
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and structure–activity relationship studies on erianin analogues as pyruvate carboxylase inhibitors in hepatocellular carcinoma cells†
Hailong
Shi‡
abc,
Jinlian
Yang‡
abc,
Zeen
Qiao‡
ab,
Lingyu
Li
ab,
Gang
Liu
ab,
Qi
Dai
c,
Li
Xu
c,
Wei
Jiao
 a,
Guolin
Zhang
a,
Fei
Wang
a,
Xiaoxia
Lu
*ac and
Xiaofeng
Ma
*a
a,
Guolin
Zhang
a,
Fei
Wang
a,
Xiaoxia
Lu
*ac and
Xiaofeng
Ma
*a
aNatural Product Research Center, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, P. R. China. E-mail: maxf@cib.ac.cn; luxx@cib.ac.cn
bUniversity of Chinese Academy of Sciences Institution Chinese Academy of Sciences Beijing, 100049, P. R. China
cNMPA Key Laboratory for Quality Monitoring and Evaluation of Traditional Chinese Medicine (Chinese Materia Medica), P. R. China
First published on 7th August 2023
Abstract
A series of novel erianin analogues were designed and synthesized based on the bioisosterism principle by altering the two aromatic rings of erianin, the substituents on the rings and the linker between them. The analogues were evaluated as pyruvate carboxylase (PC) inhibitors in hepatocellular carcinoma cells. It was found that compounds 35 and 36, where fluorine replaces a hydroxyl group, exhibited higher activity than erianin (IC50 value of 17.30 nM) in liver cancer cells with IC50 values of 15.15 nM and 10.05 nM, respectively. Additionally, at a concentration of 10 nM, compounds 35 and 36 inhibited PC with inhibitory rates of 39.10% and 40.15%, respectively, exhibiting nearly identical inhibitory activity to erianin (inhibitory rate of 40.07%). Additionally, a computer simulation docking study demonstrated the basis for better interactions between the receptors and ligands. The fluorine atom of 35 can not only form hydrogen bonds with Lys-1043 (NH⋯F, 2.04 Å), but also form fluorine bonds with the carbonyl groups of Lys-1043 (3.67 Å) and Glu-1046 (3.70 Å), due to the different orientations of the halogens on the B ring warhead. Conversely, the chlorine atom of 34 can only form alkyl hydrophobic interactions with the alkane chain in Lys-1043. Fluorinated compounds 35 and 36 also show better chemical stability and higher log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (clogP = 3.89 for 35 and 36) values than that of erianin (clogP = 3.07), and may be used as candidate compounds for further drug development.
P (clogP = 3.89 for 35 and 36) values than that of erianin (clogP = 3.07), and may be used as candidate compounds for further drug development.
1. Introduction
Accounting for 90% of liver cancers,1 hepatocellular carcinoma (HCC) is the major histological subtype of liver cancer and the third most common cause of cancer-related mortality worldwide.2 Liver transplantation is currently the most common treatment for liver cancer,3 whilst high rates of tumour recurrence and metastasis are the main reason for the poor prognosis of hepatocellular carcinoma.4 Moreover, more than 70% of patients with advanced stage HCC are not candidates for transplantation,5 either due to tumour mutation burden (TMB) or poor liver function.6 Ultimately, for most patients with advanced liver cancer, available treatments are unsatisfactory because: (i) few drugs that improve patient survival exist;7 (ii) there are high incidences of treatment-related adverse events;8 and (iii) mutated target proteins cause drug resistance, etc.9 Therefore, to change the treatment landscape of HCC management at all stages, it is now highly desirable to develop novel diagnosis and treatment methods, including new diagnostic techniques, targeted drugs, immunotherapies, and combination therapies.10Pyruvate carboxylase (PC) is a regulatory metabolic enzyme found mainly in the mitochondria that replenishes intermediates of the tricarboxylic acid cycle (TCA) and catalyses the first committed step of gluconeogenesis.11 PC has been found to be overexpressed in multiple types of tumours,12 and may also be overproduced in hepatocellular carcinoma. Targeting pyruvate carboxylase to inhibit the activity of excessively expressed PC in tumour cells is a potential target for the treatment of liver cancer via regulation of metabolic reprogramming. Over the past few decades, more than 20 PC inhibitors have been reported. However, most of these inhibitors have been used as molecular probes to elucidate various aspects of the structure and function of PC.13 ZY-444 was the first reported anticancer molecule targeting PC (Fig. 1) and is a potent and selective PC inhibitor with an IC50 value of about 1 μM that suppresses breast cancer cell viability by 90% at 5 μM.14 Despite its excellent potential in the treatment of breast cancer, no other case studies on the utilization of this inhibitor in clinical trials have been reported. Equally, no other anticancer treatments that utilize the inhibition of PC enzymes in other human cancers have been disclosed.
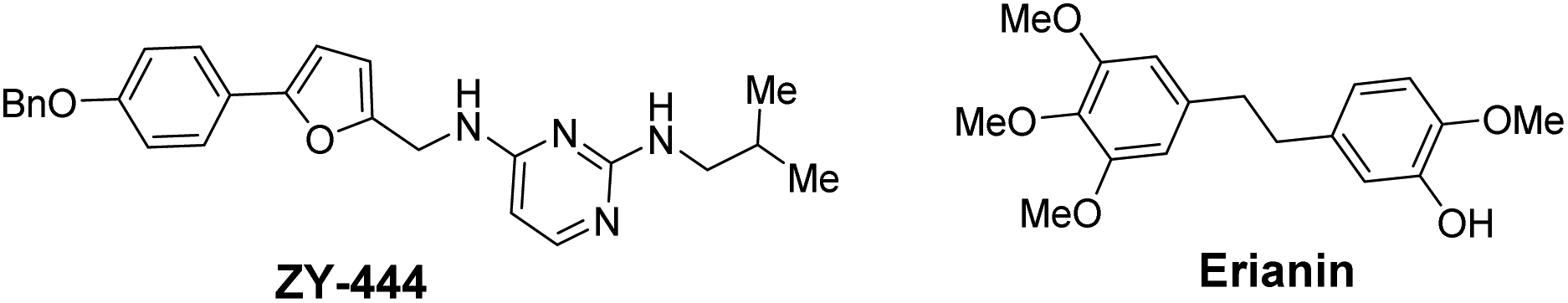 | ||
| Fig. 1 PC inhibitors: ZY-444 and erianin. | ||
Erianin is a well-known natural product with a 1,2-diphenylethane scaffold that comes from the orchid plants E. carinata,15 and has been reported as a highly efficient molecule against a variety of cancers, including hepatocellular carcinoma.16 Recently, Wang and co-workers found that erianin inhibits PC with an IC50 value of 5 nM, regulates PC-mediated metabolic reprogramming to induce oxidative stress and inhibit glycolysis, activates the adenylate-activated protein kinase (AMPK) signalling pathway, and, ultimately, causes tumour cell apoptosis and inhibits tumour metastasis.17 However, the lack of structure–activity relationship studies, its short half-life18 and low bioavailability19 have limited further drug development of erianin. These shortcomings of erianin may be related to the phenol fragment in its structure which is easily oxidized. Meanwhile, natural product-based drug development frequently uses the principle of bioisosterism to slightly alter the structure of natural products in order to enhance their affinity for the target while maintaining their biological activity. Herein, we disclose a structure–activity relationship study (SAR) of erianin to enhance its anticancer activity based on bioisosterism principles where the two aromatic rings of erianin, their ring substituents and the linker between them are changed (Fig. 2). 47 compounds were synthesized with different types of hydrophobic tail, various types and lengths of linkers, and divergent types of hydrogen bond donor (or acceptor) moieties. Biological activity against hepatocellular carcinoma cell lines was also investigated systematically. It was found that, of the 47 synthesized compounds, two exhibited better chemical stability and higher logP and enhanced activity in liver cancer cells by simply replacing easily the oxidized phenolic hydroxyl group with a fluorine atom.
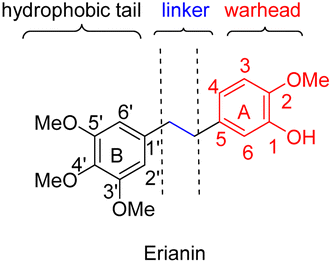 | ||
| Fig. 2 The proposed molecular pharmacophore model of erianin. | ||
2. Results and discussion
2.1 Design
Based on the analysis of the results of a preliminary activity study of the 14 natural analogues of erianin and a molecular simulation docking by Wang,17 some preliminary conclusion could be drawn: (i) the three methoxy groups on the B ring are indispensable to the maintenance of biological activity; (ii) the linear-linker between the two aromatic rings is necessary because the formation of a 6 membered ring with either the A ring or B ring resulted in a decrease in the activity; (iii) the introduction of strong hydrophilic fragment such as a sugar on the A ring or the B ring is unfavourable for biological activity; and (iv) interchanging the positions of the hydroxyl and methoxy groups on the A ring also seemed to greatly affect the activity. According to these conclusions, we hypothesized that the obvious cytotoxicity of erianin might be because its structure has a hydrophobic tail and a warhead moiety (hydrogen-bond donor or acceptor) linked by an appropriate linker (Fig. 2). Based on the principle of bioisosterism, we designed a series of compounds in which the hydroxyl group of the A ring was replaced with its classical electron–isostere halogen, the A and B rings were replaced with similar aromatic rings, and the linkers between the benzene rings were replaced with isosteres. Using these strategies, 47 erianin analogues were synthesized and then subjected to bioactive assessment.2.2 Chemistry
The compounds tested during this study were synthesized through seven different, though complementary routes (Schemes 1–7). Compounds 1–4 were synthesized according to reported literature procedures (Schemes 1 and 2).20,21 For the synthesis of 5,221423 and 15 (Scheme 3), the corresponding aryl bromides, were treated with n-butyl lithium, the obtained aryl lithium were then captured with Weinreb amides to produce intermediates S3-1 to S3-3. The products 5, 14 and 15 can be smoothly obtained via the catalytic hydrogenation of S3-1 to S3-3.
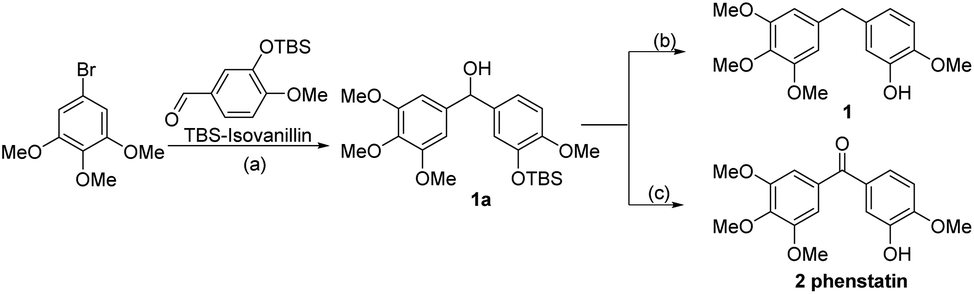 | ||
| Scheme 1 Synthetic route for compounds 1 and 2. Reagents and conditions: (a) n-BuLi, then TBS-Isovanillin, THF, −78 °C to 25 °C, 0.5 h, 86.2%; (C) (i): PDC, DCM, 25 °C, 1 h, 90.1%; (ii): TBAF, THF, 25 °C, 3 h, 89.7%; (b) (i): Et3SiH, TFA, DCM, 0 °C to 25 °C, 2.5 h, 75.4%; (ii): TBAF, THF. 25 °C, 3 h, 98.5%. | ||
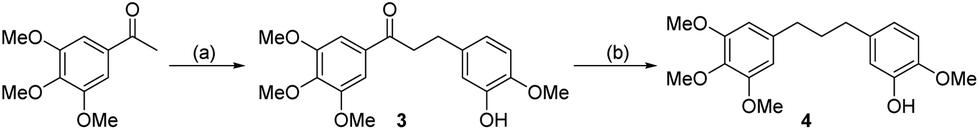 | ||
| Scheme 2 Synthetic route for compounds 3 and 4. Reagents and conditions: (a) (i): Isovanillin, Pyrrolidine, AcOH, THF, 65 °C, 8 h, 82%; (ii): Pd/C, H2, diphenyl sulfide, MeOH, 25 °C, 2 h, 86.5 %; (b) Pd/C, H2, AcOH, 25 °C,12 h, 91.4%. | ||
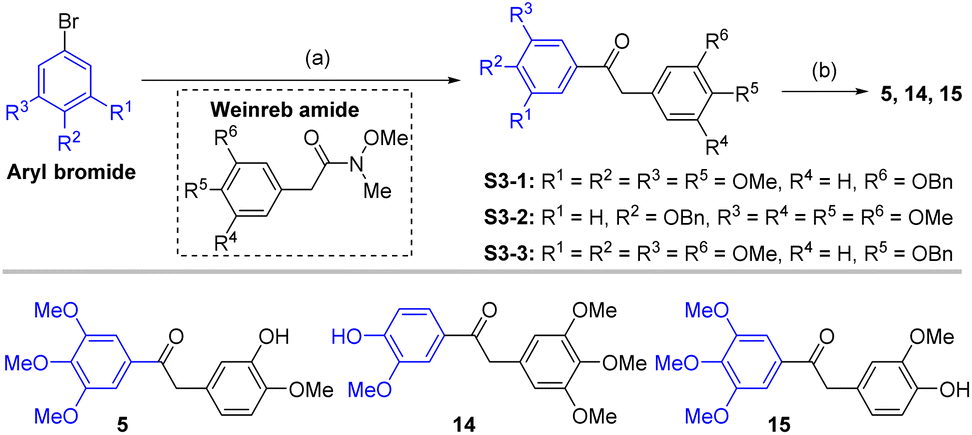 | ||
| Scheme 3 Synthetic route for compounds 5, 14 and 15. Reagents and conditions: (a) aryl bromide, n-BuLi, THF, −78 °C, 0.5 h; then Weinreb amide, −78 °C, 1 h; (b) Pd/C, H2, MeOH, 25 °C, 4 h, 85.7–92.3%. | ||
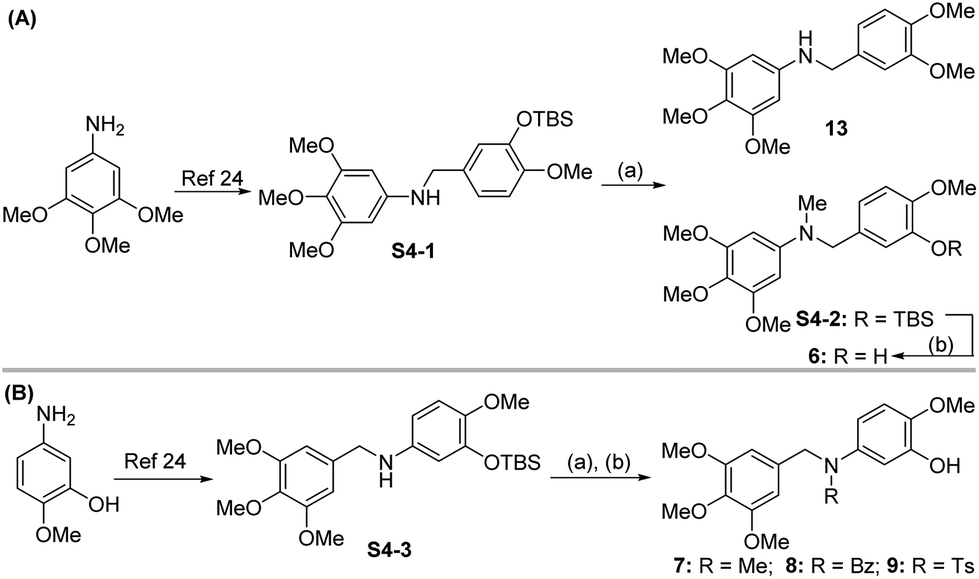 | ||
| Scheme 4 Synthetic route for compounds 6–9 and 13. Reagents and conditions: (a) MeI or BzCl or TsCl, NaH, THF, 0 °C,1.5 h, 58.6–99.1%; (b) TBAF, THF, 25 °C, 3 h, 82.7–92.1%. | ||
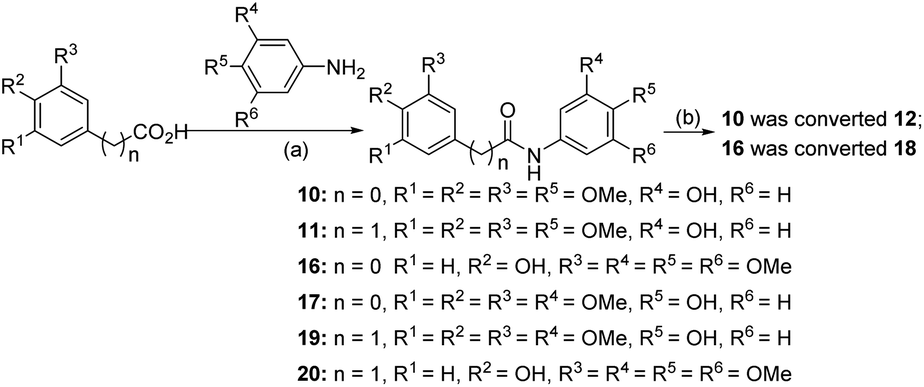 | ||
| Scheme 5 Synthetic route for compounds 10–12 and 16–20. Reagents and conditions: (a) carboxylic acids, anilines, HATU, DMF, 0 °C to 25 °C, 2 h, 73.5–83.2%; (b) Lawesson's reagent, toluene, reflux, 0.5 h, 66.3–78.2%. | ||
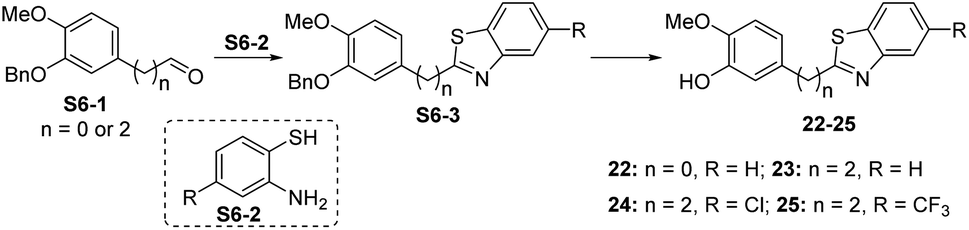 | ||
| Scheme 6 Synthetic route for compounds 22–25. Reagents and conditions: (a) S6-1, S6-2, TsOH–H2O, Na2SO4, DMF, N2, 120 °C, 10–30 min, 21–39%; (b) con. HCl/AcOH (1:2), 80 °C, 10 min, 38–78%. | ||
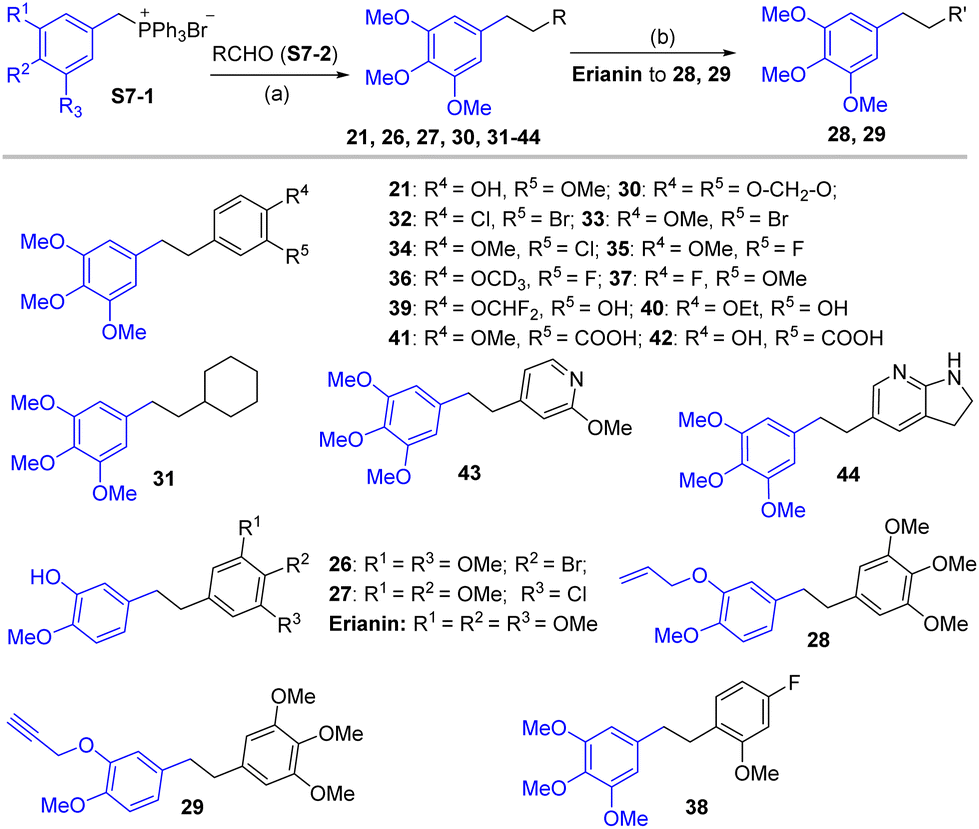 | ||
| Scheme 7 Synthetic route for compounds 26–44. Reagents and conditions: (a) (i) S7-1, S7-2, NaH, THF, 0 °C–25 °C, 12 h; (ii) Pd/C, H2, MeOH, 0 °C–25 °C, 4 h; (b) allyl bromide or 3-bromopropyne, K2CO3, THF, 25 °C, 4 h. | ||
Compound 6 was obtained by using aryl amine S4-1 as the starting material, which was obtained according to a reported literature procedure.24 Thus, S4-1 was treated with sodium hydride (NaH) and methyl iodide (MeI), and under these conditions, besides the desired methylation product S4-2, a byproduct (13) was also formed via the concurrent deprotection-methylation of the tert-butyldimethylsilyl (TBS) protected phenol hydroxyl group. The TBS protecting group in S4-2 can be smoothly removed in the presence of tetrabutylammonium fluoride (TBAF) to deliver 6 (Scheme 4A). This procedure can also be used to synthesize 7,248 and 9, in which the free amine was protected by methyl (Me), benzoyl (Bz) and toluenesulfonyl (Ts) groups (Scheme 4B). Compounds 10, 11, 16, 17, 19, and 20 were obtained directly by condensation of the corresponding carboxylic acids with amines in the presence of HATU in DMF, while the thioamide compounds 12 and 18 were produced by treatment of 1024 and 16 respectively with Lawesson's reagent (Scheme 5). The synthesis of 22–25 involves the condensation and oxidative aromatization between an aryl substituted aldehyde and a substituted 2-aminophenylthiophenol (S6-1) to give intermediate S6-3, which was followed by debenzylation in the presence of concentrated hydrochloric acid (Scheme 6). The preparations of erianin,2421,2526, 27 and 30–44 were achieved through a Wittig reaction between triphenyl (substituted benzyl)-phosphonium bromide and the corresponding aldehydes to obtained alkenes, which was then followed by catalytic hydrogenation. Compounds 28 and 29 were obtained by direct alkylation of the hydroxyl group of erianin (Scheme 7).
For those substrates such as 45–47, in which the aldehydes are not readily available, the 3,4,5-trimethoxybromobenzene derivative S8-1 was coupled with an aryl bromide S8-2 under Sonogashira cross-coupling conditions to generate S8-3, which was then hydrogenated under catalytic hydrogenation conditions (Scheme 8).
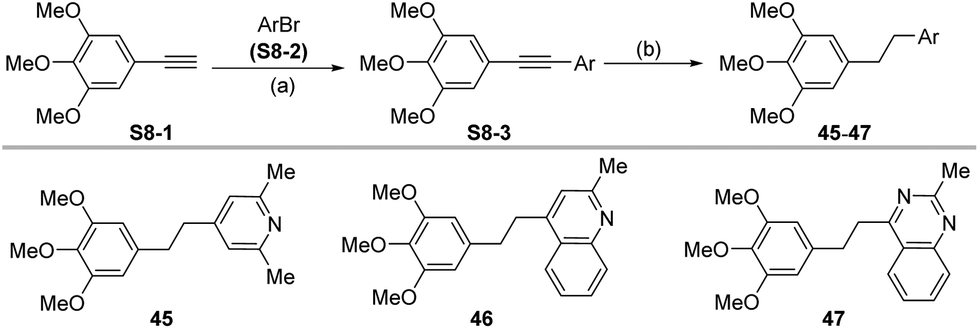 | ||
| Scheme 8 Synthetic route for compounds 45–47. Reagents and conditions: (a) S8-1, S8-2, Pd (PPh3)4 (6 mol%), CuI (6 mol%), XantPhos (6 mol%), Et3N, 1,4-dioxane, reflux,14 h; (b) Pd/C, H2, MeOH, 25 °C, 12 h. 34.3–62.5%. | ||
2.3 Structure–activity relationships
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Linkers | ArA | HCCLM3 | No. | Linkers | ArA | HCCLM3 | ||
| % (50 nM)a | % (200 nM)b | % (50 nM)a | % (200 nM)b | ||||||
| a % (50 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 50 nM. b % (200 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 200 nM. Data are mean values ± SD of n = 3. ND = NO detection, NT = NO test. | |||||||||
| 1 | –CH2– | 65.17 ± 0.72 | 74.29 ± 2.37 | 13 | –NHCH2– |
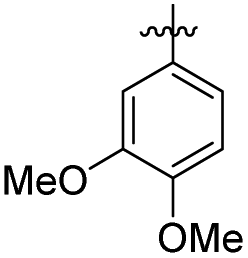
|
25.90 ± 2.22 | 23.85 ± 2.91 | |
| 2 | –CO– | 81.14 ± 0.63 | 94.18 ± 0.56 | 14 | –CH2CO– | 36.64 ± 1.64 | 31.62 ± 4.32 | ||
| 3 | –CO(CH2)2– | 39.62 ± 0.47 | 76.55 ± 2.66 | 15 | –COCH2– |
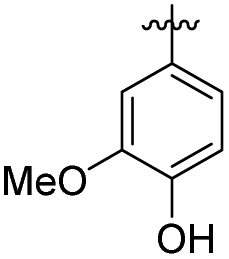
|
23.02 ± 2.53 | 28.01 ± 1.83 | |
| 4 | –(CH3)3– | 54.15 ± 3.25 | 76.91 ± 1.88 | 16 | –NHCO– | 29.66 ± 2.25 | 43.83 ± 1.50 | ||
| 5 | –COCH2– | 3.51 ± 0.16 | 41.02 ± 3.62 | 17 | –CONH– | 23.02 ± 6.70 | 28.01 ± 1.03 | ||
| 6 |
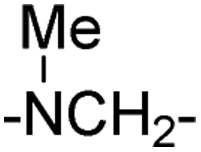
|
|
64.48 ± 1.23 | 75.96 ± 2.81 | 18 | –CSNH– | 0.60 ± 0.27 | 0.62 ± 0.11 | |
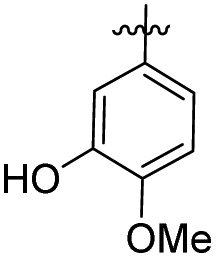
| 7 |
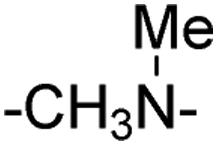
|
30.83 ± 3.26 | 44.96 ± 6.94 | 19 | –CH2CONH– | 9.44 ± 3.17 | 7.32 ± 2.41 | ||
| 8 |
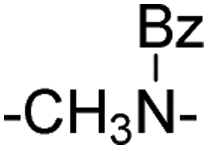
|
36.28 ± 2.21 | 46.00 ± 8.35 | 20 | –NHCOCH2– | ND | NT | ||
| 9 |
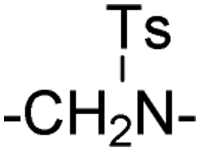
|
25.77 ± 2.46 | 41.39 ± 5.16 | 21 | –(CH2)2– | 0.45 ± 0.16 | 3.17 ± 0.56 | ||
| 10 | –CONH– | 8.54 ± 1.71 | 20.16 ± 3.70 | Erianin | 67.59 ± 2.26 | 78.86 ± 1.35 | |||
| 11 | –CH2CONH | 5.32 ± 1.45 | 8.17 ± 8.01 | ||||||
| 12 | –CSNH– | 4.27 ± 0.18 | 4.83 ± 2.17 | ||||||
Based on the preliminary screening of the activity of more than 20 erianin analogues with different linkers, we may conclude the following: the linker between the two substituted phenyl rings requires a flexible carbon chain (such as in compounds 1, 4 and erianin) or a flexible C–N chain (such as in compound 6) to maintain the bioactivity; the length of the linker should be two methylene units or a single methylene unit with an extra tertiary amine attached to ArA; elongating or shortening the length of the linker was detrimental to bioactivity; and increasing the molecular rigidity, such as with carbonyl (compounds 2, 3 and 5), amide (compounds 10, 11) and thioamide (compound 12) groups, or increasing the steric hindrance of the substituent on the N-atoms (compounds 8, 9) of the linkers reduces the bioactivities. Furthermore, it was found that the hydroxyl group at the 1-position of the A ring was essential for the bioactivity. Therefore, we then fixed the linker as two methylene units and the A ring as 1-hydroxyl-2-methoxyl phenyl and proceeded to investigate the effect that changing the substituents on the B ring has on the bioactivities.
P values are very different (Fig. 3b). From these results, it seems that possessing an appropriate logP is an important factor for the bioactivity of an analogue. To further explain the difference in bioactivity between erianin and compound 23, which have similar logP values, we extracted fragments of ArB (the other parts of these compounds were exactly the same) and calculated their clogP values (Fig. 3c). It was found that the clogP values of the B ring fragments (ArB22–27) of 22–27 were significantly larger than the B ring value of erianin. Moreover, although the clogP values of ArB-4-DMER and ArB-DMBZD were similar to that of erianin, the activity of these two compounds was seriously lost due to the hydroxyl group on the ring. In view of this, we believe that the B ring should not have hydrogen bond donors such as hydroxyl groups directly linked to the aromatic ring and should have a suitable clogP value to balance the hydrophobicity of the B ring region of the compound. At this point, we have identified the effect that the B ring composition and the linkers between the two aromatic fragments have on the bioactivity. Next, we conducted a systematic study on the effect that the A ring has on the bioactivities.
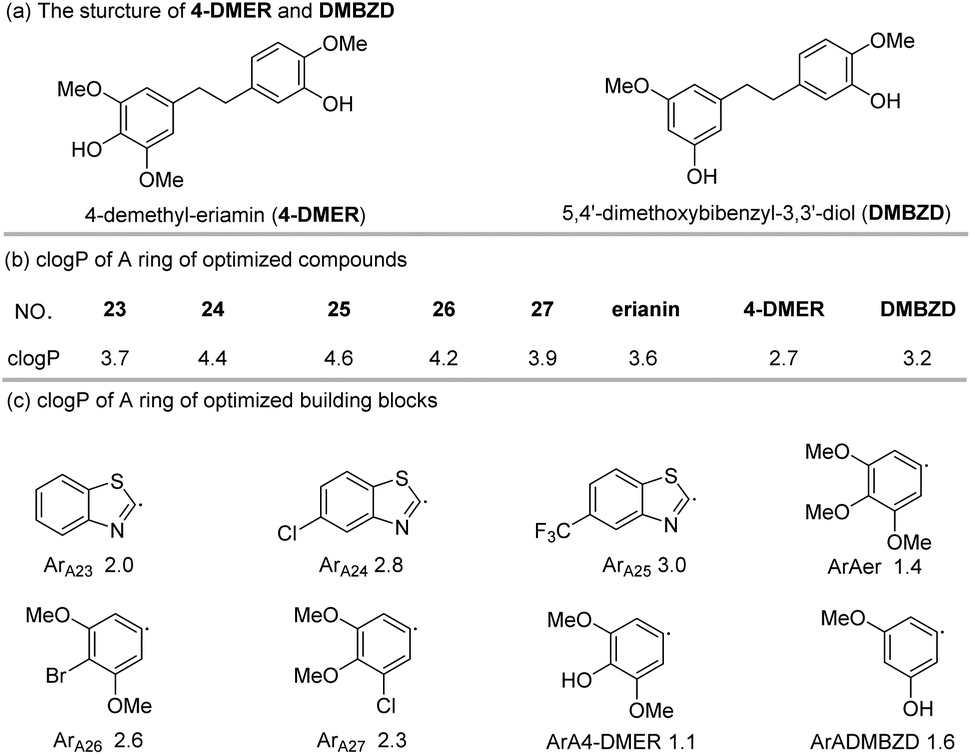 | ||
| Fig. 3 clogP values of the B rings of optimized compounds and building blocks. | ||
|
|
||||
|---|---|---|---|---|
| No. | ArB | n | HCCLM3 | |
| % (50 nM)a | % (200 nM)b | |||
| a % (50 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 50 nM. b % (200 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 200 nM. Data are mean values SD of n = 3. ND = NO detection, NT = NO test. | ||||
| 22 |
|
0 | 33.60 ± 3.53 | 25.20 ± 4.00 |
| 23 |
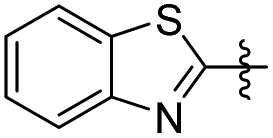
|
2 | 35.39 ± 5.91 | 44.07 ± 4.55 |
| 24 |
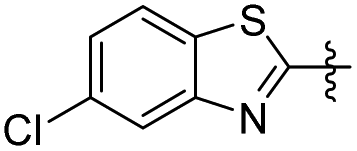
|
2 | 36.55 ± 4.94 | 43.52 ± 2.28 |
| 25 |
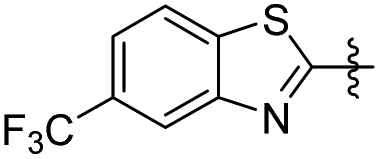
|
2 | 15.20 ± 2.41 | 12.05 ± 0.82 |
| 26 |
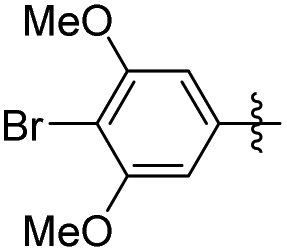
|
2 | ND | NT |
| 27 |
|
2 | 9.61 ± 3.10 | 9.86 ± 3.72 |
| Erianin |
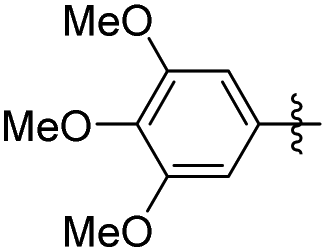
|
2 | 67.59 ± 2.26 | 78.86 ± 1.35 |
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | ArA | HCCLM3 | No. | ArA | HCCLM3 | No. | ArA | HCCLM3 | |||
| % (50 nM)a | % (200 nM)b | % (50 nM)a | % (200 nM)b | % (50 nM)a | % (200 nM)b | ||||||
| a % (50 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 50 nM. b % (200 nM) values of the erianin analogues on cell growth inhibition in HCCLM3 at a concentration of 200 nM. Data are mean values SD of n = 3. ND = NO detection, NT = NO test. | |||||||||||
| 28 |
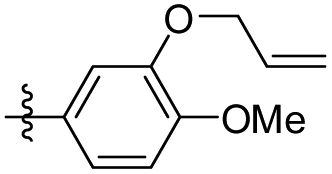
|
7.10 ± 2.85 | 30.23 ± 1.98 | 35 |
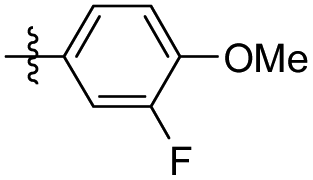
|
66.61 ± 5.45 | 64.23 ± 5.68 | 42 |
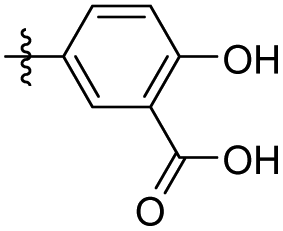
|
17.71 ± 0.72 | 20.79 ± 1.23 |
| 29 |
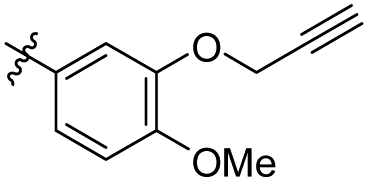
|
7.47 ± 0.70 | 32.00 ± 1.61 | 36 |
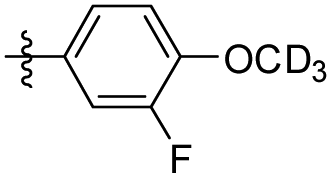
|
57.39 ± 3.70 | 60.56 ± 4.45 | 43 |
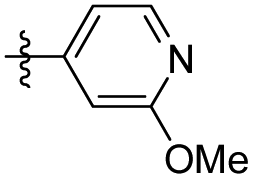
|
10.27 ± 1.49 | 13.52 ± 2.22 |
| 30 |
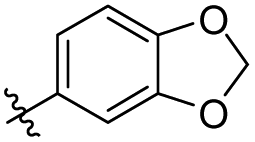
|
2.04 ± 2.32 | 3.24 ± 1.62 | 37 |
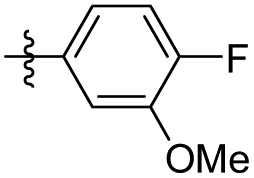
|
16.53 ± 0.63 | 6.64 ± 1.37 | 44 |
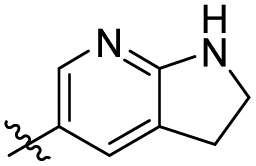
|
ND | NT |
| 31 |
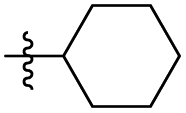
|
7.17 ± 3.01 | 8.14 ± 6.65 | 38 |
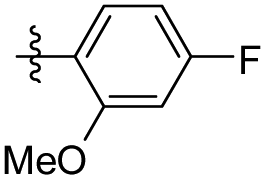
|
ND | NT | 45 |
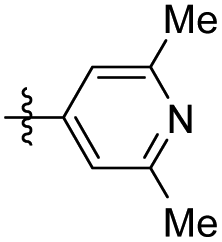
|
5.47 ± 1.19 | 18.62 ± 3.46 |
| 32 |
|
1.04 ± 0.43 | 16.23 ± 1.54 | 39 |
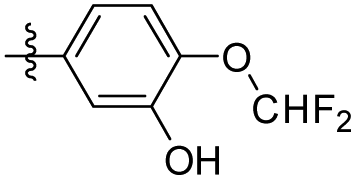
|
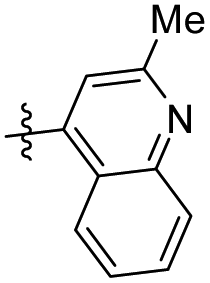
9.48 ± 4.36 | 20.59 ± 6.43 | 46 |
|
3.15 ± 1.12 | 7.89 ± 1.96 |
| 33 |
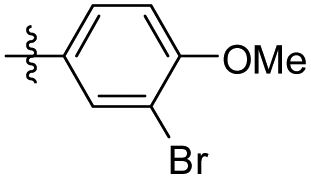
|
42.14 ± 1.62 | 19.60 ± 3.19 | 40 |
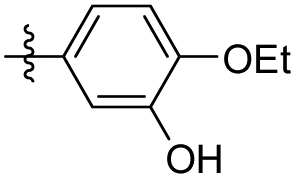
|
5.64 ± 1.10 | 10.33 ± 2.54 | 47 |
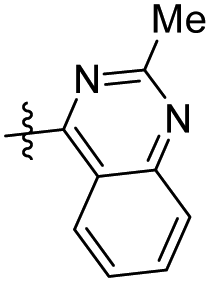
|
13.61 ± 5.29 | 11.60 ± 2.11 |
| 34 |
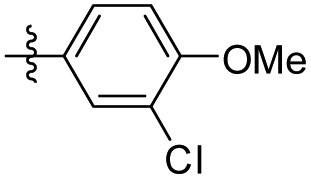
|
40.49 ± 2.81 | 72.32 ± 3.57 | 41 |
|
0.84 ± 0.36 | 3.44 ± 1.17 | Erianin | 67.59 ± 2.26 | 78.86 ± 1.35 | |
 20 and 624 as tubulin inhibitors; and 3 as a nuclear factor-κB (NF-κB) inhibitor21), erianin and compounds 34–36 have better selectivity against pyruvate carboxylase. The cytotoxic activity of compound 35 was equivalent to erianin, while 36 was stronger than erianin. However, deuterated compound 36, which has better cell-level bioactivity, exhibited almost identical PC inhibition activity to that of 35. This may be related to the fact that the deuterated methyl group has no obvious isotope effect on PC.
20 and 624 as tubulin inhibitors; and 3 as a nuclear factor-κB (NF-κB) inhibitor21), erianin and compounds 34–36 have better selectivity against pyruvate carboxylase. The cytotoxic activity of compound 35 was equivalent to erianin, while 36 was stronger than erianin. However, deuterated compound 36, which has better cell-level bioactivity, exhibited almost identical PC inhibition activity to that of 35. This may be related to the fact that the deuterated methyl group has no obvious isotope effect on PC.
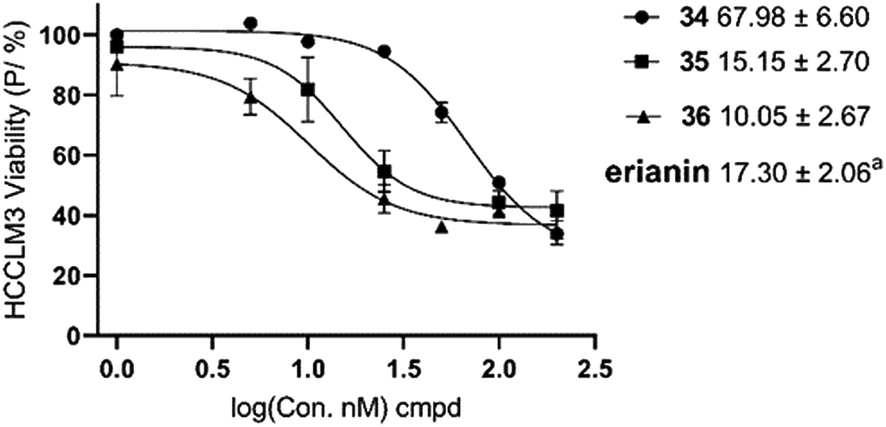 | ||
| Fig. 4 IC50 values of the erianin analogues on cell growth inhibition in HCCLM3. aIC50 value of erianin.17 Data are mean values ± SE of n = 3. | ||
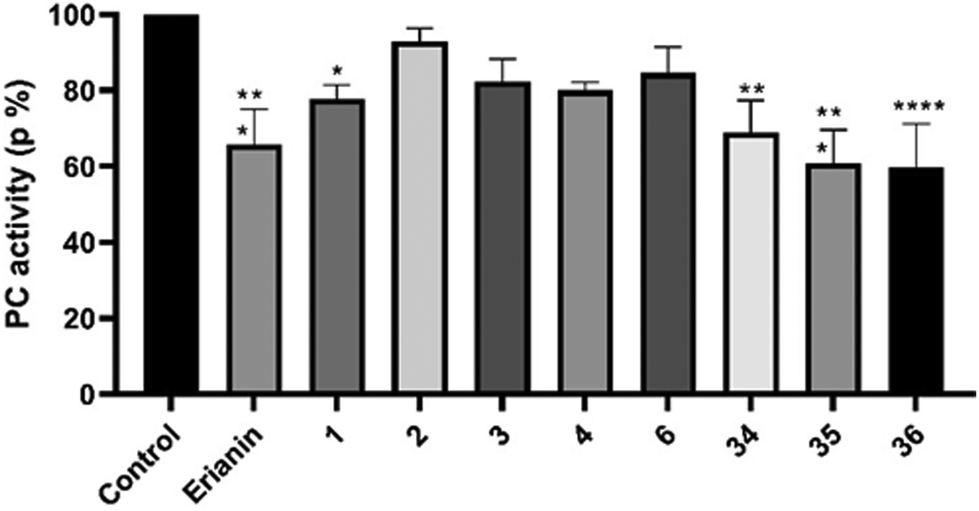 | ||
| Fig. 5 Inhibition of pyruvate carboxylase (PC) at a concentration of 10 nM. Data are mean values ± SD of n = 3. | ||
2.4 Molecular docking
The possible binding modes of the synthesized erianin analogues were studied by molecular docking calculations using the X-ray structure of human pyruvate carboxylase (PC, PDB ID: 3GB3). The results of the simulated molecular docking showed that the Libdock-scores of compounds 34, 35 and erianin were 84.47, 93.39 and 80.09, respectively. This was consistent with the order of inhibitory potency of these compounds on PC at 10 nM. Comparison of the binding modes of compound 35 with that of compound 34 and erianin (Fig. 6 and Fig. SI2–4†) showed an inversion of its binding orientation in the active site of 3GB3. The binding sites and the dominant conformation of 34 and 35 were at the same active binding site of PC. However, due to the different orientations of the halogens on the A ring warhead, the fluorine atom of 35 can not only form hydrogen bonds with Lys-1043 (NH⋯F, 2.04 Å), but also form fluorine bonds with the carbonyl groups of Lys-1043 (3.67 Å) and Glu-1046 (3.70 Å). In the optimum binding conformation of compound 34, the orientation of the chlorine atom was opposite to that of the fluorine atom in compound 35, and this chlorine atom can only form alkyl hydrophobic interactions with the alkane chain of Lys-1043. The binding region of erianin was also similar to that of compounds 34 and 35. However, the optimal binding site (Fig. SI2–4†) and conformation were different (Fig. 6, C(3D)). The hydroxyl group in the warhead of erianin can form hydrogen bonds with Gln-1073 (2.98 Å) as a hydrogen bond donor and Gln-1075 (3.07 Å) as a hydrogen bond acceptor.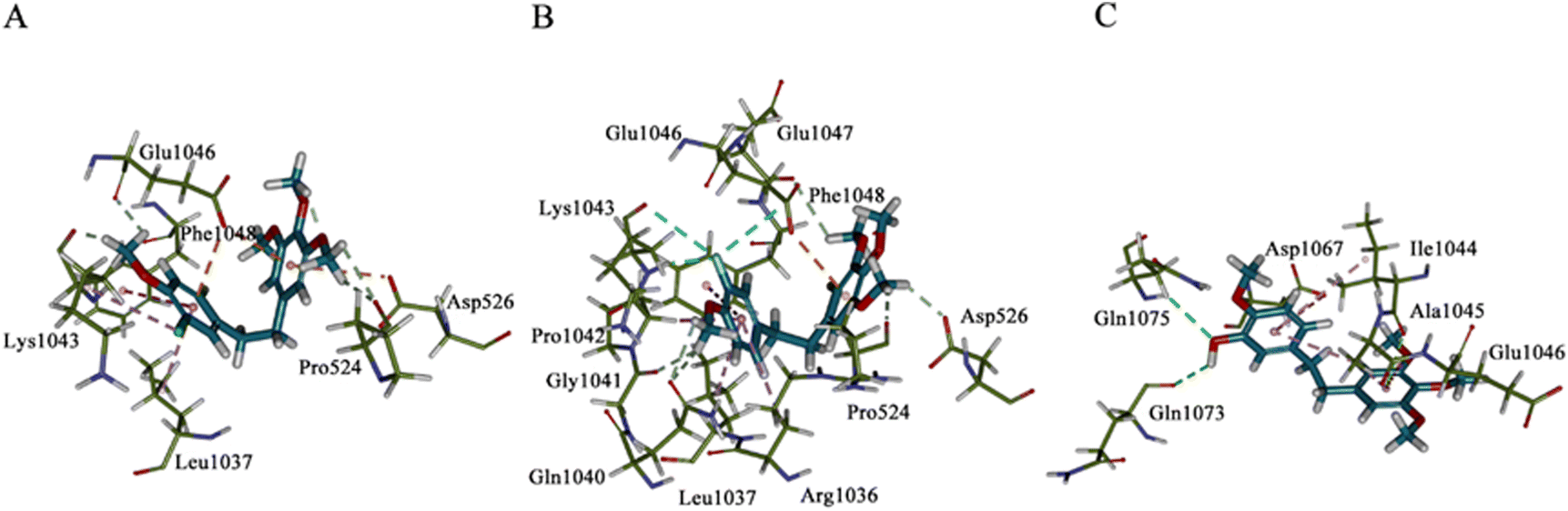 | ||
| Fig. 6 Binding conformations of 34, 35 and erianin with human pyruvate carboxylase (PDB ID: 3BG3). Panel A–C(3D): binding modes of compound 34 (panel A), 35 (panel B), and erianin (panel C) in the active site of 3BG3. | ||
3. Conclusions
In conclusion, we present a systemic structure–activity relationship study on erianin, which has previously been identified as a pyruvate carboxylase (PC)-targeting anti-hepatocellular carcinoma agent. Our studies showed that a strongly lipophilic two-atom bridge (–CH2CH2–, –NMeCH2–) linker could be optimal distance between the two rings. An essential component for strong anticancer activity of the whole molecule was the retention of amphiprotic A-ring fragments with low clogP values as a hydrophobic tail. When the B ring had a higher or lower clogP value, but a hydroxyl group substituted directly on the benzene, the activity of the molecules was greatly reduced. With respect to the A ring, the 1-hydroxyl group was found to be an important pharmacophore, and it could be replaced by halogen groups (Br, Cl, F). Additionally, compounds 35 and 36, where the hydroxyl groups were replaced by fluorine atoms, had the same or even better activity as erianin. The optimal substituents on the oxygen at the 2-position of the A ring were –Me and –CD3, while difluoromethyl and ethyl groups on the 2-position oxygen of erianin lead to a dramatic decrease in the activity.
Fluorinated compounds 35 and 36 had similar activity to erianin but had higher liposolubility (clogP 35 = clogP 36 = 3.89, @ChemDraw) and more chemical stability than erianin. These compounds may be used as candidates for the treatment or diagnosis of central nervous system (CNS) tumours and may even penetrate the brain through the blood–brain barrier. Furthermore, the research presented here would be valuable for the development of more anticancer candidates with similar core structures, higher activity and oral bioavailability. This research may even further improve the selectivity of PC inhibition and the molecules presented may serve as a chemical probe to determine whether PC can be used as a druggable target. Research is ongoing to further refine the selectivity for the PC inhibition and to identify a chemical probe for the dissection of PC-dependent tumour energy metabolism pathways and will be reported in due course.
4. Experimental section
4.1 Chemistry and synthesis
The synthesis and characterization of compounds 1–7, 10, 14, 21, 39 and 40 had already been reported in previous studies.21–27 The compounds 8, 9, 11–13, 15–20, 22–34, 36–38 and 41–47 were synthesized according to the route in Schemes 3–8. The structures of all compounds were confirmed from the analytical data obtained from 1H NMR and 13C NMR spectroscopies, and mass spectrometry. All reagents were purchased from commercial suppliers (Energy Chemical, Bide Pharma) and were of the highest commercial quality and used without further purification. All reactions were performed with magnetic stirring and were followed using thin layer chromatography (TLC) on silica gel 60 F254 precoated plates (0.25 mm, Yantai Jiangyou silica gel Development Co., Ltd) and the pure compound was visualized under UV light. Purification of the reaction products was carried out using flash column chromatography (FCC) using ultra-pure silica gel (200–300 mesh) purchased from Yantai Jiangyou silica gel Development Co., Ltd, unless otherwise stated. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance spectrometers (1H NMR at 400 MHz or 600 MHz, 13C NMR at 101 MHz or 150 MHz). High-resolution mass spectra (HRMS) were obtained using MicrOTOF-Q II mass spectrometer in ESI mode.:hexane = 1:5) to yield the corresponding product.
N-(3,4-Dimethoxybenzyl)-3,4,5-trimethoxyaniline (13). 170 mg, 44% yield. 1H NMR (400 MHz, CDCl3) δ 6.93 (t, 2H), 6.85 (d, J = 8.64 Hz, 1H), 5.90 (s, 2H), 4.23 (s, 2H), 3.88 (d, J = 2.04 Hz, 6H), 3.80 (s, 6H), 3.77 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 153.95, 149.16, 148.31, 145.08, 131.78, 130.20, 119.84, 111.18, 110.88, 90.45, 61.11, 55.96, 55.93, 55.89, 48.82; HRMS-ESI + (m/z): calcd for C18H23NO5 [M + H]+: 334.1654, found: 334.1664.
N-(3-Hydroxy-4-methoxyphenyl)-N-(3,4,5-trimethoxybenzyl) benzamide (8). 387.7 mg, 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 7.04 Hz, 2H), 7.19–7.21 (m, 3H), 6.64 (d, J = 2.36 Hz, 1H), 6.54 (t, J = 4.6 Hz, 3H), 6.31 (d, J = 8.32 Hz, 1H), 5.80 (s, 1H), 4.98 (s, 2H), 3.83 (s, 3H), 3.78 (s, 6H), 3.77 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.57, 153.13, 145.82, 145.37, 137.24, 136.86, 136.16, 133.36, 133.35, 129.54, 128.50, 127.74, 120.06, 113.96, 110.25, 105.62, 60.84, 56.09, 55.89, 54.09; HRMS-ESI + (m/z): calcd for C24H25NO6 [M + H]+: 424.1760, found: 424.1769.
N-(3-Hydroxy-4-methoxyphenyl)-4-methyl-N-(3,4,5-trimethoxybenzyl) benzenesulfonamide (9). 217.6 mg, 83% yield. 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.24 Hz, 2H), 7.27 (d, J = 8.04 Hz, 2H), 6.68 (d, J = 8.6 Hz, 1H), 6.57 (dd, J = 2.48, 8.56 Hz, 1H), 6.50 (d, J = 2.48 Hz, 1H), 6.43 (s, 2H), 5.57 (s, 1H), 4.59 (s, 2H), 3.82 (s, 3H), 3.78 (s, 3H), 3.77 (s, 6H), 2.44 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 153.06, 146.17, 145.45, 143.90, 137.26, 135.71, 132.18, 131.71, 129.51, 127.77, 121.74, 114.53, 110.23, 105.46, 60.79, 56.06, 55.90, 55.14; HRMS-ESI + (m/z): calcd for C24H27NO7S [M + Na]+: 496.1406, found: 496.1420.
N-(3-Hydroxy-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)-acetamide (11). 384.6 mg, 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.72 (s, 1H), 7.28 (d, J = 2.16 Hz, 1H), 6.93 (dd, J = 6.93, 8.48 Hz, 1H), 6.67 (d, J = 8.48 Hz, 1H), 6.64 (s, 2H), 3.77 (s, 6H), 3.72 (s, 3H), 3.63 (s, 3H), 3.50 (s, 2H), 3.35 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 168.79, 153.11, 147.63, 142.89, 136.67, 132.22, 131.87, 115.63, 112.17, 106.94, 105.15, 60.43, 56.28, 55.91, 44.01; HRMS-ESI + (m/z): calcd for C18H21NO6 [M + H]+: 348.1447, found: 348.1449.
4-Hydroxy-3-methoxy-N-(3,4,5-trimethoxyphenyl) benzamide (16). 395.5 mg, 79.2% yield. 1H NMR (400 MHz, CDCl3) δ 7.90 (s, 1H), 7.52 (d, J = 1.52 Hz, 1H), 7.34 (dd, J = 1.64, 8.24 Hz, 1H), 6.96 (s, 2H), 6.95 (d, J = 8.28 Hz, 1H), 6.07 (s, 1H), 3.93 (s, 3H), 3.84 (s, 6H), 3.83 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.29, 153.36, 149.17, 146.89, 134.77, 134.24, 126.89, 119.72, 114.02, 110.52, 97.89, 61.00, 56.09; HRMS-ESI + (m/z): calcd for C17H19NO6 [M + H]+: 334.1290, found: 334.1290.
N-(4-Hydroxy-3-methoxyphenyl)-3,4,5-trimethoxybenzamide (17). 367.3 mg, 73.5% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.81 (s, 1H), 7.39 (d, J = 2.12 Hz, 1H), 7.27 (s, 2H), 7.12 (dd, J = 2.00, 8.48 Hz, 1H), 6.76 (d, J = 8.52 Hz, 1H), 3.87 (s, 6H), 3.77 (s, 3H), 3.73 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.42, 152.31, 145.49, 141.71, 140.17, 129.75, 129.37, 113.16, 112.06, 103.83, 103.43, 59.94, 55.36, 55.00; HRMS-ESI + (m/z): calcd for C17H19NO6 [M + H]+: 334.1290, found: 334.1290.
N-(4-Hydroxy-3-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl) acetamide (19).. 384.6 mg, 83.2% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.72 (s, 1H), 7.28 (d, J = 2.16 Hz, 1H), 6.93 (dd, J = 6.93, 8.48 Hz, 1H), 6.67 (d, J = 8.48 Hz, 1H), 6.64 (s, 2H), 3.77 (s, 6H), 3.72 (s, 3H), 3.63 (s, 3H), 3.50 (s, 2H), 3.35 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 168.79, 153.11, 147.63, 142.89, 136.67, 132.22, 131.87, 115.63, 112.17, 106.94, 105.15, 60.43, 56.28, 55.91, 44.01; HRMS-ESI + (m/z): calcd for C18H21NO6 [M + H]+: 348.1447, found: 348.1445.
2-(4-Hydroxy-3-methoxyphenyl)-N-(3,4,5-trimethoxyphenyl) acetamide (20). 204.6 mg, 68.2% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 9.02 (s, 1H), 7.16 (d, J = 2.36 Hz, 1H), 6.93 (dd, J = 2.36, 8.64 Hz, 1H), 6.80 (d, J = 8.72 Hz, 1H), 6.64 (s, 2H), 3.76 (s, 6H), 3.71 (s, 3H), 3.63 (s, 3H), 3.50 (s, 2H), 3.35 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 168.86, 153.12, 146.85, 144.21, 136.66, 133.32, 132.23, 112.93, 110.38, 108.22, 106.88, 60.43, 56.33, 56.27, 44.00; HRMS-ESI + (m/z): calcd for C18H21NO6 [M + H]+: 348.1368, found: 348.1446.
:MeOH = 50:1–15:1) to yield a pale yellow solid.
N-(3-Hydroxy-4-methoxyphenyl)-3,4,5-trimethoxybenzothioamide (12). 66.3 mg, 66% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 9.16 (s, 1H), 7.46 (d, J = 2.08 Hz, 1H), 7.18 (s, 2H), 7.15 (d, J = 2.12 Hz, 1H), 6.81 (d, J = 8.44 Hz, 1H), 3.85 (s, 6H), 3.76 (s, 3H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 195.84, 152.50, 147.39, 145.33, 140.06, 138.16, 132.31, 117.54, 115.21, 109.76, 105.66, 60.58, 56.46, 56.16. HRMS-ESI + (m/z): calcd for C17H19NO5S [M + H]+: 350.1062, found: 350.1064.
N-(4-Hydroxy-3-methoxyphenyl)-3,4,5-trimethoxybenzothioamide (18). 78.2 mg, 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 9.70 (s, 1H), 7.50 (s, 1H), 7.40 (d, J = 8.16 Hz, 1H), 7.26 (s, 2H), 6.83 (d, J = 8.48 Hz, 1H), 3.84 (s, 3H), 3.76 (s, 6H), 3.68 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 196.41, 152.83, 150.23, 147.09, 136.59, 135.78, 133.97, 121.72, 114.92, 112.54, 102.59, 60.58, 56.39, 56.14; HRMS-ESI + (m/z): calcd for C17H19NO5S [M + H]+: 350.1062, found: 350.1064.
:ethyl acetate = 3:1) to obtain a white solid S3-3 (289 mg, 85% yield). The obtained ketone S3-3 (100.0 mg, 0.24 mmol) was dissolved in THF (4 mL), 10% palladium on carbon (25.2 mg) was added to the solution. The reaction vessel was evacuated and backfilled with hydrogen (H2) five times. The mixture was stirred at room temperature for 12 h and then filtered through Celite give a filtrate. This filtrate was evaporated to oil, which was purified by chromatography on silica gel (ethyl acetate:hexane = 1:3) to give 15 (63.0 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 2H), 6.89 (d, J = 8.52 Hz, 1H), 6.80 (dd, J = 1.84, 5.28 Hz, 1H), 4.19 (s, 2H), 3.93 (s, 3H), 3.91 (s, 6H), 3.88 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 196.82, 153.03, 146.75, 144.70, 142.66, 131.70, 126.54, 122.15, 114.56, 111.61, 106.32, 60.93, 56.27, 55.90, 45.30. HRMS-ESI + (m/z): calcd for C17H19NO5S [M + H]+: 350.1062, found: 350.1064.
:hexane = 1:5) to yield a pale yellow slurry. To this slurry was added a pre-prepared mixture of concentrated hydrochloric acid/acetic acid (v/v 2:1, 2 mL) and the resulting mixture was heated at 80 °C for 5–15 min. The reaction solution was cooled to room temperature and water was then added, and the acid neutralized with 1 N sodium hydroxide solution. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were then dried over sodium sulphate (Na2SO4) and evaporated under reduced pressure. The obtained residue was purified by chromatography on silica gel (10% ethyl acetate:hexane = 1:10) to give the products.
5-(Benzo[d]thiazol-2-yl)-2-methoxyphenol (22). 38 mg, 8% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.16 Hz, 1H), 7.87 (d, J = 7.96 Hz, 1H), 7.63–7.67 (m, 2H), 7.45–7.49 (m, 1H), 7.33–7.38 (m, 1H), 6.94 (d, J = 8.28 Hz, 1H), 5.73 (s, 1H), 3.97 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.83, 154.18, 149.02, 145.97, 134.98, 127.22, 126.21, 124.86, 122.93, 121.51, 120.06, 113.73, 110.72, 56.09; HRMS-ESI + (m/z): calcd for C14H11NO2S [M + H]+: 258.0589, found: 258.0588.
5-(2-(Benzo[d]thiazol-2-yl)ethyl)-2-methoxyphenol (23). 65 mg, 14% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.72 (d, J = 7.80 Hz, 1H), 7.33 (dd, J = 1.36, 8.44 Hz, 1H), 6.83 (s, 1H), 6.77 (d, J = 8.20 Hz, 1H), 6.71 (d, J = 8.12 Hz, 1H), 5.70 (s, 1H), 3.85 (s, 3H), 3.38 (t, J = 8.24 Hz, 2H), 3.10 (t, J = 8.20 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 173.13, 154.05, 145.67, 145.28, 133.41, 133.23, 131.96, 125.23, 122.49, 122.20, 119.83, 114.68, 110.77, 55.98, 36.14, 34.77; HRMS-ESI + (m/z): calcd for C16H15NO2S [M + H]+: 286.0902, found: 286.0897.
5-(2-(5-Chlorobenzo[d]thiazol-2-yl)ethyl)-2-methoxyphenol (24). 62 mg, 24% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 1.92 Hz, 1H), 7.73 (d, J = 8.52 Hz, 1H), 7.33 (dd, J = 1.96, 8.52 Hz, 1H), 6.83 (d, J = 1.92 Hz, 1H), 6.76 (d, J = 8.16 Hz, 1H), 6.71 (dd, J = 1.92, 8.20 Hz, 1H), 5.73 (s, 1H), 3.85 (s, 3H), 3.38 (t, J = 7.48 Hz, 2H), 3.11 (t, J = 8.28 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 173.13, 154.05, 145.67, 145.28, 133.41, 133.23, 131.96, 125.23, 122.49, 122.20, 119.83, 114.68, 110.77, 55.98, 36.14, 34.77; HRMS-ESI + (m/z): calcd for C16H14ClNO2S [M + H]+: 320.0512, found: 320.0514.
2-Methoxy-5-(2-(5-(trifluoromethyl)benzo[d]thiazol-2-yl)ethyl) phenol (25). 78 mg, 23% yield over two steps.1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.31 (d, 2H), 7.72 (d, J = 8.36 Hz, 1H), 6.79 (d, J = 8.16 Hz, 1H), 6.71 (s, 1H), 6.64 (d, J = 8.08 Hz, 1H), 3.72 (s, 3H), 3.43 (t, J = 15.48 Hz, 2H), 3.02 (t, J = 7.48 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 174.63, 152.73, 146.81, 146.61, 139.44, 132.97, 127.58, 127.26, 126.18, 124.02, 123.47, 121.35, 121.32, 119.41, 119.37, 116.25, 112.65, 56.05, 35.72, 34.22; HRMS-ESI + (m/z): calcd for C17H14F3NO2S [M + H]+: 354.0775, found: 354.0779.
28 (0.383 mmol, 1 eq.) in THF (8 mL) was added sodium hydride (1.5 eq.) at 0 °C. After stirring at the same temperature for 10 min, S7-2 (0.95 eq.) was added to the reaction mixture at 0 °C. The ice bath was removed, and the mixture was stirred for another 12 h. The reaction mixture was neutralized with NH4Cl and then the aqueous layer was extracted with ethyl acetate (4 × 20 mL), the combined organic layer was dried over Na2SO4 and concentrated under vacuum to give the compound as a yellow solid which was directly used as the raw material in the next reaction.
To a stirred solution of the products of the previous reaction (0.383 mmol, 1 eq.) in methanol (5 mL) was added Pd/C (0.1 eq.) at room temperature. The reaction vessel was evacuated and backfilled with hydrogen (H2) five times. The reaction mixture was stirred in a hydrogen atmosphere at rt for 4 h. After completion of the reaction, the reaction mixture was filtered, and the filtrate was evaporated under vacuum to afford the crude product which was purified by column chromatography on silica gel with ethyl acetate/hexane (1/5) to provide 26–44.
5-(4-Bromo-3,5-dimethoxyphenethyl)-2-methoxyphenol (26). 110 mg, 78% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.82 (d, J = 2.0 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 6.63 (dd, J = 8.1, 2.0 Hz, 1H), 6.38 (s, 2H), 5.58 (s, 1H), 3.89 (s, 3H), 3.87 (s, 6H), 2.86 (t, J = 3.8 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 156.76, 145.50, 144.91, 142.58, 134.60, 119.91, 114.63, 110.54, 105.13, 98.07, 56.39, 56.03, 38.43, 37.07, 29.72; HRMS-ESI + (m/z): calcd for C17H18BrO4 [M + Na]+: 389.0364; found: 389.0363.
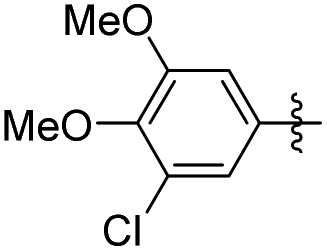
5-(3-Chloro-4,5-dimethoxyphenethyl)-2-methoxyphenol (27). 96 mg, 78% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.82 (d, J = 1.9 Hz, 1H), 6.80 (d, J = 4.6 Hz, 1H), 6.78–6.75 (m, 1H), 6.70–6.63 (m, 2H), 5.57 (s, 1H), 3.89 (d, J = 3.3 Hz, 6H), 3.87 (s, 3H), 2.84 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 148.67, 147.17, 145.43, 144.79, 135.17, 134.49, 120.21, 119.83, 114.67, 111.84, 111.14, 110.53, 56.02, 55.92, 55.79, 37.61, 37.50; HRMS-ESI + (m/z): calcd for C17H19ClO4 [M + H]+: 323.1090; found: 323.1092.
5-(3-(Allyloxy)-4-methoxyphenethyl)-1,2,3-trimethoxybenzene (28). 126 mg, 92% yield over three steps. 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 8.1 Hz, 1H), 6.75 (dd, J = 8.1, 1.8 Hz, 1H), 6.70 (d, J = 1.8 Hz, 1H), 6.38 (s, 2H), 6.15–6.03 (m, 1H), 5.41 (dd, J = 17.2, 1.5 Hz, 1H), 5.30 (dd, J = 10.5, 1.2 Hz, 1H), 4.59 (d, J = 5.4 Hz, 2H), 3.88 (s, 3H), 3.85 (d, J = 2.2 Hz, 9H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 153.02, 147.75, 137.50, 136.14, 134.13, 133.41, 120.82, 117.90, 114.14, 111.63, 105.42, 69.89, 60.89, 56.05, 38.48, 37.46; HRMS-ESI + (m/z): calcd for C21H26O5 [M + Na]+: 381.1677; found: 381.1678.
1,2,3-Trimethoxy-5-(4-methoxy-3-(prop-2-yn-1-yloxy)phenethyl) benzene (29). 127 mg, 93% yield over three steps. 1H NMR (400 MHz, CDCl3) δ 6.87 (d, J = 1.7 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.82–6.78 (m, 1H), 6.37 (s, 2H), 4.75 (d, J = 2.3 Hz, 2H), 3.88 (s, 3H), 3.84 (d, J = 2.9 Hz, 9H), 2.92–2.84 (m, 4H), 2.52 (t, J = 2.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 153.01, 147.99, 146.50, 137.40, 136.12, 134.10, 121.93, 115.10, 111.73, 105.43, 78.67, 75.72, 60.88, 56.79, 56.06, 56.00, 38.42, 37.37; HRMS-ESI + (m/z): calcd for C21H24O5 [M + Na]+: 381.1678; found: 381.1678.
5-(2-Cyclohexylethyl)-1,2,3-trimethoxybenzene (31). 77 mg, 72% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.41 (s, 2H), 3.88 (s, 6H), 3.85 (s, 3H), 2.58 (dd, J = 9.5, 6.9 Hz, 2H), 1.79 (d, J = 13.6 Hz, 2H), 1.73–1.67 (m, 2H), 1.55–1.49 (m, 2H), 1.34–1.26 (m, 3H), 1.23 (d, J = 10.1 Hz, 2H), 1.02–0.92 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 153.02, 139.09, 135.84, 105.13, 60.87, 56.04, 39.50, 37.49, 33.78, 33.35, 26.70, 26.37; HRMS-ESI + (m/z): calcd for C17H26O3 [M + H]+: 279.1960; found: 279.1956.
5-(3-Bromo-4-chlorophenethyl)-1,2,3-trimethoxybenzene (32). 106 mg, 72% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.2 Hz, 1H), 7.15–7.02 (m, 2H), 6.36 (s, 2H), 3.85 (d, J = 1.0 Hz, 6H), 3.84 (s, 3H), 2.91–2.83 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 153.08, 141.90, 140.00, 136.57, 133.75, 131.87, 130.00, 128.75, 128.41, 105.38, 60.91, 56.06, 38.19, 37.29, 36.98; HRMS-ESI + (m/z): calcd for C18H19BrClO3 [M + H]+: 385.0206; found: 385.0206.
5-(3-Bromo-4-methoxyphenethyl)-1,2,3-trimethoxybenzene (33). 118 mg, 81% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 1.7 Hz, 1H), 7.10–7.03 (m, 1H), 6.83 (d, J = 8.3 Hz, 1H), 6.37 (s, 2H), 3.89 (s, 3H), 3.85 (d, J = 2.3 Hz, 9H), 2.84 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 154.16, 153.08, 137.10, 136.26, 135.27, 133.27, 128.46, 111.83, 111.36, 105.43, 60.89, 56.30, 56.07, 38.36, 36.73; HRMS-ESI + (m/z): calcd for C18H21BrO4 [M + Na]+: 403.0521; found: 403.0520.
5-(3-Chloro-4-methoxyphenethyl)-1,2,3-trimethoxybenzene (34). 98 mg, 76% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.22 (d, J = 2.0 Hz, 1H), 7.02 (dd, J = 8.3, 2.1 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.37 (s, 2H), 3.91 (s, 3H), 3.85 (d, J = 1.8 Hz, 9H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 153.08, 137.11, 136.24, 134.79, 130.22, 127.68, 122.08, 111.98, 105.40, 60.90, 56.21, 56.07, 38.33, 36.81, 29.72; HRMS-ESI + (m/z): calcd for C18H19F3O5 [M + Na]+: 359.1026; found: 359.1025.
5-(3-Fluoro-4-methoxyphenethyl)-1,2,3-trimethoxybenzene (35). 120 mg, 98% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.97–6.93 (m, 1H), 6.88 (d, J = 6.9 Hz, 2H), 6.37 (s, 2H), 3.89 (s, 3H), 3.85 (d, J = 1.2 Hz, 9H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 153.08, 137.13, 136.25, 134.75, 124.00, 123.97, 116.22, 116.04, 113.36, 105.39, 60.89, 56.40, 56.07, 38.23, 36.95; HRMS-ESI + (m/z): calcd for C18H21FO4 [M + Na]+: 326.1368; found: 326.1367.
5-(3-Fluoro-4-(methoxy-d3) phenethyl-d3)-1,2,3-trimethoxybenzene (36). 109 mg, 88% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.95 (d, J = 12.2 Hz, 1H), 6.92–6.84 (m, 2H), 6.38 (s, 2H), 3.85 (s, 9H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 153.46, 153.07, 151.02, 137.14, 136.22, 134.68, 124.01, 123.97, 116.22, 116.04, 113.33, 105.36, 60.89, 56.06, 38.24, 36.95; HRMS-ESI + (m/z): calcd for C18H18D3FO4 [M + Na]+: 346.1510; found: 346.1509.
5-(4-Fluoro-3-methoxyphenethyl)-1,2,3-trimethoxybenzene (37). 234 mg, 36% yield over two steps. 1H NMR (400 MHz, DMSO-d6) δ 7.08 (dd, J = 8.24, 11.68 Hz, 1H), 7.02 (dd, J = 1.76, 8.52 Hz, 1H), 6.77–6.81 (m, 1H), 6.54 (s, 2H), 3.81 (s, 3H), 3.74 (s, 6H), 3.62 (s, 3H), 2.77–2.87 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 153.12, 149.23, 147.12, 138.84, 137.61, 136.07, 120.84, 115.88, 114.44, 106.10, 60.41, 56.27, 56.21, 38.0, 37.23; HRMS-ESI + (m/z): calcd for C18H21FO4 [M + H]+: 321.1502, found: 321.1508.
5-(4-Fluoro-2-methoxyphenethyl)-1,2,3-trimethoxybenzene (38). 271 mg, 42% yield over two steps. 1H NMR (400 MHz, DMSO-d6) δ 7.13 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 11.44 Hz, 1H), 6.65–6.69 (m, 1H), 6.46 (s, 2H), 3.80 (s, 3H), 3.73 (s, 6H), 3.62 (s, 3H), 2.71–2.82 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 160.90, 158.61, 153.09, 137.87, 136.85, 130.70, 125.78, 106.34, 105.94, 99.34, 60.39, 56.15, 36.16, 31.44; HRMS-ESI + (m/z): calcd for C18H21FO4 [M + H]+: 321.1502, found: 321.1501.
2-Methoxy-5-(3,4,5-trimethoxyphenethyl) benzoic acid (41). 94 mg, 71% yield over two steps. 1H NMR (400 MHz, DMSO-d6) δ 7.52 (s, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 6.54 (s, 2H), 3.78 (s, 3H), 3.74 (s, 6H), 2.87–2.74 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 168.04, 156.77, 153.09, 137.60, 136.05, 133.60, 133.19, 130.96, 121.69, 112.83, 106.14, 60.41, 56.23, 56.20, 39.97, 38.11, 36.42; HRMS-ESI + (m/z): calcd for C19H22O6 [M + Na]+: 369.1314; found: 369.1317.
2-Hydroxyl-5-(3,4,5-trimethoxyphenethyl) benzoic acid (42). 67 mg, 53% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 10.28 (s, 1H), 7.24 (d, J = 2.04 Hz, 1H), 7.33 (dd, J = 2,16, 8.48 Hz, 1H), 6.95 (d, J = 8.48 Hz, 1H), 6.37 (s, 2H), 3.84 (s, 3H), 3.83 (s, 6H), 2.81–2.91 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 174.49, 160.56, 153.10, 137.44, 137.01, 136.27, 132.69, 117.70, 111.00, 105.52, 60. 90, 56.10, 38.28, 36.84; HRMS-ESI + (m/z): calcd for C18H20O6 [M + Na]+: 355.1158; found: 355.1166.
2-Methoxy-5-(3,4,5-trimethoxyphenethyl) pyridine (43). 99 mg, 85% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 2.1 Hz, 1H), 7.38 (dd, J = 8.5, 2.4 Hz, 1H), 6.69 (d, J = 8.5 Hz, 1H), 6.37 (s, 2H), 3.94 (s, 3H), 3.85 (d, J = 2.0 Hz, 9H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 162.78, 153.11, 146.14, 139.06, 136.84, 136.31, 129.37, 110.35, 105.44, 60.89, 56.07, 53.34, 38.16, 34.01; HRMS-ESI + (m/z): calcd for C17H21NO4 [M + Na]+: 326.1368; found: 326.1367.
5-(3,4,5-Trimethoxyphenethyl)-2,3-dihydro-1H-pyrrolo[2,3-b] pyridine (44). 55 mg, 46% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.02 (s, 1H), 6.98 (s, 1H), 6.41 (s, 2H), 3.99 (s, 2H), 3.85 (d, J = 1.9 Hz, 9H), 3.08 (t, J = 8.7 Hz, 2H), 2.85 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 153.04, 137.66, 136.12, 135.57, 127.31, 114.49, 105.33, 77.35, 76.72, 60.89, 56.06, 47.66, 38.75, 37.50, 28.50; HRMS-ESI + (m/z): calcd for C18H22N2O3 [M + Na]+: 337.1528; found: 337.1527.
29 (2.96 mmol, 1.0 eq.), cuprous iodide (0.18 mmol, 0.06 eq.), Xantphos (0.18 mmol, 0.06 eq.) were placed into a Schlenk tube (20.0 mL) with a magnetic stirrer bar. The reaction vessel was evacuated and backfilled with nitrogen three times, then 1,4-dioxane (10.0 mL) was added under a positive nitrogen pressure. Subsequently, tetra(triphenylphosphine) palladium (0.18 mmol) and triethylamine (8.88 mmol, 3.0 eq.) were added. The reaction was heated at 80 °C for 12 h. After cooling to room temperature, the suspension was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography to afford the corresponding product.
To a stirred solution of the above obtained products (1,2-disubstituted acetylene, 0.66 mmol, 1.0 eq.) in THF (6 mL) was added Pd/C (0.3 eq.), and then the reaction vessel was evacuated and backfilled with hydrogen (H2) five times. The mixture was stirred at room temperature for 12 h. After completion of the reaction, the reaction mixture was evaporated under vacuum to afford the crude product which was purified by column chromatography on silica gel (ethyl acetate:hexane = 1:3) to provide the product.
2,6-Dimethyl-4-(3,4,5-trimethoxyphenethyl) pyridine (45). 114 mg, 34.3% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 6.79 (s, 2H), 6.36 (s, 2H), 3.83 (s, 3H), 3.83 (s, 6H), 3.82 (s, 3H), 2.83 (s, 4H), 2.49 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 157.60, 153.14, 150.90, 136.81, 136.42, 120.48, 105.40, 60.86, 56.07, 37.14, 24.37; HRMS-ESI + (m/z): calcd for C18H23NO3 [M + H]+: 302.1756, found: 302.1760.
2-Methyl-4-(3,4,5-trimethoxyphenethyl) quinoline (46). 156 mg, 62.6% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.36 Hz, 1H), 7.97 (d, J = 8.28 Hz, 1H), 7.68 (t, J = 7.52 Hz, 1H), 7.50 (t, J = 7.36 Hz, 1H), 7.09 (s, 1H), 6.36 (s, 2H), 3.84 (s, 3H), 3.81 (s, 6H), 3.33 (t, J = 6.36 Hz, 3H), 3.01 (t, J = 7.48 Hz, 2H), 2.70 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 158.64, 153.23, 148.11, 147.23, 136.81, 136.78, 129.49, 129.09, 125.68, 125.54, 123.15, 121.83, 105.46, 60.90, 56.12, 36.67, 34.08, 25.31; HRMS-ESI + (m/z): calcd for C21H23NO3 [M + H]+: 338.1756, found: 338.1762.
2-Methyl-4-(3,4,5-trimethoxyphenethyl) quinazoline (47). 275 mg, 51.5% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.16 Hz, 1H), 7.96 (d, J = 8.36 Hz, 1H), 7.81–7.86 (m, 1H), 6.46 (s, 2H), 3.82 (s, 3H), 3.81 (s, 6H), 3.52–3.56 (m, 2H), 3.14 (dd, J = 5.92, 8.36 Hz, 2H), 2.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.26, 163.70, 153.23, 150.35, 136.87, 136.52, 133.53, 128.46, 126.60, 124.40, 121.48, 105.50, 60.87, 56.10, 36.46, 35.31, 26.53; HRMS-ESI + (m/z): calcd for C20H22N2O3 [M + H]+: 339.1709, found: 339.1715.
4.2 Molecular docking
The interaction of the designed analogues of erianin with PC was conducted using Discovery Studio 2019. The X-ray crystal structure of PC (PDB ID: 3GB3) was retrieved from the Protein Data Bank (https://www.rcsb.org). In addition, the binding site spheres of PC that were used as the input site sphere were 18.5898, −49.3392, 24.6895, 16.7; the number of hotspots was 100. The conformations of the ligands were allowed to generate tautomers and isomers. Libdock scores were used to evaluate the binding ability with the enzyme.4.3 Bioactivity assay
Author contributions
X. M., X. L., F. W. and G. Z. designed the research. H. S., J. Y., Z. Q. and G. L. synthesized the corresponding compounds. L. L., Q. D. and L. X. evaluated the biological activity. J. W. performed the computational studies. X. M., X. L. and H. S. co-wrote the paper. All the authors discussed the results and commented on the paper.Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
We thank the NMPA Key Laboratory for Quality Monitoring and Evaluation of Traditional Chinese Medicine, Chinese Materia Medica (2023NMPA-CDDC04); the National Natural Science Foundation of China (22001246, T2192971, 21977092); the start-up grant from Chengdu Institute of Biology, Chinese Academy of Sciences; the Biological Resources Program (KFJ-BRP-008) from Chinese Academy of Sciences; and the Sichuan Science and Technology Program (2022ZYD0047, 2022YFS0001), for the financial support.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- J. M. Llovet, J. Zucman-Rossi, E. Pikarsky, B. Sangro, M. Schwartz, M. Sherman and G. Gores, Nat. Rev. Dis Primers, 2016, 2, 16018 CrossRef PubMed.
- A. Villanueva, Y. Hoshida, C. Battiston, V. Tovar, D. Sia, C. Alsinet, H. Cornella, A. Liberzon, M. Kobayashi, H. Kumada, S. N. Thung, J. Bruix, P. Newell, C. April, J. B. Fan, S. Roayaie, V. Mazzaferro, M. E. Schwartz and J. M. Llovet, Gastroenterology, 2011, 140, 1501–1512 CrossRef CAS PubMed.
- T. Li, J. Fan, L. X. Qin, J. Zhou, H. C. Sun, S. J. Qiu, Q. H. Ye, L. Wang and Z. Y. Tang, Ann. Surg. Oncol., 2011, 18, 1955–1963 CrossRef PubMed.
- Y. Y. Shao, W. Y. Shau, S. Y. Chan, L. C. Lu, C. H. Hsu and A. L. Cheng, Oncology, 2015, 88, 345–352 CrossRef CAS PubMed.
- J. Bruix and J. M. Llovet, Hepatology, 2002, 35, 519–524 CrossRef PubMed.
- J. M. Llovet and J. Bruix, Hepatology, 2008, 48, 1312–1327 CrossRef CAS PubMed.
- (a) J. M. Llovet, S. Ricci, V. Mazzaferro, P. Hilgard, E. Gane, J. F. Blanc, A. C. de Oliveira, A. Santoro, J. L. Raoul, A. Forner, M. Schwartz, C. Porta, S. Zeuzem, L. Bolondi, T. F. Greten, P. R. Galle, J. F. Seitz, I. Borbath, D. Häussinger, T. Giannaris, M. Shan, M. Moscovici, D. Voliotis and J. Bruix, N. Engl. J. Med., 2008, 359, 378–390 CrossRef CAS PubMed; (b) A. L. Cheng, Y. K. Kang, Z. Chen, C. J. Tsao, S. Qin, J. S. Kim, R. Luo, J. Feng, S. Ye, T. S. Yang, J. Xu, Y. Sun, H. Liang, J. Liu, J. Wang, W. Y. Tak, H. Pan, K. Burock, J. Zou, D. Voliotis and Z. Guan, Lancet Oncol., 2009, 10, 25–34 CrossRef CAS PubMed; (c) M. Kudo, R. S. Finn, S. Qin, K. H. Han, K. Ikeda, F. Piscaglia, A. Baron, J. W. Park, G. Han, J. Jassem, J. F. Blanc, A. Vogel, D. Komov, T. R. J. Evans, C. Lopez, C. Dutcus, M. Guo, K. Saito, S. Kraljevic, T. Tamai, M. Ren and A. L. Cheng, Lancet, 2018, 391, 1163–1173 CrossRef CAS PubMed.
- J. Chen, J. A. Gingold and X. Su, Trends Mol. Med., 2019, 25, 1010–1023 CrossRef CAS PubMed.
- J. M. Llovet, R. K. Kelley, A. Villanueva, A. G. Singal, E. Pikarsky, S. Roayaie, R. Lencioni, K. Koike, J. Zucman-Rossi and R. S. Finn, Nat. Rev. Dis. Primers, 2021, 7, 6 CrossRef PubMed.
- J. C. Wallace, Distribution and biological functions of pyruvate carboxylase in nature, Pyruvate Carboxylase, ed. D. B. Keech and J. C. Wallace, CRC Press, Boca Raton, 1985, pp. 5–64 Search PubMed.
- (a) T. Cheng, J. Sudderth, C. Yang, A. R. Mullen, E. S. Jin, J. M. Matés and R. J. DeBerardinis, Natl. Acad. Sci., 2011, 108, 8674–8679 CrossRef CAS PubMed; (b) C. T. Hensley and R. J. DeBerardinis, J. Clin. Invest., 2015, 125, 495–497 CrossRef PubMed; (c) S. Cardaci, L. Zheng, G. MacKay, N. J. F. van den Broek, E. D. Mackenzie, C. Nixon, D. Stevenson, S. Tumanov, V. Bulusu, J. J. Kamphorst, A. Vazquez, S. Fleming, F. Schiavi, G. Kalna, K. Blyth, D. Strathdee and E. Gottlieb, Nat. Cell Biol., 2015, 17, 1317–1326 CrossRef CAS PubMed; (d) T. W. M. Fan, A. N. Lane, R. M. Higashi, M. A. Farag, H. Gao, M. Bousamra and D. M. Miller, Mol. Cancer, 2009, 8, 41 CrossRef PubMed; (e) O. Warburg, Science, 1956, 123, 309–314 CrossRef CAS PubMed; (f) N. S. Forbes, A. L. Meadows, D. S. Clark and H. W. Blanch, Metab. Eng., 2006, 8, 639–652 CrossRef CAS PubMed.
- T. N. Zeczycki, M. S. Maurice and P. V. Attwood, Open Enzyme Inhib. J., 2010, 3, 8–26 CrossRef CAS PubMed.
- Q. Lin, Y. He, X. Wang, Y. Zhang, M. Hu, W. Guo, Y. He, T. Zhang, L. Lai, Z. Sun, Z. Yi, M. Liu and Y. Chen, Adv. Sci., 2020, 7, 1903483 CrossRef CAS PubMed.
- P. L. MaJumder and M. Joardar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1984, 23(11), 1040–1042 Search PubMed.
- (a) X. Zhang, Y. Wang, X. Li, A. Yang, Z. Li and D. Wang, Aging, 2019, 11, 10284–10300 CrossRef CAS PubMed; (b) X. Liu, S. Dong, M. Dong, Y. Li, Z. Sun, X. Zhang, Y. Wang, L. Teng and D. Wang, Int. J. Pharm., 2021, 607, 121034 CrossRef CAS PubMed; (c) H. Dong, M. Wang, C. Chang, M. Sun, F. Yang, L. Li, M. Feng, L. Zhang, Q. Li, Y. Zhu, Y. Qiao, T. Xie and J. Chen, Biochem. Pharmacol., 2020, 182, 114266 CrossRef CAS PubMed.
- Y. Sheng, Y. Chen, Z. Zeng, W. Wu, J. Wang, Y. Ma, Y. Lin, J. Zhang, Y. Huang, W. Li, Q. Zhu, X. Wei, S. Li, W. Wisanwattana, F. Li, W. Liu, A. Suksamrarn, G. Zhang, W. Jiao and F. Wang, J. Med. Chem., 2022, 65, 460–484 CrossRef CAS PubMed.
- H. Zhou, B. Yang, M. Hong, R. Ma and L. Sheng, Arzneim. Forsch., 2009, 59, 141–145 CAS.
- X. Yi and X. Lan, Biomed. Chromatogr., 2020, 34, e4826 CAS.
- G. R. Pettit, B. Toki, D. L. Herald, P. Verdier-Pinard, M. R. Boyd, E. Hamel and R. K. Pettit, J. Med. Chem., 1998, 41, 1688–1695 CrossRef CAS PubMed.
- H. H. Ko, L. T. Tsao, K. L. Yu, C. T. Liu, J. P. Wang and C. N. Lin, J. Med. Chem., 2003, 11, 105–111 CAS.
- M. Y. Song, Q. R. He, Y. L. Wang, H. R. Wang, T. C. Jiang, J. J. Tang and J. M. Gao, Int. J. Mol. Sci., 2020, 21, 1817 CrossRef CAS PubMed.
- M. Medarde, R. P. L. Clairac, A. C. Ramos, E. Caballero, J. L. López, D. G. Grávalos and A. S. Feliciano, Bioorg. Med. Chem. Lett., 1995, 5, 229–232 CrossRef CAS.
- R. Shirai, K. Tokuda, Y. Koiso and S. Iwasaki, Bioorg. Med. Chem. Lett., 1994, 4, 699–704 CrossRef CAS.
- A. Marset, D. Caprioglio, S. Torretta, G. Appendino and A. Minassi, Tetrahedron Lett., 2016, 57, 1540–1543 CrossRef CAS.
- Z. l. Ma, X. j. Yan, L. Zhao, J. j. Zhou, W. Pang, Z. p. Kai and F. h. Wu, J. Agric. Food Chem., 2016, 64, 746–751 CrossRef CAS PubMed.
- F. H. Wu, Z. L. Ma, Z. P. Kai, L. L. Huang, D. D. Li and J. W. Huang, CN107365248A, 2017, 11. 21.
- A. B. S. Maya, C. Pérez-Melero, C. Mateo, D. Alonso, J. L. Fernández, C. Gajate, F. Mollinedo, R. Peláez, E. Caballero and M. Medarde, J. Med. Chem., 2005, 48, 556–568 CrossRef CAS PubMed.
- R. Martín, P. Prieto, J. R. Carrillo, A. M. Rodríguez, A. de Cozar, P. G. Boj, M. A. Díaz-García and M. G. Ramírez, J. Mater. Chem. C, 2019, 7, 9996–10007 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01114c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |