
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoisomeric enrichment of 1,4-enediols and H2-splitting inhibition on Pd-supported catalysts†
Jordi
Ballesteros-Soberanas
,
Marta
Mon
and
Antonio
Leyva-Pérez
 *
*
Instituto de Tecnología Química (UPV-CSIC), Universidad Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: anleyva@itq.upv.es
First published on 16th August 2023
Abstract
Pd-supported catalysts are fundamental tools in organic reactions involving H2 splitting. Here we show that 1,4-enediols enriched in one diastereoisomer are produced from the classical Pd-catalyzed semi-hydrogenation reaction with H2, starting from the corresponding, widely available 1,4-diacetylenic diols. The semi-hydrogenation reaction proceeds concomitantly with the desymmetrization of the meso/racemic form of the enediol. We also show that these products, if added in advance to H2, completely inactivate the Pd catalyst (only when added before H2). These results provide a simple way not only to produce 1,4-enediols enriched in one diastereoisomer by a classical catalytic method but also to stop H2 dissociation on Pd nanoparticles.
1 Introduction
Pd supported catalysts such as Pd on carbon (Pd/C) and the Lindlar catalyst (Pd–Pb–CaCO3) are recurrently used in organic synthesis to catalyze the hydrogenation of reducible functional groups.1 The challenge usually consists in stopping the reaction at the desired functionality, since the Pd catalyst splits and transfers H2 so efficiently that over-hydrogenation reactions occur (alkyne → alkene → alkane, nitro → oxime → amine,…).2,3 Thus, the strong ability of Pd catalysts to activate H2 is largely presupposed by the scientific community.We report here that the semi-hydrogenation reaction of 1,4-acetylene diols 1 on simple Pd catalysts proceeds in a diastereoselective way to give 1,4-enediols 2 (meso form) preferentially and 3 (racemic form) and, furthermore, that the 1,4-enediol products 2 and3 completely poison the Pd catalyst inhibiting further splitting of H2, as shown in Fig. 1. The drastic inhibition of H2 reactivity on Pd-supported catalysts by complex molecules is extremely rare and opens new ways to manage H2 gas in the presence of Pd catalysts. This effect may also explain the spontaneous diastereoisomeric enrichment found during the semi-hydrogenation reaction of these substrates.
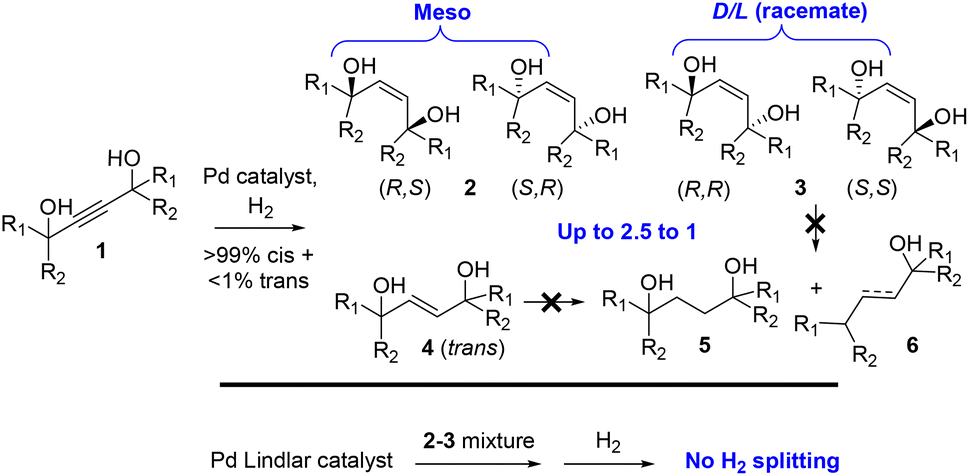 | ||
| Fig. 1 The spontaneous diastereo-enriched formation of alkenediols 2 during the semi-hydrogenation reaction of alkynes 1 catalyzed by classical Pd catalysts (i.e. Lindlar catalyst) and the complete inhibition of H2 splitting when the Pd catalyst is previously impregnated with 2 and3. | ||
1,4-Enediols constitute a family of gemini-type compounds with wide use in industry as emulsifiers, fragrances and drugs, among other applications.4–6 Their synthesis relies on two methodologies: the classical cis-selective Pd-catalyzed semi-hydrogenation of 1,4-acetylene diols, studied in this work in depth for the first time as far as we know, and the trans-selective semi-hydrogenation of the same alkynes with stoichiometric LiAlH4.7,8
