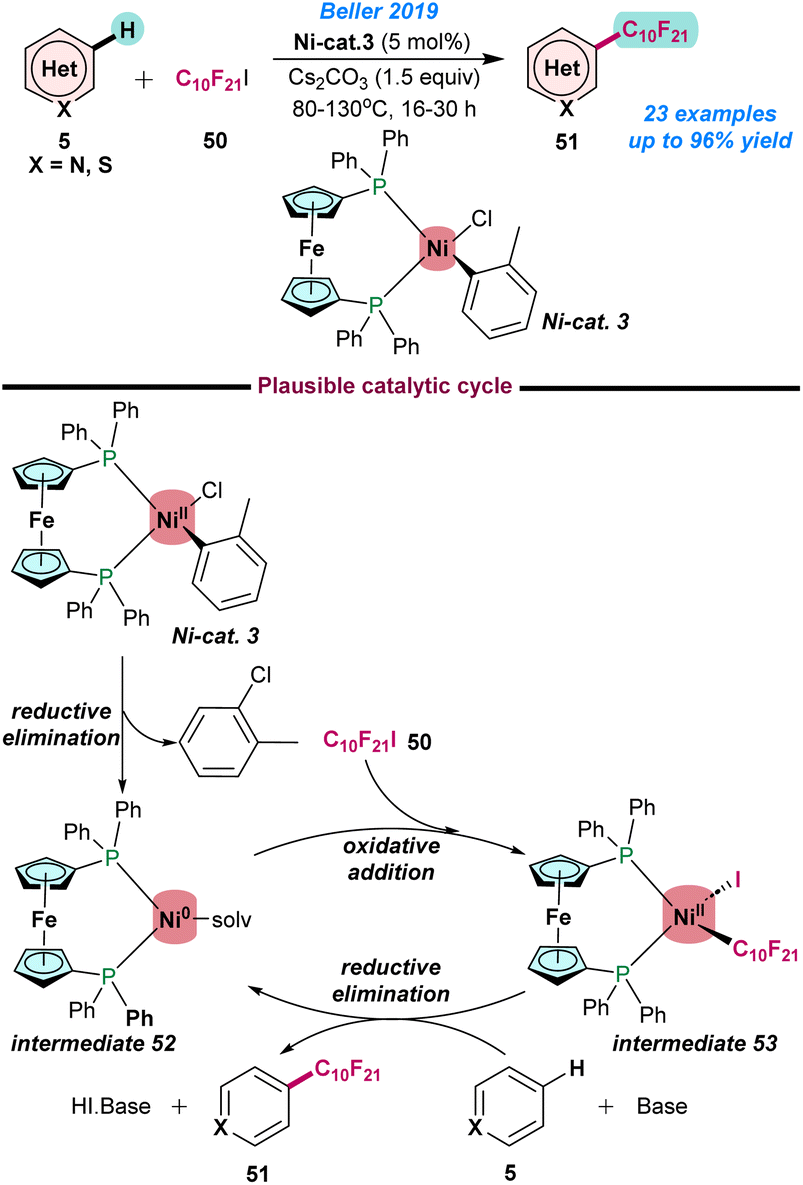
In 2019, Beller and co-workers demonstrated the perfluoroalkylation of various N-heteroarenes using an air and moisture-stable (dppf)Ni(o-tol)Cl complex (Scheme 20).82 The use of Ni-cat. 3 and C10F21I (50) as a perfluoroalkylated source resulted in the formation of the desired product (51) in moderate to excellent yields using Cs2CO3 as a base. The synthesis of various perfluoroalkylated heteroarenes with sensitive functional groups (aldehydes, free amino groups, etc.) is noteworthy. It was proposed that Ni-cat. 3 undergoes reductive elimination to afford the Ni(0) intermediate 52, which upon oxidative addition with perfluoroalkyl iodide gives intermediate 53. Subsequently, activation of heteroarenes in the presence of a base resulted in the formation of the desired product 51 and intermediate 52 being regenerated to continue the catalytic cycle (Scheme 20).
