
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid molecules based on an emodin scaffold. Synthesis and activity against SARS-CoV-2 and Plasmodium†
Youzhi
Li
abc,
Franck
Touret
d,
Xavier
de Lamballerie
d,
Michel
Nguyen
bc,
Marion
Laurent
bc,
Françoise
Benoit-Vical
bc,
Anne
Robert
 *bc,
Yan
Liu
*a and
Bernard
Meunier
*abc
*bc,
Yan
Liu
*a and
Bernard
Meunier
*abc
aEducation Mega Center, Guangdong University of Technology, School of Chemical Engineering and Light Industry, No. 100 Waihuan Xi Road, Guangzhou, P. R. China. E-mail: yanliu@gdut.edu.cn
bLaboratoire de Chimie de Coordination du CNRS, 205 route de Narbonne, BP 44099, 31077 Toulouse Cedex 4, France. E-mail: anne.robert@lcc-toulouse.fr; bernard.meunier@lcc-toulouse.fr
cNew Antimalarial Molecules and Pharmacological Approaches, MAAP, Inserm ERL 1289, 205 Route de Narbonne, BP 44099, 31077 Toulouse Cedex 4, France
dUnité des Virus Émergents (UVE), Aix Marseille Univ, IRD 190, Inserm 1207, 27 Boulevard Jean Moulin, 13005 Marseille Cedex 05, France
First published on 24th August 2023
Abstract
Since the Covid-19 epidemic, it has been clear that the availability of small and affordable drugs that are able to efficiently control viral infections in humans is still a challenge in medicinal chemistry. The synthesis and biological activities of a series of hybrid molecules that combine an emodin moiety and other structural moieties expected to act as possible synergistic pharmacophores in a single molecule were studied. Emodin has been reported to block the entry of the SARS-CoV-2 virus into human cells and might also inhibit cytokine production, resulting in the reduction of pulmonary injury induced by SARS-CoV-2. The pharmacophore associated with emodin was either a polyamine residue (emodin-PA series), a choice driven by the fact that a natural alkyl PA like spermine and spermidine play regulatory roles in immune cell functions, or a diphenylmethylpiperazine derivative of the norchlorcyclizine series (emoxyzine series). In fact, diphenylmethylpiperazine antagonists of the H1 histamine receptor display activity against several viruses by multiple interrelated mechanisms. In the emoxyzine series, the most potent drug against SARS-CoV-2 was (R)-emoxyzine-2, with an EC50 value = 1.9 μM, which is in the same range as that of the reference drug remdesivir. However, the selectivity index was rather low, indicating that the dissociation of antiviral potency and cytotoxicity remains a challenge. In addition, since emodin was also reported to be a relatively high-affinity inhibitor of the virulence regulator FIKK kinase from the malaria parasite Plasmodium vivax, the antimalarial activity of the synthesized hybrid compounds has been evaluated. However, these molecules cannot efficiently compete with the currently used antimalarial drugs.
Introduction
Scientific and medical context of antiviral drugs. Drug targets to fight SARS-CoV-2
In the absence of an efficient vaccine, the therapeutic arsenal to fight the AIDS virus is based on a large spectrum of nearly thirty different drugs that are able to control the disease. At the beginning of this century, several drugs were approved against the hepatitis C flavivirus (HCV) that infected more than 70 million people in the world in 2015, including the efficient sofosbuvir.1 In both cases, these efficient drugs, based on modified nucleosides, demonstrate that innovative medicinal chemistry is needed to create antiviral drugs to complement other treatments (vaccines or antibodies) or offer a therapeutic solution when they are not available as preventive or curative tools.Despite alerts on epidemics due to the Ebola virus in West Africa in 2013–2016 or to coronaviruses, SARS-CoV-1 in 2003 and MERS in 2012, limited attention has been paid to the search for efficient antivirals to treat these emerging viruses. When SARS-CoV-2 became a pandemic in early 2020, no antiviral agent was available to control infection in humans. The spike glycoprotein (S) of SARS-CoV-2 mediates the entry of the virus into host cells by binding to the host cellular receptor angiotensin-converting enzyme 2 (ACE2) which is present on the surface of vascular endothelial cells lining the entire circulatory system, from the heart to the smallest capillaries.2,3 The entry of the virus, by fusion at the plasma membrane surface, is facilitated by the proteolytic cleavage of the S protein, mediated by human transmembrane serine protease (TMPRSS2). So, molecules that are able to block the protein S mediated entry of the virus by preventing the S–ACE2 interaction or by inhibiting human TMPRSS2 might be potentially active against SARS-CoV-2. The high affinity interaction of the spike protein S and ACE2 involves polar amino acids from both proteins; so, synthetic oligopeptides were proposed as potential inhibitors of the S–ACE2 interaction.4 However, smaller and more druggable molecules are expected to be more suitable to inhibit the entry of SARS-CoV-2 into host cells.
Emodin (6-methyl-1,3,8-trihydroxyanthraquinone, Scheme 1), a natural product isolated from rhubarb, is a possible candidate among the “small molecules” exhibiting some capacity to block the entry of the SARS-CoV-2 virus into human cells.5 In fact, the activity of this anthraquinone derivative against SARS-CoV-1 was evidenced in 2003.6 Inhibition of the binding of the S-protein to ACE2 by emodin is dose-dependent (IC50 = 200 μM).6 Emodin also inhibits a transmembrane ion-channel of SARS-CoV-1 that is essential for the release of the virus from the infected cell.7 Emodin has a significant inhibitory effect on the entry, the replication, or the nuclease activity of several viruses, suggesting that it might be a potent antiviral drug with a broad spectrum.7 In addition, emodin reduces pulmonary inflammation in rats with acute lung injury, by inhibiting the mTOR/HIF-1α/VEGF signaling pathway.8 It also exerts an immunosuppressive activity that might be mediated by the inhibition of cytokine production.7 Therefore, emodin has three major advantages as a potential broad antiviral agent: (i) inhibition of the virus entry into host cells; (ii) inhibition of the ion channel involved in virus release from cells; and (iii) reduction of pulmonary inflammation in lung injuries. Unfortunately, emodin exhibits poor intestinal absorption, low bioavailability and fast elimination due to an active hydroxylation and/or glucuronidation metabolism, which are the main limitations for its development as a drug candidate.9In vitro, emodin disturbs glutathione and fatty acid metabolism in human liver cells, a feature that suggests potential toxic effects of the drug.10
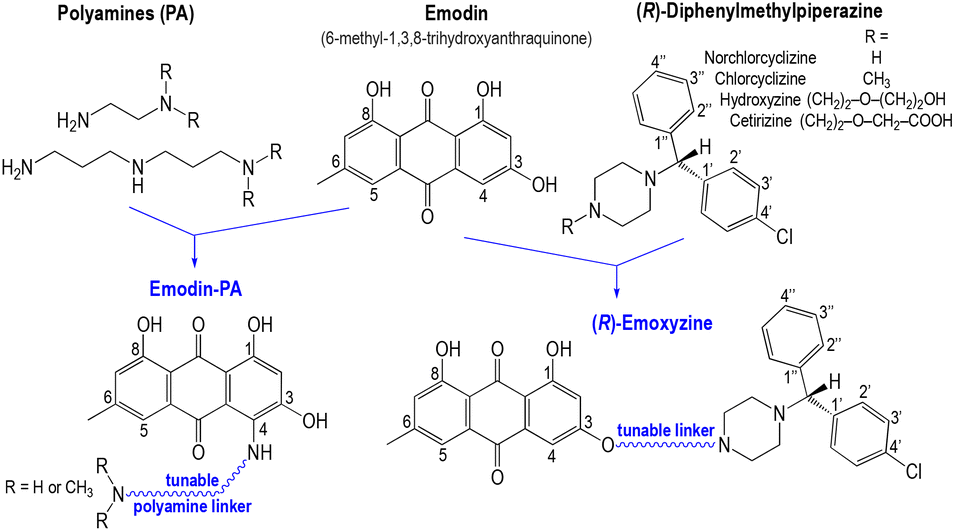 | ||
| Scheme 1 Emoxyzine and emodin-PA hybrid molecules containing emodin and norchlorcyclizine, or emodin and polyamine pharmacophores, respectively. The structures of the clinically used hydroxyzine and cetirizine are given for comparison. | ||
Design of hybrid molecules potentially active against SARS-CoV-2
Taking into consideration all these different aspects, we designed new emodin-based hybrid molecules modified by the attachment of a second molecular entity that is able to improve the pharmacokinetics of emodin and/or to enhance its pharmacological activity in order to enlarge its safety window. We previously developed the strategy of hybrid molecules for the preparation of antimalarial agents with a dual mode of action.11Diphenylmethylpiperazine derivatives of the norchlorcyclizine series (Scheme 1) are the first-generation histamine receptor (H1) antagonists belonging to the benzylpiperazine class. Chlorcyclizine (Scheme 1) was shown to have virucidal activity against HIV and several other RNA or DNA viruses, and also in a mouse model of HCV infection.12,13 In this series, hydroxyzine, a derivative bearing an ethoxyethanol side chain, was also found to be active against hepatitis C virus (HCV) infection in cell culture and was able to inhibit a SARS coronavirus proteinase in vitro.14,15 Moreover, in a multicenter observational study of patients hospitalized with Covid-19, the use of hydroxyzine was associated with decreased mortality,16 an effect that may be mediated by the anti-inflammatory properties of H1 antihistamines but remains to be firmly established in a random clinical trial.16 Besides other antihistamine drugs, hydroxyzine was reported to exhibit direct antiviral activity against SARS-CoV-2 in vitro by multiple interrelated mechanisms, especially the on-target effects of antihistamines inhibiting inflammatory responses that increase mortality. It also inhibits the acid sphingomyelinase (ASM) that is activated by pro-inflammatory cytokines.17,18 Alternatively, hydroxyzine exhibits off-target ACE2 inhibitory activity and consequently has the potential to disrupt the interaction of the SARS-CoV-2 spike glycoprotein S with ACE2, thus preventing cell infection.19
For all these reasons, we decided to synthesize hybrid small molecules named emoxyzines that combine norchlorcyclizine, which is the pharmacophore of diphenylmethylpiperazine anti-histamine drugs, and emodin structures connected through a potentially active linker (Scheme 1) connected at the C3 of the emodin residue. Hybrid molecules may combine in defined chemical entities two or more structural domains having complementary biological functions and activities, thus acting as possibly synergistic pharmacophores.11 As a linker between emodin and norchlorcyclizine moieties, we have considered ethoxyethyl linkers closely related to the structure of hydroxyzine (emoxyzine-1, -2, and -3 Scheme 2). We also synthesized a series of emodin–polyamine conjugates (named emodin-PA) having the polyamine (PA) residue linked at the C4 of the emodin cycle (Scheme 3), a choice driven by the fact that natural alkyl PA like spermine and spermidine play regulatory roles in immune cell functions.20,21 Spermine inhibits proinflammatory cytokine synthesis in human mononuclear cells and downregulates the monocyte proinflammatory cytokine response.22 In fact, during SARS-CoV-2 infection, the T cell response may provide early control of the cytokine storm triggered by acute viral infection.23,24 In addition, we also considered an emoxyzine derivative having an alkyldiamine (piperazine) linker (emoxyzine-7, Scheme 2).
 | ||
| Scheme 2 General scheme for the synthesis of emoxyzine derivatives bearing a linker at the C3 of emodin moiety. (a) K2CO3, DMF, 60 °C, 6 h. (b) Et3N, DMF, 80 °C, overnight; then HCl in diethyl ether, RT, 30 min. (c) 1-Bromo-2-chloroethane, Et3N, DMF, 80 °C, 6 h; then 1-Boc-piperazine, 100 °C, overnight. (d) HCl in H2O/CH3OH, 60 °C, 1.5 h. (e) Dimethylamine hydrochloride, Et3N, DMF, 80 °C, 6 h; then HCl in Et2O, RT, 30 min. | ||
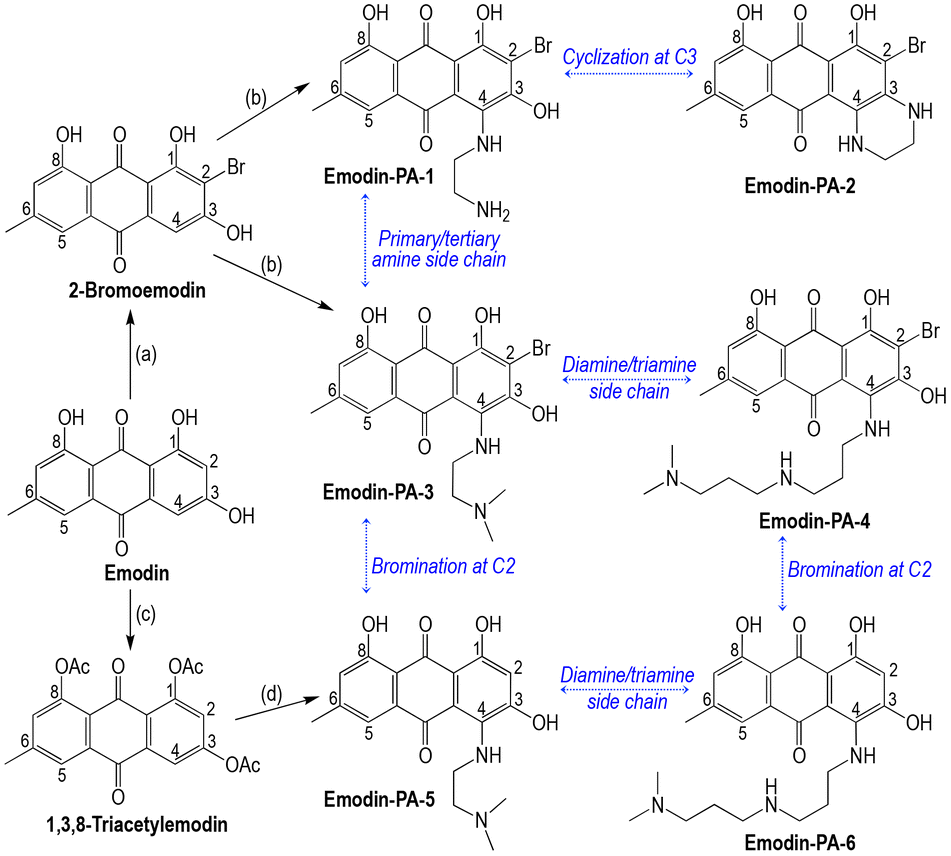 | ||
| Scheme 3 General scheme for the synthesis of emodin-PA derivatives bearing a polyamine substituent at C4 of the emodin residue. (a) NBS in THF, overnight. (b) Ethylenediamine or N,N-dimethylethylenediamine or N,N-dimethyldipropylenetriamine, RT, 5–24 h. (c) Ac2O, H2SO4 in pyridine, reflux, 2.5 h. (d) N,N-dimethylethylenediamine or N,N-dimethyldipropylenetriamine, RT, overnight. Comparisons of the structures of emodin-PA-n are emphasized by blue labels. | ||
Due to their different antiviral targets, these pharmacophores might provide synergistic antiviral effects when associated in such hybrid compounds. The polyamine linkers may have a combined activity with emodin and norchlorcyclizine due to their own activity on the immune system. In addition, substitution of the linker at the C4 of emodin is expected to slow down the P450 mediated metabolism of the emodin fragment and, consequently, should optimize the pharmacological activity of these hybrid molecules.
The potential anti-Covid-19 activity of these hybrid molecules was evaluated on VeroE6 cells expressing the transmembrane serine protease TMPRSS2, infected with the SARS-CoV-2 strain BavPat1.25–27
In addition, emodin was also reported to be a relatively high-affinity inhibitor of FIKK kinase from the malaria parasite Plasmodium vivax (IC50 value of emodin against PvFIKK = 1.9 μM).28 So the activity of emodin, emoxyzine and emodin-PA hybrids has been evaluated against the major human malaria parasite Plasmodium falciparum, as potential lead compounds for the development of antimalarial drugs targeting the parasite FIKK kinase, without interfering with host kinases since FIKK kinases are found exclusively in Apicomplexa.29 In fact, Plasmodium FIKK kinases play a species-specific role in the virulence of the parasite by controlling the rigidity and cyto-adhesion of host erythrocytes.30
Results and discussion
Syntheses of emoxyzine derivatives (substitution at the C3 of emodin)
Hybrid compounds containing emodin and norchlorcyclizine moieties bound by ethoxyethyl tethers of variable lengths (emoxyzine-1, -2 and -3, respectively) were prepared by substitution of the ω-brominated side chain of emodin-link-n intermediates by the secondary amine norchlorcyclizine in DMF/triethylamine at 80 °C. After protonation by hydrochloric acid, these emoxyzines were obtained as hydrochloride salts in high yields (>95%, Scheme 2, step b). The putative role of the tether for biological activity was investigated by preparing emoxyzine-7, a compound whose linker contained a protonable piperazinyl residue. This hybrid compound was synthesized by the reaction of emodin-link-1 with (R)-norchlorcyclizine-link-1, which was itself prepared by the functionalization of (R)-norchlorcyclizine with a piperazinyl residue (Scheme 2, steps c and d).It is noteworthy that although the norchlorcyclizine derivatives cetirizine and hydroxyzine are used as racemates in clinical practice, their antihistaminic properties are attributable to the R configuration of the benzylic chiral carbon.31 Then, in order to evaluate the putative role of the chiral center in the biological activity of these molecules, we synthesized the racemic derivatives (R,S)-emoxyzine-1 and (R,S)-emoxyzine-2 using (R,S)-norchlorcyclizine as the starting material. We also checked that the chiral center of the (R)-norchlorcyclizine residue did not undergo racemization under the basic conditions of the coupling reaction between emodin and norchlorcyclizine moieties. For this purpose, (R)-norchlorcyclizine was incubated in DMF/triethylamine at 80 °C overnight. After work-up, the (R)-norchlorcyclizine was detected as a single peak by chiral HPLC (Rt = 4.8 min), while the (S)-norchlorcyclizine expected at 4.3 min was not detected [analysis of (R,S)-norchlorcyclizine showed 2 peaks at 4.3 and 4.8 min in a 50/50 ratio, ESI, S2†]. In addition, when deuterated water was added after heating of (R)-norchlorcyclizine in DMF/triethylamine at 80 °C, the 1H NMR spectrum of the product was the same as that of the starting (R)-norchlorcyclizine, with the benzylic proton detected at 4.21 ppm (integral = 1H) (ESI, S3†). This confirms that triethylamine in DMF at 80 °C was inefficient to abstract this proton, a pathway required for the racemization of the chiral center. Similarly, chiral HPLC of (R)-emoxyzine-2 synthesized from (R)-norchlorcyclizine exhibited a single peak at Rt = 11.2 min, while the analysis of its racemic analogue (R,S)-emoxyzine-2 synthesized using (R,S)-norchlorcyclizine exhibited two peaks at 9.6 min and 11.1 min (50/50), thus confirming the stability of the chiral center under these experimental conditions (ESI, S4†).
Emoxyzines were stable upon storage in the solid state at room temperature under air for at least six months, and at 60 °C under air for at least four months, without any detectable change in NMR analyses. Emoxyzines were also stable after two weeks in solution in DMSO at room temperature. The solubility of emoxyzines-1–3 was in the range of 41–44 mg mL−1 in DMSO at room temperature. The solubility of emoxyzine-7 was 28 mg mL−1 under the same conditions. All these hybrid compounds were almost insoluble in pure water.
Syntheses of emodin-PA derivatives (substitution at the C4 of emodin)
Emodin–polyamine conjugates were synthesized by substitution of a polyamine side chain at the C4 of 2-bromoemodin (emodin-PA-n, with n = 1–4, Scheme 3) or 1,3,8-triacetylemodin (emodin-PA-5 and emodin-PA-6). 2-Bromoemodin and 1,3,8-triacetylemodin were preliminarily synthesized by the reaction of emodin with N-bromosuccinimide in THF or with acetic anhydride in pyridine, respectively. The reaction of 1,2-ethylene diamine with 2-bromoemodin at room temperature afforded the monosubstituted derivative at C4, emodin-PA-1, in 70% yield after purification. Under the same conditions but for a longer time (overnight), nucleophilic substitution of the distal primary amine of the side chain onto C3–OH afforded the cyclic 3,4-diethylaminoemodin derivative emodin-PA-2 in 37% yield (Scheme 3). The reaction of N,N-dimethylethylenediamine or N,N-dimethyldipropylenetriamine with 2-bromoemodin afforded emodin-PA-3 and emodin-PA-4 (90–96% yield), respectively, thus allowing the investigation of (i) the role of the distal amine substitution pattern (emodin-PA-1 vs. emodin-PA-3) and (ii) the role of the length and the number of amine functions of the side chain (emodin-PA-3 vs. emodin-PA-4). Starting from 1,3,8-triacetylemodin, the substitution of N,N-dimethylethylenediamine or N,N-dimethyldipropylenetriamine afforded emodin-PA-5 (97% yield) and emodin-PA-6 (85% yield), respectively. These compounds are the non-brominated counterparts of emodin-PA-3 and emodin-PA-4, respectively. It is noteworthy that whether C2 was brominated or not, the substitution of the primary amine occurred selectively at C4. Conversely, the C3–OH position was substituted as the secondary site only under the drastic conditions of the synthesis of emodin-PA-2.The stability of emodin-PA derivatives in solution in DMSO was evaluated by NMR. Emodin-PA-2 was found to be stable at 75 °C for at least one day, and it was stable when exposed to daylight at room temperature for at least one week. Conversely, all other emodin-PAs exhibited significant degradation in DMSO solution after one week at room temperature under day light. Under these conditions, the product loss was 52–54 mol% (emodin-PA-3 and -5) or 30–35 mol% (emodin-PA-4 and -6). At 4 °C in DMSO in the dark, for one week, degradation was 12% or 17% for emodin-PA-3 and -4, respectively, while it was below 5% for emodin-PA-5 and -6. All emodin-PAs were found to be stable when stored at 4 °C for 5 months in the solid state.
Anti-SARS-CoV-2 activity of emodin-PA and emoxyzine dual molecules
The antiviral activity of emodin-based dual molecules was evaluated using VeroE6 TMPRSS2 cells infected with SARS-CoV-2, as previously described.26,32 We applied a standardised methodology for evaluating antiviral compounds against RNA viruses, based on RNA yield reduction. This assay is based on qRT-PCR quantification to determine the concentration of the molecule that is able to inhibit the concentration of the viral RNA in the supernatant by 50% (EC50). The monophosphate adenosine analogue remdesivir was used as the positive control and the reference compound.26,33 In addition, this assay uses a cell line expressing TMPSS2 to avoid identifying compounds with antiviral activity linked to the endosomal replication pathway34 or inducing phospholipidosis.35 The results are reported in Table 1 and Fig. 1. Under these conditions, (R)-emoxyzine-1 and all emodin-PA derivatives showed no antiviral activity with concentrations that inhibit viral RNA replication (EC50 values) higher than 10 μM, about five times higher than the EC50 value of remdesivir (EC50 = 2.6 μM). Emodin itself exhibited a slightly lower EC50 value of 6.3 μM, but its cytotoxicity (CC50) on VeroE6 TMPSSR2 cells, under the same culture conditions but without viral infection, was 13.5 μM, resulting in a poor selectivity index (SI = CC50/EC50) of 2.1, consistent with the reported cytotoxicity of this anthraquinone drug. Interestingly, the CC50 values of emodin-PA-2, emodin-PA-3 and emodin-PA-5 on VeroE6 TMPSSR2 cells were higher than 50 μM, i.e. higher than that of emodin itself (13.5 μM), indicating that simple substitutions of the anthraquinone structure may be efficient to decrease the cytotoxicity of this basic drug skeleton and, possibly, to increase the selectivity indexes of emodin derivatives.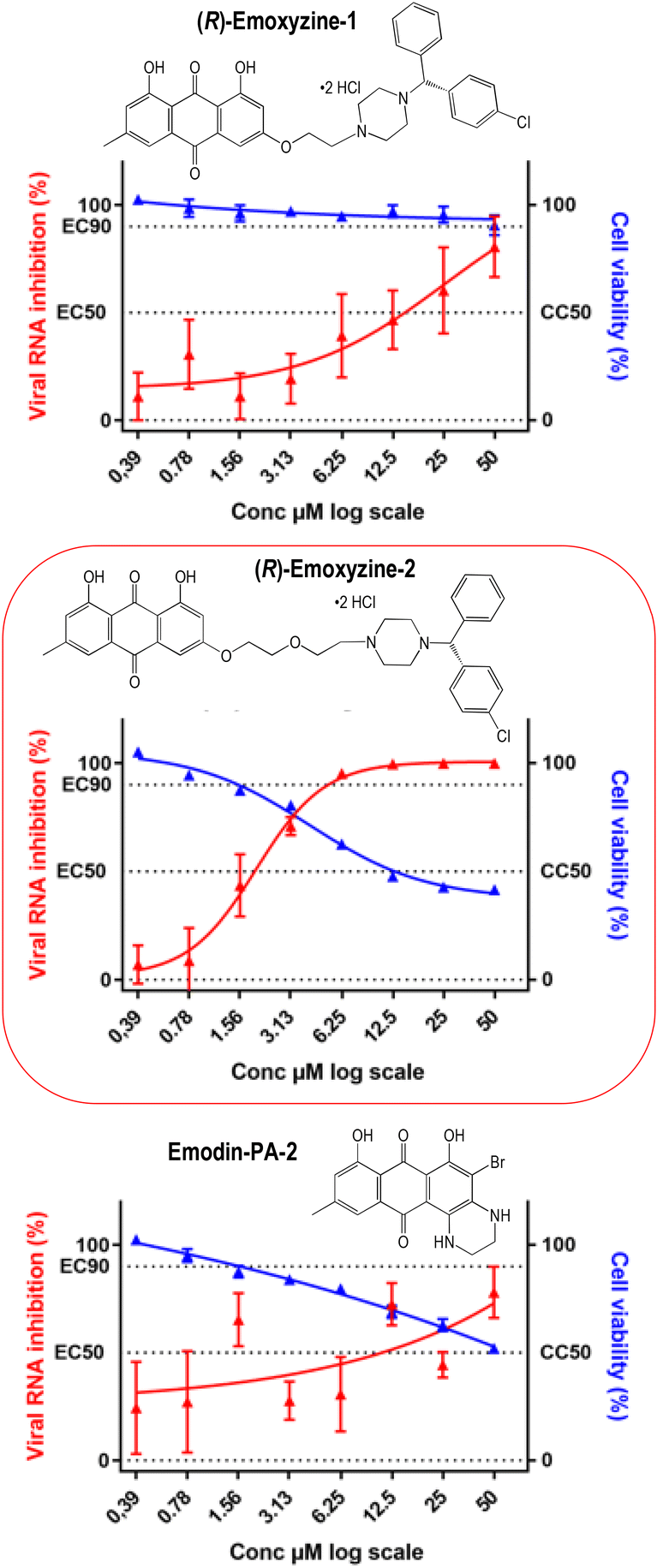 | ||
| Fig. 1 SARS-CoV-2 RNA inhibition (red traces) and VeroE6 TMPSSR2 cell viability (blue traces) in the presence of (R)-emoxyzine-1 or (R)-emoxyzine-2, or emodin-PA-2. | ||
| Molecule | EC50 (μM) (SARS-CoV-2) | CC50 (μM) (VeroE6 TMPSSR2) | SIa |
|---|---|---|---|
| a Selectivity index = CC50/EC50. b Ref. 27. | |||
| (R)-Emoxyzine-1 | 14.8 | >50 | >3.4 |
| (R)-Emoxyzine-2 | 1.9 | 12.9 | 6.8 |
| (R,S)-Emoxyzine-2 | 12.2 | 35.2 | 2.9 |
| (R)-Emoxyzine-3 | 5.0 | 16.9 | 3.4 |
| (R)-Emoxyzine-7 | 2.9 | 4.3 | 1.5 |
| Emodin-PA-2 | 10.9 | >50 | 4.6 |
| Emodin-PA-3 | 49.6 | >50 | >1 |
| Emodin-PA-4 | 12.5 | 14.7 | >1.2 |
| Emodin-PA-5 | 46.3 | >50 | >1 |
| Emodin | 6.3 | 13.5 | 2.1 |
| (R)-Norchlorcyclizine | 12.3 | 14.7 | 1.2 |
| (R,S)-Hydroxyzine | 31.7 | 61.6 | 1.9 |
| Remdesivir | 2.6 | >20b | >8 |
Among the compounds with promising antiviral activity, i.e. with EC50 values lower than 5 μM, (R)-emoxyzine-2 inhibited SARS-CoV-2 replication with EC50 of 1.9 μM (Table 1), which is in the same range as the EC50 value of remdesivir (2.6 μM). The cytotoxicity CC50 of (R)-emoxyzine-2 was 12.9 μM, resulting in a selectivity index (SI = CC50/EC50) of 6.8. In addition, the inhibition of viral RNA replication higher than 90% was obtained with an (R)-emoxyzine-2 concentration of 6.25 μM. Inhibition of SARS-CoV-2 replication by the racemic analogue (R,S)-emoxyzine-2 was also evaluated. Its EC50 value was 12.2 μM, 6 times higher than that of the optically active (R)-emoxyzine-2, and its SI value was lower than 3. These results indicate that (i) both emodin and (R)-norchlorcyclizine fragments are required to optimize CC50 and SI values and (ii) a longer link between the two pharmacophores allowed to drastically decrease the EC50 values [1.9 μM for (R)-emoxyzine-2 compared to 14.8 μM for (R)-emoxyzine-1]. So, we decided to synthesize and evaluate emoxyzine derivatives with a longer ethoxyethyl linker [(R)-emoxyzine-3, Scheme 2] or with a linker containing additional amine functions [(R)-emoxyzine-7, Scheme 2]. However, since these compounds exhibited a rather low value of selectivity index, it appears that dissociation of antiviral potency and cytotoxic activity remains a challenge.
Antimalarial activity of emodin-PA and emoxyzine dual molecules
The antimalarial activity of hybrid drugs based on the emodin skeleton has been evaluated in vitro against the artemisinin resistant strain F32-ART of Plasmodium falciparum. The results are reported in Table 2. All the tested emodin-PA and emoxyzine derivatives exhibited IC50 values in the micromolar range, the lowest being those of (R)-emoxyzine-7 and emodin-PA-6, measured at 1.0 and 1.2 μM, respectively. However, these molecules were largely less potent by three orders of magnitude compared to the clinically used antimalarial drugs such as atovaquone, here used as the reference drug control on this Plasmodium strain [the IC50 value of atovaquone routinely tested was found to be 1 nM]. In addition, the compound emodin-link-5 (Scheme 2), a derivative of emodin functionalized by a short diethylamine chain at C3, exhibited an IC50 value of 4.8 μM. So, it appears that the norchlorcyclizine moiety was not required to get IC50 values in the range of 1–10 μM.| Molecule | IC50 (μM) ± SEM (P. falciparum) | CC50 (μM) (Vero) | SIa |
|---|---|---|---|
| a Selectivity index calculated as CC50 (Vero)/IC50 (Plasmodium). b The rather poor solubility of emodin-PA-2 might be responsible for erratic results. c Not determined. | |||
| (R)-Emoxyzine-1 | 28 ± 1.5 | >50 | >1.8 |
| (R,S)-Emoxyzine-1 | 27 ± 6 | >50 | >1.9 |
| (R)-Emoxyzine-2 | 12 ± 0.5 | >50 | >4.2 |
| (R,S)-Emoxyzine-2 | 13 ± 0.5 | >50 | >3.8 |
| (R)-Emoxyzine-3 | 7.5 ± 0.6 | >50 | >7 |
| (R)-Emoxyzine-7 | 1.0 ± 0.1 | 16 | 16 |
| Emodin-PA-1 | 2.5 ± 0.1 | >50 | >20 |
| Emodin-PA-2b | >50 | 5 | 0.1 |
| Emodin-PA-3 | 4 ± 0.1 | >50 | >12 |
| Emodin-PA-4 | 5 ± 0.5 | 24 | 5 |
| Emodin-PA-5 | 4.0 ± 0 | >50 | >12 |
| Emodin-PA-6 | 1.2 ± 0.2 | 23 | 19 |
| Emodin | >50 | 34 | <0.7 |
| Emodin-link-5 | 4.8 ± 0.5 | Ndc | — |
| (R,S)-Hydroxyzine | 33 | >50 | >1.5 |
| Atovaquone | 0.001 | 0.45 | 450 |
It is noteworthy that the cytotoxicity of these hybrid molecules against Vero cells was low, with CC50 values higher than 50 μM, in most cases (SI ranging from 0.1 to >20), contrary to emodin itself which exhibited a lower antimalarial activity compared to its cytotoxicity [IC50 (Plasmodium) > 50 μM, CC50 (Vero cells) = 34 μM, SI < 0.7]. These results indicated that the selectivity of emodin derivatives can be modulated to some extent by the substitution pattern of the anthraquinone entity. Moreover, it is to be noted that the emodin activity revealed on the protein kinase PvFIKK28 is not found against the parasite P. falciparum. One should note that the standard assay used to determine the capacity of these compounds to inhibit the proliferation of the P. falciparum parasite is probably not an appropriate way to evaluate the potential benefit of these molecules. In fact, Plasmodium FIKK inhibitors are expected to decrease virulence factors, contributing to a better adaptability of the parasite to survive in the circulation of the human host.30
Conclusion
Two series of hybrid molecules based on an emodin skeleton covalently linked to a diphenylmethylpiperazine moiety or an alkyl polyamine were synthesized and their activity against both SARS-CoV-2 and P. falciparum was evaluated. In these series, (R)-emoxyzine-2 was as potent as remdesivir in inhibiting SARS-CoV-2 replication with a selectivity index for the virus of 6.8. However, the dissociation of antiviral activity and cytotoxicity is still a challenge for these hybrid molecules.Emoxyzine and emodin-PA hybrid molecules, exhibiting IC50 values equal or higher than 1 μM on P. falciparum, cannot be considered as efficient antimalarial drugs to target P. falciparum parasite proliferation.
Nevertheless, the results showed that combining two or more pharmacophores into a single hybrid molecule may provide modulation or significant improvement of biological properties.
Experimental
Materials and equipment
All solvents and commercially available reagents were purchased from Sigma-Aldrich, Fluka, Acros, TCI, or BLD Pharmatech and were used without further purification. ESI+-mass spectra were obtained using a Thermo Fisher or an Xevo G2 QTOF (Waters) instrument. The characterization of all hybrid compounds was based on the consistency of high resolution 1H- and 13C-NMR 2D correlations and mass spectrometry analyses. The consistency of elemental analyses and the absence of detected impurities in the NMR spectra indicated a purity higher than 98%. Purity below 98% was evaluated on the basis of 1H NMR analysis. 1H- and 13C-NMR spectra were recorded on Brucker Avance Neo 600 or Bruker Avance 400 and Avance 300 spectrometers, at 298 K unless otherwise stated. Chemical shift values are given in ppm, using tetramethylsilane (TMS) as the external standard. The NMR spectra of final products have been completely assigned using 2D COSY, HMQC, HMBC and NOESY correlations (600 MHz for 1H and 151 MHz for 13C). The spectra of intermediate compounds were recorded at 400 MHz for 1H NMR and at 100 MHz for 13C NMR, and the assignment is a proposal based on comparison with the established spectra/assignment of final products. Chiral HPLC analyses of norchlorcyclizine and emoxyzine-2 were performed using an Acquity UPC equipment (Waters) with a Chiralpack IJ column (3 μm, 4.6 mm × 100 mm, Daicel chiral technologies), flow rate was 2 mL, UV detection at λ = 210 nm. Elution was carried out with the following gradient: (A) = supercritical CO2, (B) = diethylamine 0.1 vol% in CH3OH. Gradient for norchlorcyclizine: A/B = 95/5 for 1 min, then from A/B = 95/5 to A/B = 60/40 at 6 min, then A/B = 60/40 for 2 min; gradient for emoxyzine-2: from initial A/B = 85/15 to A/B = 50/50 at 10 min, followed by A/B = 50/50 for 4 min.Syntheses and characterization of emodin-PA derivatives
Syntheses of emoxyzine derivatives
(R,S)-Emoxyzine-1 was prepared exactly in the same way as (R)-emoxyzine-1, but using the racemate (R,S)-norchlorcyclizine as the starting material. The yield was 99%. NMR analysis provided the same result as for (R)-emoxyzine-1 within experimental error (≤0.04 ppm in 1H). ESI+-MS: m/z (relative intensity) = 583.3 (100), 584.2 (38), 585.2 (40), 586.2 (14), 587.2 (3) [M + H]+. Elemental analysis calcd (%) for C34H31ClN2O5·2.0HCl·4.3H2O (apparent MW = 742.27): C 55.68, H 5.72, N 3.82, found: C 55.56, H 4.97, N 3.83. Purity > 98%.
(R,S)-Emoxyzine-2 was prepared exactly in the same way as (R)-emoxyzine-2, but using the racemate (R,S)-norchlorcyclizine as the starting material. The yield was 91%. NMR analysis provided the same result as for (R)-emoxyzine-2 within experimental error (≤0.02 ppm in 1H). ESI+-MS: m/z (relative intensity) = 583.3 (100), 584.2 (38), 585.2 (40), 586.2 (14), 587.2 (3) [M + H]+. Elemental analysis calcd (%) for C34H31ClN2O5·2.0HCl·4.3H2O (apparent MW = 742.27): C 55.68, H 5. 72, N 3.82, found: C 55.56, H 4.97, N 3.83. Purity > 98%. HPLC (Chiralpack IJ): 9.6 min (S enantiomer, 50%), 11.1 min (R enantiomer, 50%).
Stability of the chiral center of (R)-norchlorcyclyzine
To a solution of (R)-norchlorcyclizine (60 mg, 0.21 mmol, 1.0 equiv.) in dry DMF (2 mL), triethylamine was added (64 mg, 0.63 mmol, 3.0 equiv.). The reaction mixture was stirred at 80 °C overnight under argon. Deuteroxide was then added and the mixture was stirred for 30 min at room temperature. The resulting solution was poured into ethyl acetate (60 mL). The organic layer was washed with water (5 × 30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was obtained as a colorless oil (59 mg) and analyzed by 1H NMR in CDCl3. 1H NMR analysis of the product was the same as that of the starting (R)-norchlorcyclizine, with the benzylic proton detected at 4.21 ppm (1H) (ESI, S3†).Anti-SARS-CoV-2 assays
Antiviral assays were carried out according to ref. 26. Briefly, one day prior to infection, 5 × 104 VeroE6 TMPRSS2 cells per well were seeded in 100 μL assay medium (containing 2.5% FCS) in 96 well culture plates. The next day, eight 2-fold serial dilutions of compounds (from 0.39 μM to 50 μM in triplicate were added to the cells (25 μL per well, in assay medium). Four virus control wells were supplemented with 25 μL of assay medium. After 15 min, 25 μL of a virus mix diluted in medium was added. The amount of virus working stock used was calibrated prior to the assay, based on a replication kinetics, so that the viral replication was still in the exponential growth phase for the readout. On each culture plate, a control compound (Remdesivir, BLD pharm) was added in duplicate with eight 2-fold serial dilutions (0.08 μM to 10 μM). Plates were incubated for 2 days at 37 °C prior to quantification of the viral genome by real-time RT-PCR. To do so, 100 μL of viral supernatant was collected in S-Block (Qiagen) previously loaded with VXL lysis buffer containing proteinase K and RNA carrier. RNA extraction was performed using the Qiacube HT automat and the QIAamp 96 DNA kit HT following manufacturer instructions. Viral RNA was quantified by real-time RT-qPCR (GoTaq 1-step qRT-PCR, Promega) using 3.8 μL of extracted RNA and 6.2 μL of RT-qPCR mix and standard fast cycling parameters, i.e., 10 min at 50 °C, 2 min at 95 °C, and 40 amplification cycles (95 °C for 3 s followed by 30 s at 60 °C). Quantification was provided by four 2 log serial dilutions of an appropriate T7-generated synthetic RNA standard of known quantities (102 to 108 copies per reaction). RT-qPCR reactions were performed on QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems) and analyzed using QuantStudio 12K Flex Applied Biosystems software v1.2.3. Primers and probe sequences, which target SARS-CoV-2 N gene, were: Fw: GGCCGCAAATTGCACAAT; rev: CCAATGCGCGACATTCC; probe: FAM-CCCCCAGCGCTTCAGCGTTCT-BHQ1. For the evaluation of the 50% cytotoxic concentrations (CC50), the same culture conditions as for the determination of the EC50 were used, without addition of the virus, and cell viability was measured using CellTiter Blue® (Promega) following manufacturer's instructions. The 50% effective and cytotoxic concentrations EC50 and CC50 were determined using logarithmic interpolation as previously described.36 All data obtained were analyzed using GraphPad Prism 9 software (Graphpad software).Antimalarial and cytotoxicity assays
Antimalarial proliferative assay (IC50 evaluation) and cytotoxicity assay (CC50 evaluation) were performed as previously described.37 Briefly, for each experiment, extemporaneous solutions of compounds were prepared in DMSO. The antimalarial activity of compounds was evaluated on the F32-ART artemisinin-resistant strain of P. falciparum. Parasites were treated with doses of compounds ranging from 50 nM to 50 μM (with 0.5 vol% of final DMSO concentration) and their viability, related with DNA degradation, was measured using the SYBR Green fluorescence-based method after 48 h of incubation at 37 °C. The molecule concentration able to inhibit the parasite growth by 50% (IC50 value) was determined from the dose-inhibition curves drawn with GraphPad Prism software. All assays were performed in triplicate in 2 to 4 independent experiments. The cytotoxicity of the hybrid molecules was evaluated on regular Vero cells using the MTT method to quantify cell metabolic activity [MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]. Vero cells were firstly plated for 24 h, then exposed for 48 h at 37 °C to compounds with doses tested in duplicate from 5 nM to 50 μM, with 0.5 vol% of final DMSO concentration. Cytotoxicity assays were carried out in one or two independent experiments. The CC50 values were determined in the same way as for the antimalarial experiments [cell growth inhibition = f(log compound concentration)]. For antimalarial and cytotoxicity assays, atovaquone was introduced as the comparative reference drug, and the selectivity index (SI) was calculated from the cytotoxicity/antimalarial activity ratio.Author contributions
Youzhi Li: investigation and writing. Franck Touret: methodology, investigation, validation, writing. Xavier de Lamballerie: funding acquisition. Michel Nguyen: investigation. Marion Laurent: investigation. Françoise Benoit-Vical: methodology, supervision and validation, writing, funding acquisition. Anne Robert: methodology, supervision, validation and writing. Yan Liu: conceptualization, funding acquisition. Bernard Meunier: conceptualization, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work has been financially supported by the Centre National de la Recherche Scientifique (CNRS) France, the Institut National de la Sante et de la Recherche Médicale (Inserm) France, the Fondation pour la recherche médicale (FRM, “Équipe EQU202103012596”), and by the University of Technology of Guangdong (GDUT), China. Y. Li is indebted for a scholarship from the Department of Education of Guangdong Province. The mass spectrometry facilities of the Institute of Chemistry of Toulouse (ICT, UAR 2599, Catherine Claparols), and Christian Bijani (LCC-CNRS) and Isabelle Fabing (UMR 5068 CNRS-University of Toulouse, PICT, IBiSA) are acknowledged for technical assistance with MS, NMR and chiral HPLC analyses, respectively.References
- C. W. Spearmann, G. M. Dusheiko and M. Sonderup, Lancet, 2019, 394, 1451–1466 CrossRef PubMed.
- A. G. Harrison, T. Lin and P. Wang, Trends Immunol., 2020, 41, 1100–1115 CrossRef CAS PubMed.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444–1448 CrossRef CAS PubMed.
- J. Yang, S. J. L. Petitjean, M. Koehler, Q. Zhang, A. C. Dumitri, W. Chen, S. Derclaye, S. P. Vincent, P. Soumillion and D. Alsteens, Nat. Commun., 2020, 11, 4541 CrossRef CAS PubMed.
- D. L. McKee, A. Sternberg, U. Stange, S. Laufer and C. Naujokat, Pharmacol. Res., 2020, 157, 104859 CrossRef CAS PubMed.
- T. Y. Ho, S. L. Wu, J. C. Chen, C. C. Li and C. Y. Hsiang, Antiviral Res., 2007, 74, 92–101 CrossRef CAS PubMed.
- X. Dong, J. Fu, X. Yin, S. Cao, X. Li, L. Lin, Huyiligepi and J. Ni, Phytother. Res., 2016, 30, 1207–1218 CrossRef CAS PubMed.
- X. Li, C. Shan, Z. Wu, H. Yu, A. Yang and B. Tan, Inflammation Res., 2020, 69, 365–373 CrossRef CAS PubMed.
- C. H. Shia, Y. C. Hou, S. Y. Tsai, P. S. Huieh and Y. L. Leu, J. Pharm. Sci., 2010, 99, 2185–2195 CrossRef CAS PubMed.
- X. Liu, Y. Liu, Y. Qu, M. Cheng and H. Xiao, Toxicol. Res., 2015, 4, 948–955 CrossRef CAS.
- B. Meunier, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS PubMed.
- A. N. Chakrabarty, M. Moojerjee and S. G. Dastidar, Int. J. Antimicrob. Agents, 2000, 14, 215–220 CrossRef CAS PubMed.
- S. He, B. Lin, V. Chu, Z. Hu, X. Hu, J. Xiao, A. Q. Wang, C. J. Schweitzer, Q. li, M. Imamura, N. Hiraga, N. Southall, M. Ferrer, W. Zheng, K. Chamaya, J. J. Marugan and T. J. Liang, Sci. Transl. Med., 2015, 282, 282ra49 Search PubMed.
- L. Mingorance, M. Friesland, M. Coto-Llerena, S. Pérez-del-Pulgar, L. Boix, J. M. López-Oliva, J. Bruix, X. Forns and P. Gastaminza, Antimicrob. Agents Chemother., 2014, 58, 3451–3460 CrossRef PubMed.
- Y. Liu, T. F. Zheng, F. Jin, L. Zhou, Z. M. Liu, P. Wei and L. H. Lai, Acta Chim. Sin., 2007, 65, 1707–1712 CAS.
- (a) N. Hoertel, M. Sánchez, R. Vernet, N. Beeker, A. Neuraz, C. Blanco, M. Olfson, C. Lemogne, P. Meneton, C. Daniel, N. Paris, A. Gramfort, G. Lemaitre, E. Salamanca, M. Bernaux, A. Bellamine, A. Burgun and F. Limosin, medRxiv, 2020 DOI:10.1101/2020.10.23.20154302; (b) M. Sánchez-Rico, F. Limosin, R. Vernet, N. Beeker, A. Neuraz, C. Blanco, M. Olfson, C. Lemogne, P. Meneton, C. Daniel, N. Paris, A. Gramfort, G. Lemaitre, P. De La Muela, E. Salamanca, M. Bernaux, A. Bellamine, A. Burgun, N. Hoertel and on behalf of AP-HP/Université de Paris/INSERM COVID-19 Research Collaboration/AP-HP COVID CDR Initiative/“Entrepôt de Données de Santé” AP-HP Consortium, J. Clin. Med, 2021, 10, 5891 Search PubMed.
- J. Kornhuber, N. Hoertel and E. Gulbins, Mol. Psychiatry, 2022, 27, 307–314 CrossRef CAS PubMed.
- M. Maceyka and S. Spiege, Nature, 2014, 510, 58–67 CrossRef CAS.
- L. R. Reznikov, M. H. Norris, R. Vashisht, A. P. Bluhm, D. Li, Y.-S. J. Liao, A. Brown, A. J. Butte and D. A. Ostrov, Biochem. Biophys. Res. Commun., 2021, 538, 173–179 CrossRef CAS PubMed.
- R. S. Hesterberg, J. L. Cleveland and P. K. Epling-Burnette, Med. Sci., 2018, 6, 22 Search PubMed.
- G. M. Carriche, L. Almeida, P. Stüve, L. Velasquez, A. Dhillon-LaBrooy, U. Roy, M. Lindenberg, T. Strowig, C. Plaza-Sirvent, I. Schmitz, M. Lochner, A. K. Simon and T. Sparwasser, J. Allergy Clin. Immunol., 2021, 147, 335–348 CrossRef CAS PubMed.
- M. Zhang, T. Caragine, H. Wang, P. S. Cohen, G. Botchkina, K. Soda, M. Bianchi, P. Ulrich, A. Cerami, B. Sherry and K. J. Tracey, J. Exp. Med., 1997, 185, 1759–1768 CrossRef CAS PubMed.
- A. C. Karlsson, M. Humbert and M. Buggert, Sci. Immunol., 2020, 5, eabe8063 CrossRef PubMed.
- P. Doshi, Br. Med. J., 2020, 370, m3563 CrossRef PubMed.
- S. Matsuyama, N. Nao, K. Shirato, M. Kawase, S. Saito, I. Takayama, N. Nagata, T. Sekizuka, H. Katoh, F. Kato, M. Sakata, M. Tahara, S. Kutsuna, N. Ohmagari, M. Kuroda, T. Suzuki, T. Kageyama and M. Takeda, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7001–7002 CrossRef CAS.
- F. Touret, M. Gilles, K. Barral, A. Nougairède, J. van Helden, E. Decroly, X. de Lamballerie and B. Coutard, Sci. Rep., 2020, 10, 13093 CrossRef CAS PubMed.
- F. Touret, J.-S. Driouich, M. Cochin, P. R. Petit, M. Gilles, K. Barthélémy, G. Moureau, F.-X. Mahon, D. Malvy, C. Solas, X. de Lamballerie and A. Nougairède, Antiviral Res., 2021, 193, 105137 CrossRef CAS.
- B. C. Lin, D. R. Harris, L. M. D. Kirkman, A. M. Perez, Y. Qian, J. T. Schermerhorn, M. Y. Hong, D. S. Winston, L. Xu and G. S. Brandt, ACS Omega, 2017, 2, 6605–6612 CrossRef CAS PubMed.
- K. T. Osman, H. J. Lou, W. Qiu, V. Brand, A. M. Edwards, B. E. Turk and R. Hui, Mol. Biochem. Parasitol., 2015, 201, 85–89 CrossRef CAS PubMed.
- (a) M. C. Nunes, J. P. D. Goldring, C. Doerig and A. Scherf, Mol. Microbiol., 2007, 63, 391–403 CrossRef CAS; (b) H. Davies, H. Belda, M. Broncel, X. Ye, C. Bisson, V. Introini, D. Dorin-Semblat, J.-P. Semblat, M. Tibúrcio, B. Gamain, M. Kaforou and M. Treeck, Nat. Microbiol., 2020, 5, 848–863 CrossRef CAS PubMed.
- D. Y. Wang, F. Hanotte, C. De Vos and P. Clement, Allergy, 2001, 56, 339–343 CrossRef CAS PubMed.
- A. Weiss, F. Touret, C. Baronti, M. Gilles, B. Hoen, A. Nougairède, X. de Lamballerie and M. O. A. Sommer, PLoS One, 2021, 16, e0260958 CrossRef CAS PubMed.
- Remdesivir was considered as the reference for in vitro studies, in the absence of any efficient anti-SARS-CoV-2 drug. One should note that the WHO does not recommend the use of remdesivir as a Covid-19 treatment and has excluded it from the list of priority drugs due to the lack of benefit for patients and the numerous adverse effects. See: J. Hsu, Br. Med. J., 2020, 371, m4457 CrossRef PubMed.
- M. Hoffmann, K. Mösbauer, H. Hofmann-Winkler, A. Kaul, H. Kleine-Weber, N. Krüger, N. C. Gassen, M. A. Müller, C. Drosten and S. Pöhlmann, Nature, 2020, 585, 588–590 CrossRef CAS PubMed.
- T. A. Tummino, V. V. Rezelj, B. Fischer, A. Fischer, M. J. O'Meara, B. Monel, T. Vallet, K. M. White, Z. Zhang, A. Alon, H. Schadt, H. R. O'Donnell, J. Lyu, R. Rosales, B. L. McGovern, R. Rathnasinghe, S. Jangra, M. Schotsaert, J.-R. Galarneau, N. J. Krogan, L. Urban, K. M. Shokat, A. K. Kruse, A. García-Sastre, O. Schwartz, F. Moretti, M. Vignuzzi, F. Pognan and B. K. Shoichet, Science, 2021, 373, 541–547 CrossRef CAS.
- F. Touret, C. Baronti, O. Goethals, M. Van Loock, X. de Lamballerie and G. Querat, Antiviral Res., 2019, 168, 109–113 CrossRef CAS.
- T. Reyser, T. H. To, C. Egwu, L. Paloque, M. Nguyen, A. Hamouy, J.-L. Stigliani, C. Bijani, J.-M. Augereau, J.-P. Joly, J. Portela, J. Havot, S. R. A. Marque, J. Boissier, A. Robert, F. Benoit-Vical and G. Audran, Molecules, 2020, 25, 3838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data and spectra. (1) Chiral HPLC of (R)-norchlorcyclizine and (R,S)-norchlorcyclizine, (2) retention of configuration of (R)-norchlorcyclizine after reaction in DMF/Et3N, 80 °C, (3) chiral HPLC of (R)-emoxyzine-2 and (R,S)-emoxyzine-2, (4) 1H and 13C NMR spectra of emodin-PA and emoxyzine derivatives, (5) mass spectra of emodin derivatives. See DOI: https://doi.org/10.1039/d3ob01122d |
| This journal is © The Royal Society of Chemistry 2023 |