We could not find specific studies for the semi-hydrogenation reaction of 1,4-acetylene diols in the literature, except for a seminal study published 70 years ago,9 which focused on the competing full hydrogenation and hydrogenolysis reactions of 2–4, to give products 5 and 6. However, synthetic researchers recurrently employ the stoichiometric method with LiAlH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,8 and also the classical catalytic hydrogenation to prepare a variety of 1,4-enediols, from industrial compounds10 to more elaborate academic molecules.11–17 However, a significant diastereo enrichment (>1.5:1) of the meso form is not reported yet, to our knowledge.9,11
7,8 and also the classical catalytic hydrogenation to prepare a variety of 1,4-enediols, from industrial compounds10 to more elaborate academic molecules.11–17 However, a significant diastereo enrichment (>1.5:1) of the meso form is not reported yet, to our knowledge.9,11
2 Results and discussion
2.1 Pd-catalyzed semi-hydrogenation reaction of 1,4-diacetylenic diols
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (metal mol%) | 2a (%) | 3a (%) | 5a/6a (%) | 2a/3a |
| a 10 bar H2, 12 h reaction time, 1 M EtOH. b 1 M toluene as a solvent, 12 h reaction time. | |||||
| 1 | Lindlar (0.15) | 53 (58)a (64)b | 34 (29)a (25)b | 13 (0)a (0)b | 1.55 (2.00)a (2.50)b |
| 2 | c-Pd/TiS (0.015) | 59 | 34 | 7 | 1.44 |
| 3 | Pd-(CaCO3)n (0.003) | 53 | 42 | 5 | 1.26 |
| 4 | Pd/C (0.04) | 15 | 9 | 76 | 1.60 |
| 5 | Pt/C (0.018) | 28 | 20 | 52 | 1.37 |
| 6 | Ni-RANEY® (25) | 25 | 21 | 54 | 1.15 |

The catalytic results show that small amounts of the different Pd catalysts (0.003–0.15 mol%, Table 1, entries 1–3) give on average an ∼1.5 to 1 ratio of the meso form 2a with respect to the D/L form 3a, with low formation of trans4a or alkane products 5a–6a. However, according to the experiments performed with the Lindlar catalyst (entry 1), the diastereoisomeric ratio can be increased by changing the reaction conditions (pressure, reaction time, and solvent): the diastereoisomeric meso:D/L ratio (2a/3a) improves to 2 when increasing H2 pressure and concentration and to 2.5 when changing the solvent from ethanol to toluene (see also Table S1†). The quantification of the different regioisomers is based on the assignation of the corresponding signals by combined gas-chromatography-mass spectrometry (GC-MS, Fig. S1†), 1H, 13C and distortionless enhancement by polarization transfer (DEPT) nuclear magnetic resonance (NMR, Fig. S2†), including 2D-nuclear Overhauser enhancement spectroscopy (NOESY, Fig. S3†), the independent synthesis of the trans product 4a by the LiAlH4 method, and comparison with literature data.9,14 The commercial non-selective catalysts exhibit prominent alkane formation along with undesired hydrogenolytic reactions (entries 4–6 in Table 1, see also Table S2†). The significant diastereo enrichment found here with Pd catalysts (up to 2.5:1) is much higher than any results previously reported.9,11
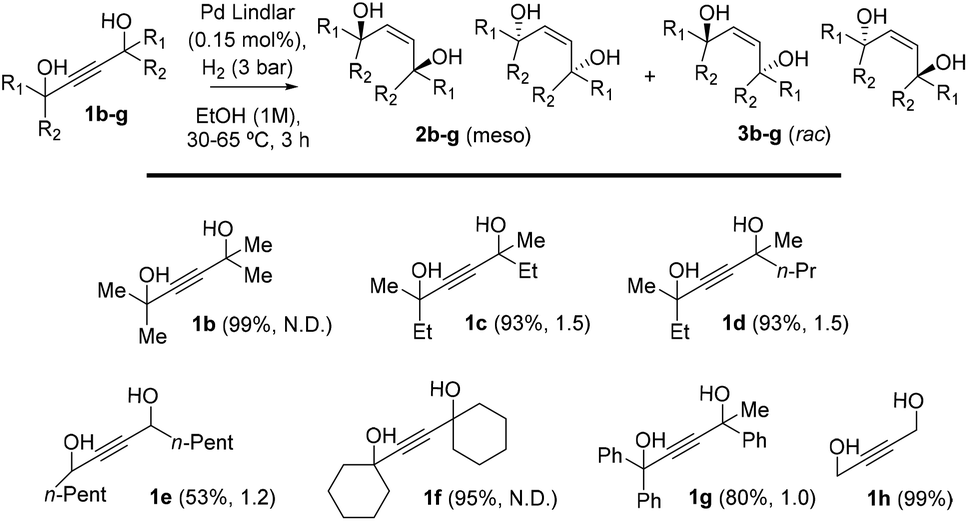 | ||
| Fig. 2 Catalytic results for different 1,4-acetylene diols 1b–h during the semi-hydrogenation reaction under the indicated reaction conditions (see also Table S3†). Within parentheses are shown the reaction yield and diastereoisomeric ratio. N.D.: isomers not distinguishable by GC and NMR. | ||
 | ||
| Fig. 3 The proposed mechanism for the diastereo-differentiation found in products 2a/3a. Energy values correspond to the minimum stabilization energy found for the corresponding molecules and intermediates according to molecular mechanics calculations in a vacuum (MM2). | ||
To check that the epimerization reaction occurs in alkyne 1a without requiring the participation of H2, the semi-hydrogenation products 2a/3a were also placed in the presence of the Lindlar catalyst and H2O(18O) (as above), and the results (see again Table S4 and Fig. S4†) show that the quaternary C–OH bonds in 2a/3a incorporate just 6% of 18O atoms. This is congruent with the higher stability of the propargyl cation. Molecular mechanics calculations (MM2) show that the stabilization energy for the propargyl cation is just 12 kcal mol−1, and that the minimum stabilization energy for the meso form of 1a is slightly higher than that of the rac form, although product meso2a is slightly less stable than rac3a (see Fig. 3). These results support the fact that the epimerization occurs on the alkyne products rather than on the alkene products. The incorporation of 18O atoms under H2 could not be conveniently assessed by the coincidence of masses in the hydrogenated products and the isotopically-labelled products.
It is true that the reaction does not canonically produce water, but water is not strictly necessary for the epimerization, since the simple formation of the highly stabilized propargyl cation of alkyne 1a generated a nucleophilic OH− to carry out the epimerization reaction (see Fig. 3). Indeed, H2O must be present in traces during the reaction, supplied by the reactants, solvent or catalyst support. Even any trace of air in the reaction system will produce water after reaction with H2. Thus, some water molecules must be present during the reaction anyway.
This mechanism is in line with the substrate scope results shown in Fig. 2. There, it can be seen that tertiary alcohols that are able to stabilize better the carbocation in the tertiary carbon atom, after H2O release, are indeed the higher yielding substrates (i.e. products 1a–d and 1f), while secondary alcohols and tertiary alcohols that are not so prone to stabilize the carbocation only gave moderate results (products 1e and 1g, respectively).
2.2 Inhibition of H2 splitting on the Pd catalyst
:40, Fig. S5†), which is extremely rare considering that the reaction proceeds from 1a to 2a/3a. Fig. 4 shows that this lack of catalytic activity of the Lindlar catalyst when 1a and 2a/3a are present from the beginning of the reaction also occurs when the semi-hydrogenation reaction is halted by simply releasing the H2 atmosphere and letting air seep into the reaction mixture, at ∼50–70% conversion (>99% selectivity to 2a/3a). Notice that pressurizing back with H2 does not re-start the reaction and, indeed, the inhibition persists if the Lindlar catalyst is filtered off and washed with ethanol (Fig. S5†). The inhibition effect also occurs in toluene (Fig. S6 and S7†), and thus it seems to be independent of the solvent used, and the trans enediol 4a also inhibits the catalyst. In contrast, the reaction proceeds to completion if H2 is present from the beginning. Moreover, if the reaction time is extended further and more Pd catalyst is added in order to achieve an over-hydrogenation reaction and get the alkane product 5a/6a, the solid then retains some of the catalytic activity after recovery (Fig. S5†). These combined results suggest that the enediol products 2a–4a poison the Lindlar catalyst in the hydrogenation reaction, provided that H2 is not introduced before, since in that case the semi-hydrogenation reaction proceeds normally.
 | ||
| Fig. 4 Kinetics for the semi-hydrogenation reaction of TMDD 1 catalyzed by the Lindlar catalyst (0.5 mol%) at 10 bar H2, 1 M EtOH and 65 °C, either continuous (left) or interrupted after H2 release and re-filling (right). | ||
 | ||
| Fig. 5 Results for the hydrogenation reaction of nitrobenzene 7 (top) and acetophenone 9 (bottom) in the presence of different Pd-supported catalysts (Lindlar or Pd/C) with or without impregnation with enediols 2a/3a, at 3 bar H2 and 0.6 M toluene and under the indicated reaction conditions, optimized for each catalyst and reaction. Selectivity towards the indicated products is >95% in all cases, and nitroso or oxime intermediates were not found for 7. Notice that for 9, the fully hydrogenated product 11 is the major product without 2a/3a. | ||
 | ||
| Fig. 6 H2–D2 isotopic exchange experiments, at 30 °C, for the fresh Lindlar catalyst (top) and poisoned Lindlar catalyst, with the 2a + 3a mixture (bottom). | ||
Fourier-transform infrared (FT-IR) spectroscopy supports the strong binding of the diol to the support (Fig. S10†). It can be seen that the original signal at 1441 cm−1, corresponding to the carbonate groups of the Lindlar catalyst, decreases in intensity after adding the 1,4 enediols 2a/3a, with the appearance of a new band at 1404 cm−1. This new band does not correspond to free 2a/3a, as indicated by the relative intensity of the main 2a/3a signals (i.e. 2951 vs. 1364 cm−1). A fast evaluation of the adsorption mode of alkyne 1a and alkene 2a on the surface of different Pd crystalline slabs [(100), (110) and (111)] was carried out with a graph neural network called GAME-Net.39 The results (Fig. S11†) show that the adsorption process is exothermic in all cases and that the adsorption energy for the alkenes is systematically higher than for the alkyne, >25 kcal mol−1 in the case of the more common Pd(111) surface. By modelling the latter (Fig. S12†), it can be seen how alkenes 2a/4a are disposed parallelly to the Pd(111) surface, regardless of the cis/trans configuration, with the alkene and alcohol groups pointed towards the Pd atoms. These results explain that both 2a and 4a poison the Pd surface. The simplest gemini alkynediol 2-butyn-1,4-diol 1h also shows similar energetic values on Pd surfaces (Fig. S11†). The above results, together, confirm the strong binding of 1,4-enediols to Pd-supported catalysts, inhibiting the H2 activation, which could also have application in the synthesis of ligand (1,4-enediols)-modified Pd nanoparticles for a variety of applications.
3 Conclusions
We have shown here that the semi-hydrogenation reaction of 1,4-diacetylenic diols catalyzed by different supported Pd catalysts preferentially gives the meso form of the enediol, probably by a selective C–OH bond epimerization process, also catalyzed by Pd. The resulting 1,4-enediol products inhibit the dissociation of H2 on the Pd-supported catalysts if added in the beginning of the hydrogenation reaction, when H2 is not present yet in the reaction mixture, not only for alkynes but also for nitro and ketone groups. The adsorption mode and strength of 1,4-enediols seem to be the key factors here. These results are of interest to design hydrogenation reactions with classical Pd catalysts.Author contributions
J. B.-S. performed most of the experimental work and analysis, M. M. studied the scope of functional groups and catalysts, and A. L.-P. supervised the project and wrote the manuscript.Conflicts of interest
Two patents covering some of these results have been filed: PCT/ES2022/070672 (co-authored by J. B.-S. and A. L.-P.) and P202330474 (co-authored by J. B.-S., M.-M. and A. L.-P.).Acknowledgements
Financial support by the projects PID2020-115100GB-I00 (funded by Spanish MCIINN, MCIN/AEI/10.13039/501100011033MICIIN), Severo Ochoa Centre of Excellence Program (CEX2021-001230-S) and “La Caixa” Foundation grant (ID 100010434, code LCF/BQ/DI19/11730029) is acknowledged. M. M. thanks MICIIN for support from a contract under the Juan de la Cierva program (FJC2019-040523-I).References
- A. O. King, R. D. Larsen and E. Negishi, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, 2002, pp. 2719–2752 Search PubMed.
- B. Bridier, N. López and J. Pérez-Ramírez, Dalton Trans., 2010, 39, 8363–8376 RSC.
- A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112–1115 CrossRef CAS PubMed.
- C. S. Poss and S. L. Schreiber, Acc. Chem. Res., 1994, 27, 9–17 CrossRef CAS.
- M. Hans, M. Charpentier, V. Huch, J. Jauch, T. Bruhn, G. Bringmann and D. Quandt, J. Nat. Prod., 2015, 78, 2381–2389 CrossRef CAS PubMed.
- F. M. Menger, J. S. Keiper and V. Azov, Langmuir, 2000, 16, 2062–2067 CrossRef CAS.
- R. K. Hill, S. L. Pendalwar, K. Kielbasinski, M. F. Baevsky and P. N. Nugara, Synth. Commun., 1990, 20, 1877–1884 CrossRef CAS.
- L. Grigorjeva, A. Kinens and A. Jirgensons, J. Org. Chem., 2015, 80, 920–927 CrossRef CAS PubMed.
- R. J. Tedeschi, J. Org. Chem., 1962, 27, 2398–2402 CrossRef CAS.
- Y. Qiu, X. Gao, J. Kass, H. W. Cao, X. Li, X. Peng, B. Suh and Y. S. Or, US20190060258A1, 2018.
- D. Finnegan, B. A. Seigal and M. L. Snapper, Org. Lett., 2006, 8, 2603–2606 CrossRef CAS PubMed.
- H. Mouhib, W. Stahl, M. Lüthy, M. Büchel and P. Kraft, Angew. Chem., Int. Ed., 2011, 50, 5576–5580 CrossRef CAS PubMed.
- K. B. Bazhykova, Russ. J. Org. Chem., 2021, 57, 203–211 CrossRef CAS.
- X. Ariza, N. Fernández, J. Garcia, M. López, L. Montserrat and J. Ortiz, Synthesis, 2004, 128–134 CAS.
- M. J. Burk, J. E. Feaster and R. L. Harlow, Tetrahedron: Asymmetry, 1991, 2, 569–592 CrossRef CAS.
- J. Bach, R. Berenguer, J. Garcia, T. Loscertales, J. Manzanal and J. Vilarrasa, Tetrahedron Lett., 1997, 38, 1091–1094 CrossRef CAS.
- M. Amador, X. Ariza and J. Garcia, Heterocycles, 2006, 67, 705–720 CrossRef CAS PubMed.
- A. A. Guedez, S. Frömmel, P. Diehl and W. Püttmann, Environ. Sci. Pollut. Res., 2010, 17, 321–330 CrossRef CAS PubMed.
- K. Vincze, M. Gehring and T. Braunbeck, Environ. Sci. Pollut. Res., 2014, 21, 8233–8241 CrossRef CAS PubMed.
- E. Minnich, EP0940169A1, 1999.
- G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J., 2014, 20, 5926–5937 CrossRef PubMed.
- P. T. Witte, P. H. Berben, S. Boland and J. G. Donkervoort, Top. Catal., 2012, 55, 505–511 CrossRef CAS.
- J. A. Delgado, O. Benkirane, C. Claver, D. Curulla-Ferré and C. Godard, Dalton Trans., 2017, 46, 12381–12403 RSC.
- J. Ballesteros-Soberanas, J. C. Hernández-Garrido, J. P. Cerón-Carrasco and A. Leyva-Pérez, J. Catal., 2022, 408, 43–55 CrossRef CAS.
- J. Ballesteros-Soberanas, J. A. Carrasco and A. Leyva-Pérez, J. Org. Chem., 2023, 88, 18–26 CrossRef CAS PubMed.
- M. Tejeda-Serrano, M. Mon, B. Ross, F. Gonell, J. Ferrando-Soria, A. Corma, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2018, 140, 8827–8832 CrossRef CAS PubMed.
- P. McNeice, M. A. Müller, J. Medlock, W. Bonrath, N. Rockstroh, S. Bartling, H. Lund, K. Junge and M. Beller, ACS Sustainable Chem. Eng., 2022, 10, 9787–9797 CrossRef CAS.
- S. Mao, B. Zhao, Z. Wang, Y. Gong, G. Lü, X. Ma, L. Yu and Y. Wang, Green Chem., 2019, 21, 4143–4151 RSC.
- H. Fernández-Pérez, P. Etayo, J. R. Lao, J. L. Núñez-Rico and A. Vidal-Ferran, Chem. Commun., 2013, 49, 10666–10675 RSC.
- W. Hong, W. A. Swann, V. Yadav and C. W. Li, ACS Catal., 2022, 12, 7643–7654 CrossRef CAS.
- H. J. Hu, Q. Q. Wang, D. X. Wang and Y. F. Ao, Adv. Synth. Catal., 2021, 363, 4538–4543 CrossRef CAS.
- D. E. Ward, Y. Liu and D. How, J. Am. Chem. Soc., 1996, 118, 3025–3026 CrossRef CAS.
- X. Jia, J. Ma, F. Xia, Y. Xu, J. Gao and J. Xu, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- T. R. Hoye and J. C. Suhadolnik, J. Am. Chem. Soc., 1985, 107, 5312–5313 CrossRef CAS.
- H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 15411–15414 CrossRef CAS PubMed.
- P. W. Albers, K. Möbus, C. D. Frost and S. F. Parker, J. Phys. Chem. C, 2011, 115, 24485–24493 CrossRef CAS.
- M. Armbrüster, M. Behrens, F. Cinquini, K. Föttinger, Y. Grin, A. Haghofer, B. Klötzer, A. Knop-Gericke, H. Lorenz, A. Ota, S. Penner, J. Prinz, C. Rameshan, Z. Révay, D. Rosenthal, G. Rupprechter, P. Sautet, R. Schlögl, L. Shao, L. Szentmiklósi, D. Teschner, D. Torres, R. Wagner, R. Widmer and G. Wowsnick, ChemCatChem, 2012, 4, 1048–1063 CrossRef.
- M. García-Mota, B. Bridier, J. Pérez-Ramírez and N. López, J. Catal., 2010, 273, 92–102 CrossRef.
- S. Pablo-García, S. Morandi, R. A. Vargas-Hernández, K. Jorner, Ž. Ivković, N. López and A. Aspuru-Guzik, Nat. Comput. Sci., 2023, 3, 433–442 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S12, Tables S1–S5, compound characterization and NMR copies. See DOI: https://doi.org/10.1039/d3ob01025b |
| This journal is © The Royal Society of Chemistry 2023 |