 | ||
| Scheme 20 Ni-catalysed C–H perfluoroalkylation of heteroarenes (ref. 82). | ||
5.2 Cobalt-catalysed perfluoroalkylation
In 2020, Beller and co-workers further investigated the perfluoroalkylation of hetero(arenes) using a cobalt-based nano-catalyst. After screening a series of different catalysts, Co@N/C-800 was proven to be the best catalyst in combination with cesium carbonate as a base. Catalytic reactions of p-methyl pyridine (52) with perfluoroalkyl bromide (53) afforded a library of perfluoro-alkylated products. Notably, the catalyst can be recycled up to four times without having much effect on the product yield (Scheme 21a).83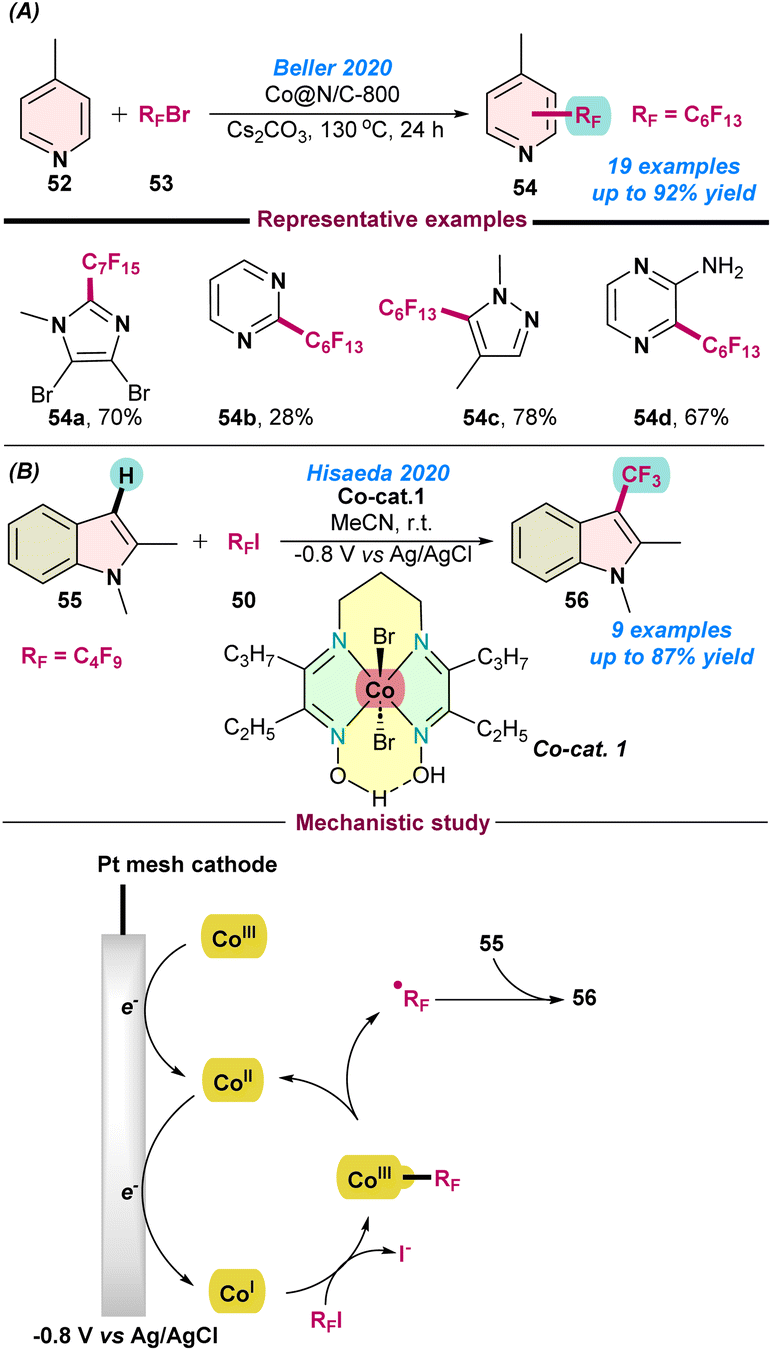 | ||
| Scheme 21 (a) Perfluoroalkylation of hetero(arenes) using a cobalt nano-catalyst (ref. 83) and (b) the electro-catalysed direct perfluoroalkylation of indoles (ref. 84). | ||
In the same year (2020), Hisaeda and co-workers reported perfluoroalkylation of indoles derivatives, using a vitamin B12 based-model complex following an electrochemical approach (Scheme 21b).84 Catalytic reactions using a cobalt complex, [Co(III){(C2C3)(DO)(DOH)pn}Br2] were performed using controlled-potential electrolysis at −0.8 V vs. Ag/AgCl in an organic solvent. The turnover number (TON) was 87, indicating the sufficient stability of the catalyst for the perfluoroalkylation reaction. Radical quenching experiments suggested a radical pathway and the homolytic cleavage of Co–C bond during the progress of the reaction. Based on the control experiments and previously reported studies, a catalytic cycle was proposed, as presented in Scheme 21b. Initially, the Co(III)-species transformed into a super-nucleophilic Co(I)-species under electrolysis conditions and readily attached to RF–I and formed a Co(III)–RF intermediate, which produced an RF-radical after homolytic cleavage and generated a Co(II) species. Finally, the RF-radical attacked the indole derivatives (55), and the desired product was formed after one-electron oxidation, as shown in Scheme 21b.
5.3 Copper-catalysed perfluoroalkylation
In 2015, Mikami and co-workers reported the Cu(I)-catalysed perfluoroalkylation of N-heteroarenes using a bis(perfluoroalkyl)zinc reagent (Scheme 22a).85 The protocol has significant advantages, such as: (a) it is air-stable and uses a readily accessible perfluoroalkylating agent, (b) requires no use of ligand and perfluoro activators, and (c) the methodology is applicable to both trifluoromethylation and perfluoroalkylation with good product yield. The proposed catalytic cycle reveals that the first step is transmetallation, where the perfluoroalkyl group is transferred to CuI from Zn(RF)2(DMPU)2 and generates [Cu(RF)I]− and Zn(RF)I(DMPU)2. However, the complex Zn(RF)I(DMPU)2 was found to be in Schlenk equilibrium with Zn(RF)2(DMPU)2 and ZnI2. Next, consecutive oxidative addition and reductive elimination generate the desired product from the Cu species (Scheme 22a).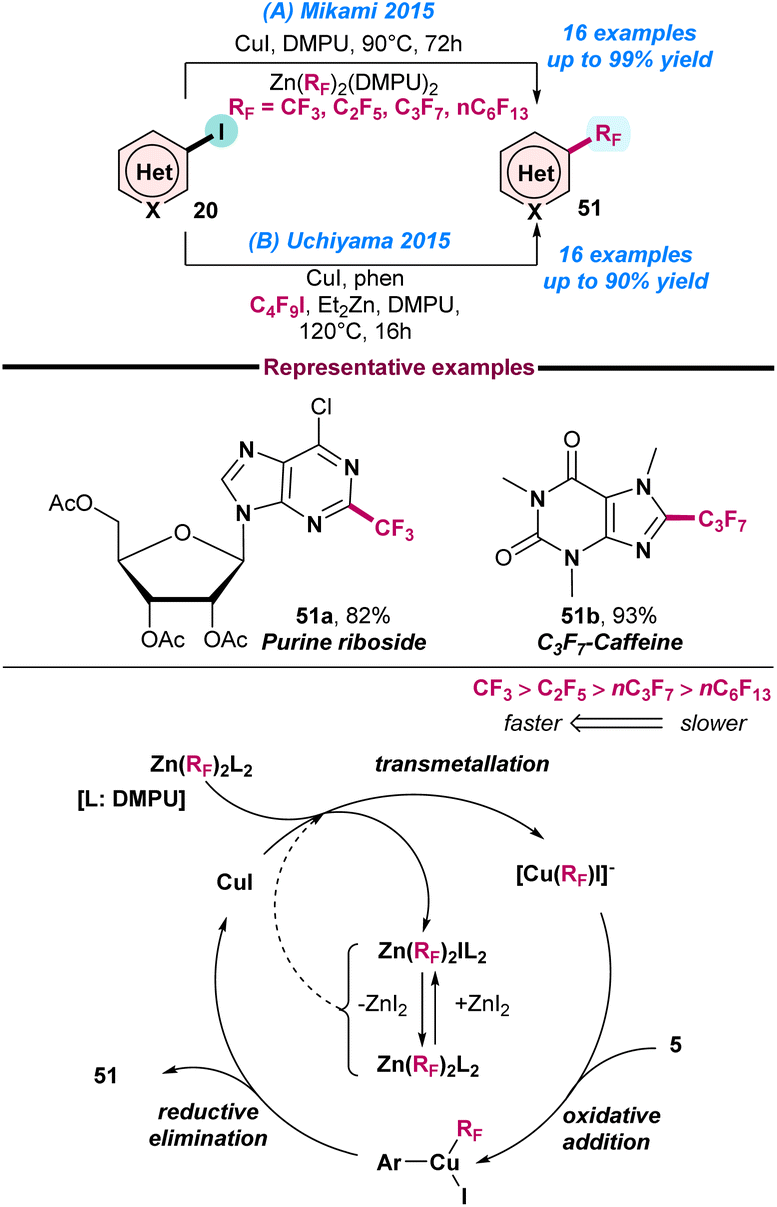
In the same year, Uchiyama and co-workers also reported a similar Cu(I)-catalysed cross-coupling reaction between perfluoroalkyl iodide and heteroaryl iodide (Scheme 22b).86 The method was highly chemo- and regioselective, and it was proposed that a rapid halogen-zinc exchange reaction occurred between the organozinc species and perfluoroalkyl iodide and generated a thermally stable perfluoroalkylzinc reagent. This perfluoroalkylzinc species was coupled with heteroaryl iodide to give the desired product in good yield, with excellent functional group tolerance.
In 2016, Weng and co-workers demonstrated the perfluoroalkylation of heteroaryl halides using a Cu(I)-catalyst (Scheme 23a).87 They synthesized moisture-stable and readily soluble (in organic solvents) copper(I)perfluorocarboxylato complexes, [(phen)2Cu](O2CRF) via the fusion of copper tert-butyloxide, phenanthroline ligands, and perfluorocarboxylic acids. Interestingly, the presence of the second phenanthroline ligand facilitated the perfluoroalkylation by controlling the activation energy required for the decarboxylation of perfluorocarboxylate.
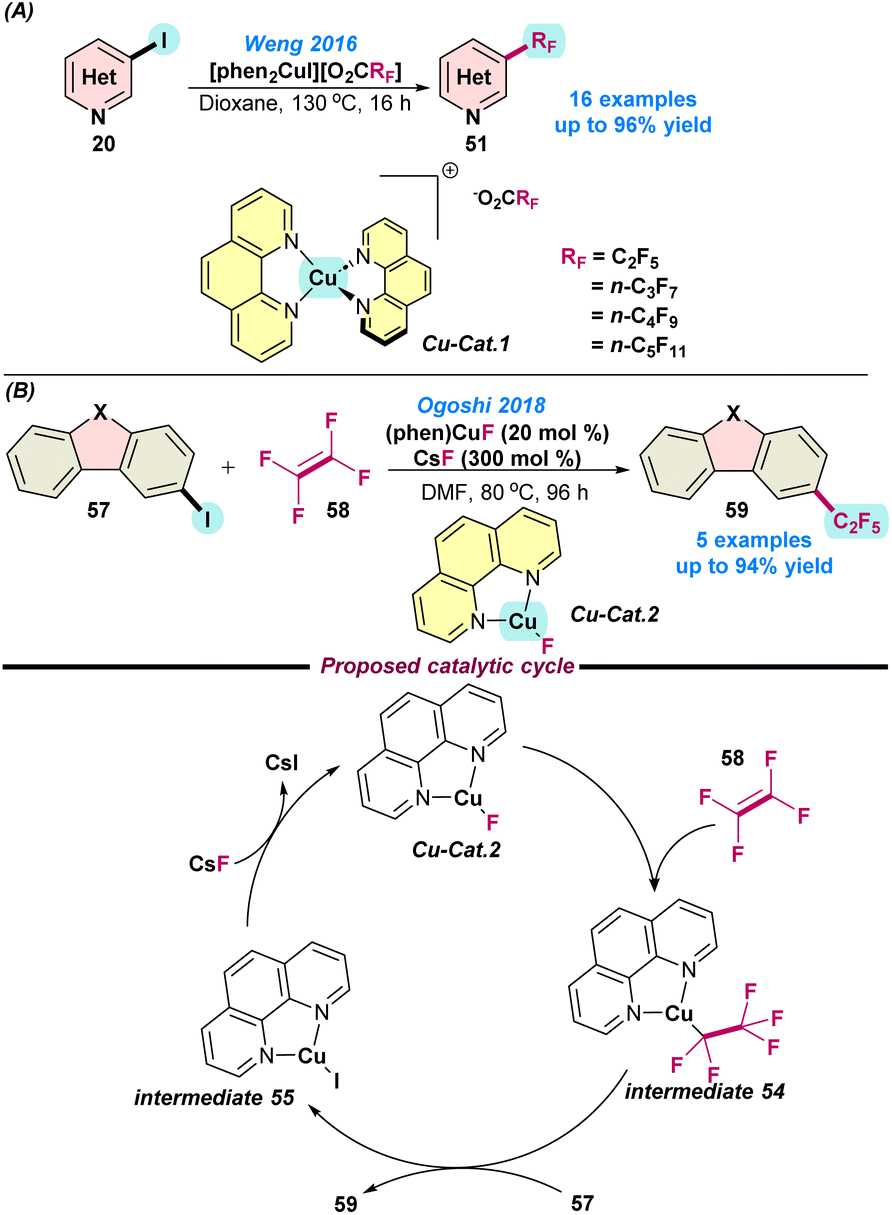 | ||
| Scheme 23 (a) Perfluoroalkylation using copper(I)perfluoro-carboxylato complexes (ref. 87) and (b) the Cu(I)-catalysed perfluoroalkylation of heteroarenes using TFE (ref. 88). | ||
In 2018, Ogoshi and co-workers reported the Cu(I)-catalysed pentafluoroalkylation of N-heteroarenes using tetra-fluoroethylene (TFE) as a fluorine source (Scheme 23b).88 It was postulated that (a) TFE was used as a monomer, and the tetrafluoroethylene-based polymer can be easily prepared and (b) TFE is cost efficient and can be prepared from an environmentally benign feedstock. However, other Cu-based complexes could not participate in the fluorocupration reaction. Mechanistic studies showed that, initially, TFE was fluorocuprated smoothly at room temperature, leading to the formation of intermediate 54. This intermediate readily reacted with 57 and transformed into intermediate 55, along with the generation of penta-fluorinated product 59. Finally, Cu-cat. 2 was regenerated from intermediate 55via abstracting the fluoride ion from CsF. Here, the fluorocupration did not follow the outer-sphere mechanism because the fluoride ion did not restrict the fluorocupration of TFE (Scheme 23b).
In the same year, Cai and co-workers proposed the Cu(II)Br catalysed regioselective C–H perfluoroalkylation of 8-aminoquinoline amides with RFSO2Na (RF = CF3, CF2H, C4F9, C5F11, and C6F13) (Scheme 24a).89 After screening the various parameters, CuBr in the presence of AIBN as an oxidant gave the best results. This methodology delivered the direct fluorine-containing six-membered heteroatom core structure and was well tolerated by various functional groups. The reaction follows comparable catalytic pathways to those presented in Scheme 1.
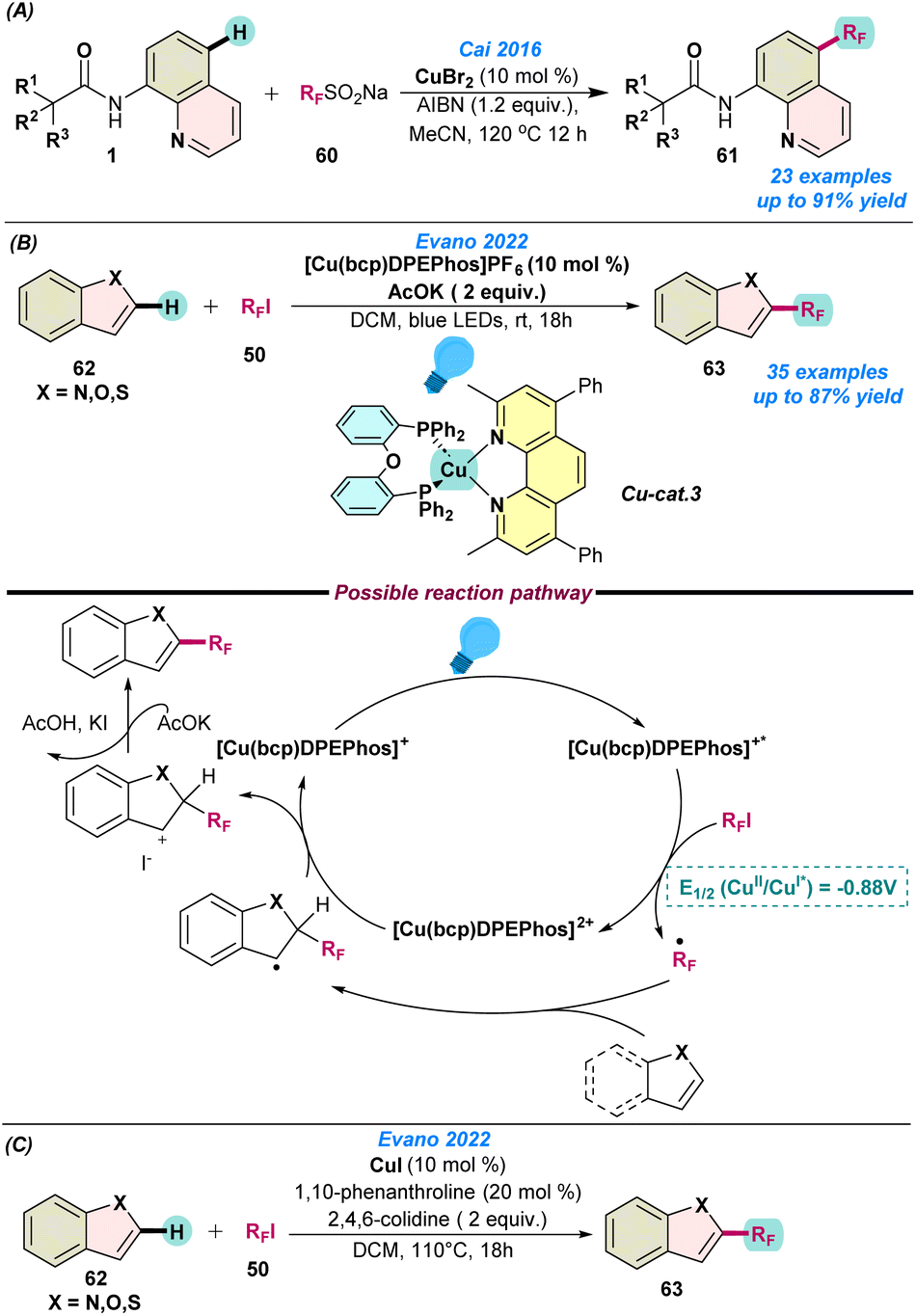 | ||
| Scheme 24 (a) Cu-catalysed perfluoroalkylation of 8-aminoquinoline amides (ref. 89), (b) the Cu-catalyzed perfluoroalkylation of heteroarenes using photoinduced and thermal conditions (ref. 90 and 91) and (c) Cu-catalysed perfluoroalkylation of heteroarenes using thermal conditions (ref. 91). | ||
In 2022, Evano and co-workers reported the Cu-catalysed perfluoro-alkylation of N-heteroarenes with perfluoroalkyl iodide using both photoinduced (Scheme 24b)90 and thermal conditions (Scheme 24c).91 Under thermal conditions, CuI, phenanthroline and 2,4,6-colidine were used as a catalyst, ligand, and base, respectively, for a duration of 18 h at 110 °C. To overcome the temperature restrictions, they performed the reaction under mild photoinduced conditions, where [Cu(bcp)DPEPhos]PF6 was used as a photo-redox catalyst in the presence of potassium acetate. However, in both cases, DCM was used as a solvent. Both methodologies showed broad substrate scope with a wide range of functional group tolerance. To understand the reaction pathway, they performed the standard reaction with 1.0 equiv. of TEMPO, which suppressed the product formation, indicating the formation of a radical species during the progress of the reaction.
Based on the control experiments, they proposed a catalytic cycle under photoinduced conditions. Initially, adsorption of a photon by [Cu(bcp)DPEPhos]+ transforms it into [Cu(bcp)DPEPhos]+*, which triggers single-electron transfer and generates the Cu(II) species [Cu(bcp)DPEPhos]2+ along with the formation of an electrophilic RF radical from RFI. Next, the transient RF-radical attaches to the N-heteroarenes to form a benzylic/allylic radical intermediate species, which further oxidizes in the presence of the Cu(II) species to complete the catalytic cycle and deliver the desired product after re-aromatization (Scheme 24c).
6. Conclusion
The direct incorporation of fluorine, trifluoromethyl, and perfluoroalkyl groups into N-heteroarenes is a topic of current interest in chemistry and biochemistry because of its inclusive utilization in the agrochemicals and pharmaceutical industries. The advances in the synthetic methods for fluorination and fluoroalkylation over the last few years are a result of the introduction of cost-effective reagents and techniques. Precious metal-catalysed direct Csp2–H fluorination and fluoroalkylation is one of the most remarkable and potential processes known, however, the use of 3d-transitional metals has been extensively amplified in the last few years because of their high natural abundance, low cost and relatively low toxicity. This review focusses on direct Csp2 fluorination and fluoroalkylation functionalisations via the Csp2–H bond, cross-coupling reactions, and decarboxylation reactions using non-precious-metal catalysts. We have highlighted the thermally-mediated methodologies, photoredox catalyses, and electrocatalyses that have evolved effectively for the fluorination, trifluoromethylation, and perfluoroalkylation of N-heteroarenes. A wide range of heteroarenes have been utilized for fluorination, trifluoromethylation, and perfluoroalkylation reactions using diverse 3d-metal catalysed conditions. Besides, varied fluorinated building blocks are used as coupling partners in these reactions. The newly established fluorinated moieties, which have never been explored previously, are discussed. We have discussed the mechanistic pathways of fluorination reactions, wherein most of them proceeded via radical intermediate formation to initiate the reaction with an un-activated organic moiety. We have discussed the synthesis procedures of a couple of fluorine and fluorine moieties widely used as important drugs and natural products.From this review, it is evident that fluorination, particularly C(sp2)–F bond formation, has been predominantly explored under homogeneous metal catalysis. Nevertheless, heterogeneous metal catalysed C(sp2)–F bond formation has shown quite promising results and has a very promising future scope. Furthermore, asymmetric synthesis using fluorinated substrates or fluorine-based drug discovery require more attention, importantly, trifluoromethylation and perfluoroalkylation are still underdeveloped and need considerable attention. Overall, there is a lot of scope to develop newer technologies based on fluorination, trifluoromethylation, and perfluoroalkylation reactions. We strongly envisage that more detailed mechanistic studies, including DFT investigations, will assist such fluorination and perfluoroalkylation technologies in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CSIR-India (02(0373)/19/EMR-II) for financial support. IIT Roorkee (SMILE-32) is gratefully acknowledged for instrument facilities. P. thanks UGC-India and A. B. thanks IIT-R for financial support.References
- S. Dehnen, L. L. Schafer, T. Lectka and A. Togni, Org. Lett., 2021, 23, 9013–9019 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- S. Caron, Org. Process Res. Dev., 2020, 24, 470–480 CrossRef CAS.
- Application of N-Fluorobenzenesulfonimide for the direct fluorination, see: Q. Gu and E. Vessally, RSC Adv., 2020, 10, 16756–16768 RSC.
- G. Tarantino and C. Hammond, Green Chem., 2020, 22, 5195–5209 RSC.
- Application of organofluorine compounds to the agrochemical industry, see: Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- For a selective review on precious metal-based C–H functionalization using fluorinated building blocks, see: H. Sindhe, B. Chaudhary, N. Chowdhury, A. Kamble, V. Kumar, A. Lad and S. Sharma, Org. Chem. Front., 2022, 9, 1742–1775 RSC.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed.
- K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817–829 CrossRef CAS PubMed.
- A. D. Dilman and V. V. Levin, Eur. J. Org. Chem., 2011, 831–841 CrossRef CAS.
- P. T. Nyffeler, S. G. Duron, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- S. Stavber, Molecules, 2011, 16, 6432–6464 CrossRef CAS PubMed.
- B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585–2634 CAS.
- Q. Cheng and T. Ritter, Trends Chem., 2019, 1, 461–470 CrossRef CAS.
- I. Hussain and T. Singh, Adv. Synth. Catal., 2014, 356, 1661–1696 CrossRef CAS.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- G. Bertuzzi, L. Bernardi and M. Fochi, Catalysts, 2018, 8, 632 CrossRef.
- B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- Review on fluorination methods in drug discovery, see: D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC.
- Review on fluoroalkylation reactions in aqueous media, see: H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Green Chem., 2018, 20, 1662–1731 RSC.
- Review on remote fluorination and fluoroalkyl(thiol)ation reactions, see: F.-G. Zhang, X.-Q. Wang, Y. Zhou, H.-S. Shi, Z. Feng, J.-A. Ma and I. Marek, Chem. – Eur. J., 2020, 26, 15378–15396 CrossRef CAS PubMed.
- Review on selective radiofluorination (organofluorine-18) compounds, see: T. T. Bui and H.-K. Kim, Chem. – Asian J., 2021, 16, 2155–2167 CrossRef CAS PubMed.
- For a selective review on Cu-based C(sp2)–F and C(sp3)–F bond constructions, see: T. Dong and G. C. Tsui, Chem. Rec., 2021, 21, 4015–4031 CrossRef CAS PubMed.
- Review on nucleophilic fluorination with potassium fluoride (KF), see: M. Khandelwal, G. Pemawat and R. K. Khangarot, Asian J. Org. Chem., 2022, 11, e202200325 CrossRef CAS.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem. – Asian J., 2012, 7, 1744–1754 CrossRef CAS PubMed.
- S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2013, 257, 3051–3069 CrossRef CAS.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS PubMed.
- C. Zhang, Adv. Synth. Catal., 2014, 356, 2895–2906 CrossRef CAS.
- C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed.
- K. Natte, R. V. Jagadeesh, L. He, J. Rabeah, J. Chen, C. Taeschler, S. Ellinger, F. Zaragoza, H. Neumann, A. Bruckner and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 2782–2786 CrossRef CAS PubMed.
- S. Barata-Vallejo, B. Lantano and A. Postigo, Chem. Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed.
- A. Prieto, O. Baudoin, D. Bouyssi and N. Monteiro, Chem. Commun., 2016, 52, 869–881 RSC.
- L. Mudarra, S. M. de Salinas and M. H. P. Temprano, Synthesis, 2019, 2809–2820 Search PubMed.
- G. Baishya and N. B. Dutta, ChemistrySelect, 2021, 6, 13384–13408 CrossRef CAS.
- D. Mandal, S. Maji, T. Pal, S. K. Sinha and D. Maiti, Chem. Commun., 2022, 58, 10442–10468 RSC.
- F. L. Qing, X. Y. Liu, J. A. Ma, Q. Shen, Q. Song and P. Tang, CCS Chem., 2022, 4, 2518–2549 CrossRef CAS.
- S. Barata-Vallejo, S. M. Bonesi and A. Postigo, RSC Adv., 2015, 5, 62498–62518 RSC.
- D. J. C.-Pazos, J. D. Lasso and C.-J. Li, Org. Biomol. Chem., 2021, 19, 7116–7128 RSC.
- H. Baguia and G. Evano, Chem. – Eur. J., 2022, 28, e202200975 CrossRef CAS PubMed.
- Examples on the synthesis of fluoroalkylated compounds using fluoroalkyl anhydrides, see: W. Wu, Y. You and Z. Weng, Chin. Chem. Lett., 2022, 33, 4517–4530 CrossRef CAS.
- C. Zhang, Adv. Synth. Catal., 2017, 359, 372–383 CrossRef CAS.
- D. E. Yerien, S. B.-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- N. Levi, D. Amir, E. Gershonov and Y. Zafrani, Synthesis, 2019, 4549–4567 CAS.
- J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC.
- J. Ding, Y. Zhang and J. Li, Org. Chem. Front., 2017, 4, 1528–1532 RSC.
- N. J. Taylor, E. Emer, S. Preshlock, M. Schedler, M. Tredwell, S. Verhoog, J. Mercier, C. Genicot and V. Gouverneur, J. Am. Chem. Soc., 2017, 139, 8267–8276 CrossRef CAS PubMed.
- M. S. McCammant, S. Thompson, A. F. Brooks, S. W. Krska, P. J. H. Scott and M. S. Sanford, Org. Lett., 2017, 19, 3939–3942 CrossRef CAS PubMed.
- S.-S. Luo, L.-J. Su, Y. Jiang, X.-B. Li, Z.-H. Li, H. Sun and J.-K. Liu, Synlett., 2018, 29, 1525–1529 CrossRef CAS.
- P. Xu, P. L.-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- W.-T. Fan, Y. Li, D. Wang, S.-J. Ji and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 20524–20530 CrossRef CAS PubMed.
- Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837–5841 CrossRef CAS PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536–2539 CrossRef CAS PubMed.
- X.-P. Fu, Y.-L. Xiao and X. Zhang, Chin. J. Chem., 2018, 36, 143–146 CrossRef CAS.
- C. Xu, W.-H. Guo, X. He, Y.-L. Guo, X.-Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170 CrossRef PubMed.
- J. Sheng, H.-Q. Ni, K.-J. Bian, Y. Li, Y.-N. Wang and X.-S. Wang, Org. Chem. Front., 2018, 5, 606–610 RSC.
- V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernandez, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS PubMed.
- Z. Zou, H. Li, M. Huang, W. Zhang, S. Zhi, Y. Wang and Y. Pan, Org. Lett., 2021, 23, 8252–8256 CrossRef CAS PubMed.
- X. Li, J. Zhao, M. Hu, D. Chen, C. Ni, L. Wang and J. Hu, Chem. Commun., 2016, 52, 3657–3660 RSC.
- H. Serizawa, K. Ishii, K. Aikawa and K. Mikami, Org. Lett., 2016, 18, 3686–3689 CrossRef CAS PubMed.
- S.-Q. Zhu, Y.-L. Liu, H. Li, X.-H. Xu and F.-L. Qing, J. Am. Chem. Soc., 2018, 140, 11613–11617 CrossRef CAS PubMed.
- Y. Li, Z. Yang, Y. Liu, Y. Liu, Y. Gu and P. Liu, Mol. Catal., 2021, 511, 111747 CrossRef CAS.
- Y. Wu, H. R. Zhang, R. X. Jin, Q. Lan and X. S. Wang, Adv. Synth. Catal., 2016, 358, 3528–3533 CrossRef CAS.
- E. A. Meucci, S. N. Nguyen, N. M. Camasso, E. Chong, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 12872–12879 CrossRef CAS PubMed.
- S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, M. C. Roy and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2021, 60, 24620–24629 CrossRef CAS PubMed.
- P. Z. Zhang, C. K. Li, G. Y. Zhang, L. Zhang, Y. J. Jiang and J. P. Zou, Tetrahedron, 2016, 72, 3250–3255 CrossRef CAS.
- X.-M. Ji, L. Wei, F. Chen and R.-Y. Tang, RSC Adv., 2015, 5, 29766–29773 RSC.
- C. Shen, J. Xu, B. Ying and P. Zhang, ChemCatChem, 2016, 8, 3560–3564 CrossRef CAS.
- X. Shi, X. Li, L. Ma and D. Shi, Catalysts, 2019, 9, 278–292 CrossRef.
- X. Shi, X. Li, X. Li and D. Shi, Front. Chem., 2019, 7, 613–623 CrossRef CAS PubMed.
- R. Wang, J. Wang, Q. Tang, X. Zhao, J. Wang, J. Chang, Y. Wu, Z. Zhang, S. Wang, Y. Wu and Y. Leng, Tetrahedron Lett., 2019, 60, 586–590 CrossRef CAS.
- M. Sandeep, B. Shantharjun, G. P. Kumar and K. R. Reddy, Eur. J. Org. Chem., 2021, 246–252 CrossRef CAS.
- Y. Li, Y. Liu, D. Hao, C. Li, Y. Liu, Y. Gu, L. Vaccaro and P. Liu, Tetrahedron, 2022, 105, 132610 CrossRef CAS.
- H.-R. Zhang, C.-C. Feng, N. Chen and S.-L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202209029 CrossRef CAS PubMed.
- L. Zhang, Y. Yang, P. Zhang, C. Chen and C. Shen, Eur. J. Org. Chem., 2022, e202200696 CrossRef CAS.
- M. Khrizanforov, S. Strekalova, V. Khrizanforova, V. Grinenko, K. Kholin, M. Kadirov, T. Burganov, A. Gubaidullin, T. Gryaznova, O. Sinyashin, L. Xu, D. A. Vicicb and Y. Budnikova, Dalton Trans., 2015, 44, 19674–19681 RSC.
- M. N. Khrizanforov, S. V. Fedorenko, S. O. Strekalova, K. V. Kholin, A. R. Mustafina, M. Ye Zhilkin, V. V. Khrizanforova, Y. N. Osin, V. V. Salnikov, T. V. Gryaznovaa and Y. H. Budnikova, Dalton Trans., 2016, 45, 11976–11982 RSC.
- S. Zhang, N. R. Loria, F. Weniger, J. Rabeah, H. Neumann, C. Taeschlerc and M. Beller, Chem. Commun., 2019, 55, 6723–6726 RSC.
- S. Zhang, F. Weniger, C. R. Kreyenschulte, H. Lund, S. Bartling, H. Neumann, S. Ellinger, C. Taeschler and M. Beller, Catal. Sci. Technol., 2020, 10, 1731–1738 RSC.
- L. Cui, T. Ono, Y. Morita and Y. Hisaeda, Dalton Trans., 2020, 49, 7546–7551 RSC.
- K. Aikawa, Y. Nakamura, Y. Yokota, W. Toya and K. Mikami, Chem. – Eur. J., 2015, 21, 96–100 CrossRef CAS PubMed.
- H. Kato, K. Hirano, D. Kurauchi, N. Toriumi and M. Uchiyama, Chem. – Eur. J., 2015, 21, 3895–3900 CrossRef CAS PubMed.
- Y. Huang, M. J. Ajith, K.-W. Huang, Z. Zhang and Z. Weng, Dalton Trans., 2016, 45, 8468–8474 RSC.
- M. Ohashi, N. Ishida, K. Ando, Y. Hashimoto, A. Shigaki, K. Kikushima and S. Ogoshi, Chem. – Eur. J., 2018, 24, 9794–9798 CrossRef CAS PubMed.
- L.-K. Jin, G.-P. Lua and C. Cai, Org. Chem. Front., 2016, 3, 1309–1313 RSC.
- H. Baguia, J. Beaudelot, C. Moucheron and G. Evano, Chem. Commun., 2022, 58, 9080–9083 RSC.
- H. Baguia and G. Evano, Chem. – Eur. J., 2022, 28, e202103599 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |