
Synthetic strategies for the construction of C3-fluorinated oxindoles
Deeksha
,
Bittu
and
Ritesh
Singh
 *
*
Department of Chemistry, Central University of Rajasthan, Ajmer, Rajasthan 305817, India. E-mail: ritesh.singh@curaj.ac.in
First published on 28th July 2023
Abstract
C3-fluorinated oxindoles are important scaffolds known to demonstrate various biological properties. As bio-isosteres of oxindoles, these compounds have shown tremendous potential in drug research discovery programs. Besides, they also serve as starting materials for synthesizing other fluorine-containing new architectures, thus launching research for developing new methods for their synthesis. Consequently, various approaches have been developed over the years to synthesize C3-fluorinated oxindoles. This review highlights the strategies developed to date to access C3-difluoro and monofluorooxindoles via intermolecular and intramolecular approaches. The key findings of the strategies developed are discussed along with the prevailing mechanism.
 Deeksha | Deeksha was born in 1994 at Bhiwani (Haryana), India. She obtained her B.Sc. degree in 2016 from Maharshi Dayanand University, Rohtak, India, and M.Sc. (2018) in organic chemistry from Guru Jambheshwar University of Science and Technology, Hisar, India. Currently, she is pursuing her Ph.D. in the Department of Chemistry at the Central University of Rajasthan under the supervision of Dr Ritesh Singh. Her research work focuses on C–H bond functionalization and aryne chemistry. |
 Bittu | Bittu was born in 2003 at Badalwas, Jhunjhunu (Rajasthan), India. She graduated from Vivekananda Public School, Khetri, Rajasthan, India. She obtained her B.Sc. degree in 2023 from the Central University of Rajasthan, Ajmer, India. |
 Ritesh Singh | Dr Ritesh Singh earned his doctoral degree from the CSIR-Central Drug Research Institute, Lucknow. He then moved to the University of Rochester, USA for his postdoctoral stay with Prof. Rudi Fasan working on chemoenzymatic synthesis (bio-catalysis). He started his independent career at the CSIR-IICT, Hyderabad as INSPIRE faculty. He also received the prestigious JSPS postdoctoral fellowship to conduct research at the Kyoto Prefectural University of Medicine (KPUM), Kyoto, Japan, with Prof. Takayoshi Suzuki. After serving at the NIPER-Raebareli for a year as a faculty, he joined the Central University of Rajasthan in 2019 as an Assistant Professor. His research interest lies in developing novel synthetic strategies to access biologically relevant scaffolds and natural products via C–H bond functionalization and in developing metal-free approaches to synthesize important pharmaceutical molecules. |
1. Introduction
Organofluorine compounds have garnered significant attention from synthetic and medicinal chemists due to their widespread application in pharmaceuticals and agrochemicals. The introduction of fluorine atoms into organic compounds significantly influences the compounds’ electronic, physical, and biological properties. Owing to the unique properties that the fluorine atom brings into these molecules, like increased lipophilicity, lower pka, and increased bioavailability, it has become the kingpin in the drug discovery program.1 Therefore, introducing fluorine atoms into privileged scaffolds is an active and important research area. In this context, oxindole constitutes a ‘privileged’ core found in numerous pharmaceuticals, natural products, and agrochemicals.2 However, its bio-isosteres, 3,3-difluorinated/3-fluorinated oxindoles, have shown tremendous potential in drug research discovery programs and have created a niche in medicinal chemistry.3 For instance, C3-difluorinated oxindoles act as an apoptosis inhibitor (A)3a and EP3 receptor antagonist (B),3b and are also used for the treatment of abnormal cell growth (C),3c whereas C3-mono-fluorinated oxindoles like MaxiPost (D) treat acute ischemic stroke,3d and behave as a cannabinoid receptor CB2 agonist (E)3e and a kinase activator (F)3f (Fig. 1).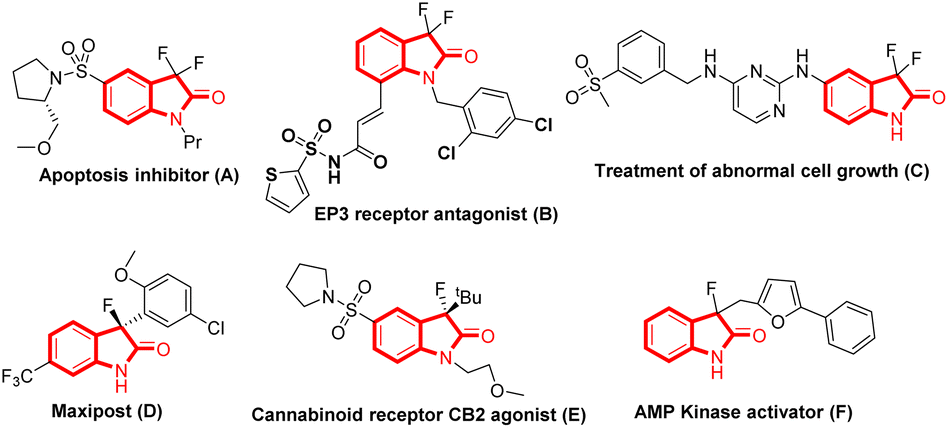 | ||
| Fig. 1 Bio-active C3-difluoro/monofluoro oxindoles. | ||
Though simple oxindoles were first synthesized in 1866 by reducing isatins,2d synthesis of C3-fluorinated oxindoles remained elusive for a long time, perhaps due to the lack of any available general method. Thus, developing new and efficient strategies is highly desirable owing to the immense biological significance of C3-mono/difluoro oxindoles and the limited methods available to access such scaffolds. Consequently, various research groups have reported several new approaches for constructing C3-fluorinated oxindoles. Although recently a review article describing the approaches to access stereogenic 3-fluorinated carbon centres from 3-fluorooxindoles has been published,3g a comprehensive review enumerating the strategies developed to date for the construction of C3-di/monofluoro oxindoles is not available, which will be highly beneficial for researchers in this area. We have organized this review based on the intermolecular and intramolecular strategies developed for C3-difluorinated and C3-monofluorinated oxindoles, wherein the strategies have been discussed along with the analysis of the prevailing mechanism.
2. C-3 difluorinated oxindoles
2.1 Intermolecular approach
 | ||
| Scheme 1 Synthesis of C3 difluorinated oxindoles from isatins using DAST as the fluorinating reagent. | ||
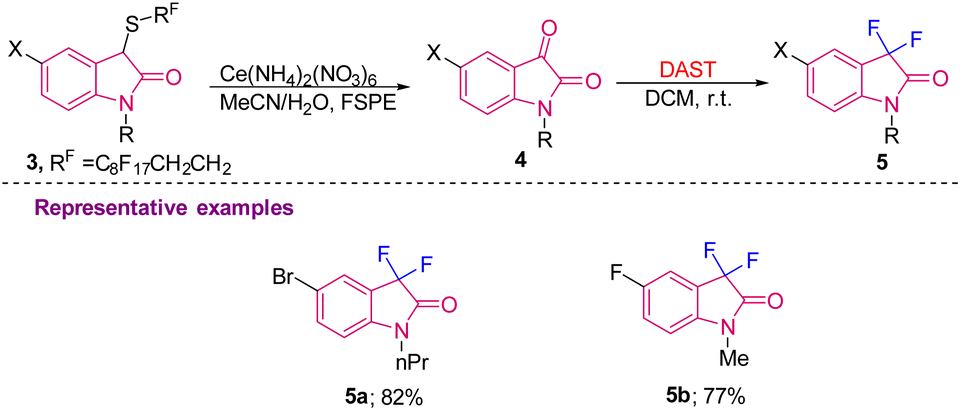
The above strategy was later used by Procter's group in 2007 to generate difluorinated oxindoles 5 using DAST in DCM at room temperature from oxindole derivatives 4 prepared via sequential oxidative cleavage of 3via the Pummerer process, which were further separated by the FSPE (fluorous solid-phase extraction) method. 5-F/Br substituted difluoro oxindoles were synthesised in 77 and 82% yields, respectively (Scheme 2).10
 | ||
| Scheme 2 Synthesis of C3-difluorinated oxindoles using DAST via FSPE (fluorous solid-phase extraction). | ||
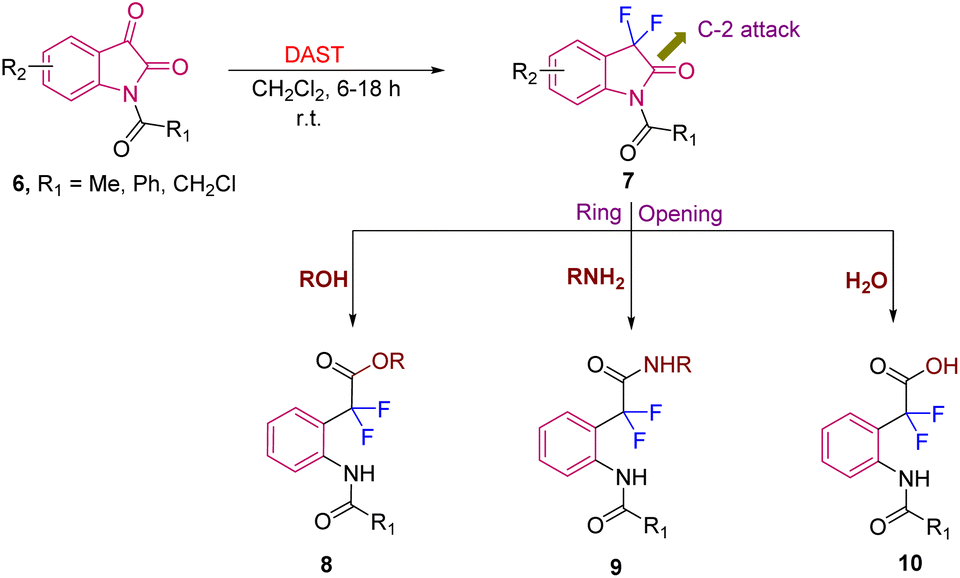
Furthermore, Boechat et al., in 2008, demonstrated the versatility of the obtained difluorinated oxindoles 7 (synthesized from N-acyl isatin 6 (>90% yield) using DAST) by converting them into other difluorinated scaffolds. The difluorinated oxindoles 7 in the presence of nucleophiles like ROH, RNH2 and H2O readily underwent ring opening via attack at the C-2 position of 7, due to their enhanced electrophilicity because of the presence of two-fluorine atoms at C-3 and the acyl group at N-1, leading to esters 8 (>75% yield), amides 9 (49–80% yields), and acids 10 (>94% yield), respectively (Scheme 3).11
 | ||
| Scheme 3 Synthesis of C3-difluorinated oxindoles using DAST followed by ring opening. | ||
To increase the efficiency of the method, Ley's group used a modular flow microreactor to synthesize the desired C3-difluorooxindoles 12 in 71% yield using isatin as the starting material, DAST as a fluorinating agent in DCM/MeCN, wherein isatin was injected from Pump 1, and the mixture of DAST in DCM and MeCN was injected from Pump 2, and both were finally mixed at the T point, from where the solution moved to a CFC (convection flow coil) at 80 °C for 45 minutes and further passed through an Omnifit glass column filled with CaCO3 and SiO2 to give the pure product 12 (Scheme 4).12
 | ||
| Scheme 4 Synthesis of C3 difluorinated oxindoles using DAST via a modular/continuous flow microreactor. | ||
Later in the same year, Ley's group again developed the method for fluorinating 11 using a continuous-flow microreactor. Since DAST is volatile, corrosive, reacts explosively with water at temperatures >90 °C and undergoes dissociation to give side products, such as SF4 and (Et2N)2SF2, to overcome these limitations and to improve yield, Ley came up with a better strategy that includes the use of a Vapourtec R2+/R4 modular device, which can load corrosive DAST mounted on the R2+ module. The substrate and DAST in DCM and MeCN were mixed at the T point and then transferred to the CFC at 90 °C for 27 min; high temperature led to boiling of the DCM solvent, which was suppressed by a BPR (back pressure regulator) at 100 psi and then the mixture was passed through a glass column for quenching of residual DAST and removal of side products that provided the pure product 12 in 73% yield.13
Later, Haufe et al. in 2009 used the same strategy of reacting isatin with nucleophilic fluorinating agent DAST for the synthesis of substituted C3-difluorinated oxindoles that possess caspase inhibitor activity. They synthesised caspase inhibitor 15, which was obtained from the lead structure (S)-5-(1-(2-methoxymethylpyrodidinyl) sulfonyl) isatin 13 that was alkylated with 3-bromopropanol using K2CO3 as base to obtain 14. Compound 14 was then treated with DAST in DCM at −78 °C, which furnished difluoro 5-pyrrolidinyl sulfonyl isatin 15 in moderate yields (Scheme 5).14
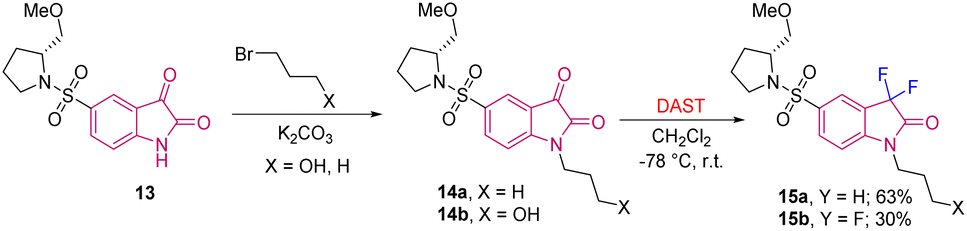 | ||
| Scheme 5 Synthesis of difluoro 5-pyrrolidinyl sulfonyl isatin (caspase inhibitor) using DAST. | ||
The medicinal utility of the C3-difluorooxindoles was further demonstrated by Singh's group, in 2010, where they used methyl ester derivatives of isatin 16a–c to synthesize 1,7-disubstituted oxindoles 19a–c, potent EP3 receptor inhibitors. The synthesis started from 16a–c, which were converted into gem-difluoro derivatives 17a–c in 90–99% yields using DAST in DCM. Saponification of 17, followed by EDCI-mediated coupling of 18a–c provided the final bio-active compounds 19a–c in 62–80% yields (Scheme 6).15
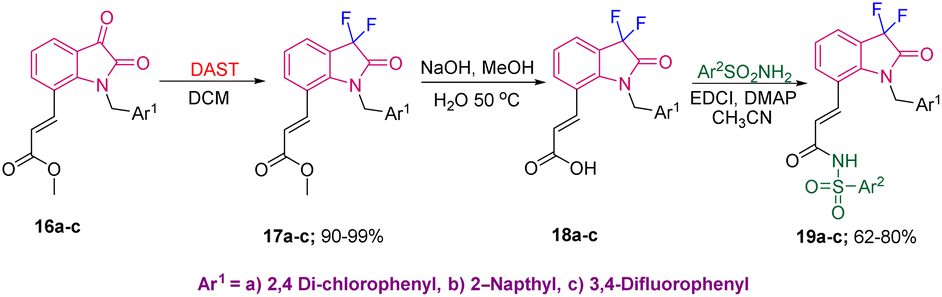 | ||
| Scheme 6 Synthesis of aryl sulfonamide-derived difluoro amides. | ||
Several chemists synthesised C3-fluorinated oxindoles using DAST as the fluorinating reagent; however, it was determined that it is thermally unstable and too explosive. To overcome these disadvantages, an alternative fluorinating agent, i.e. Deoxo-Fluor [(CH3OCH2CH2)2NSF3], was chosen by the group of Shreeve to provide a safer reagent for C3-difluorination of isatins. The requirement of the catalytic amount of HF (generated in situ by adding a few drops of ethanol) was essential to obtain the desired 3,3-difluoro oxindoles (21a–c) in 88–90% yield (Scheme 7).16
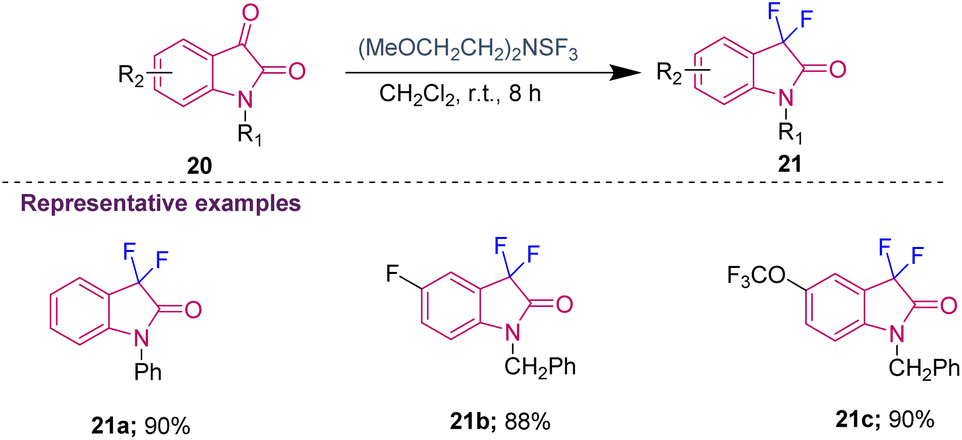 | ||
| Scheme 7 Nucleophilic fluorination of isatins using Deoxo-Fluor. | ||
In 2018, Tang's group developed a method to synthesize C3-monofluoro and C3-difluoro oxindoles from the same strategy using isatinhydrazones and Selectfluor. Difluorooxindoles were synthesized from isatinhydrazones and Selectfluor (2 equiv.) in MeCN at room temperature with a reaction time of 10 h. Furthermore, the substrate scope for 24 was explored to ascertain the versatility of the method. Yield was observed to improve on increasing the size and steric hindrance of the protecting group of N-substituted isatinhydrazones 24b–e. Aryl-substituted isatin hydrazones with halogen and EWG gave the desired products in good to excellent yields (24f–g). An NO2-substituted isatin required high temperature for the reaction to happen due to its poor solubility (24h). However, EDGs like Me or OMe gave either poor yield (24i) or no desired product (24j) (Scheme 8).17
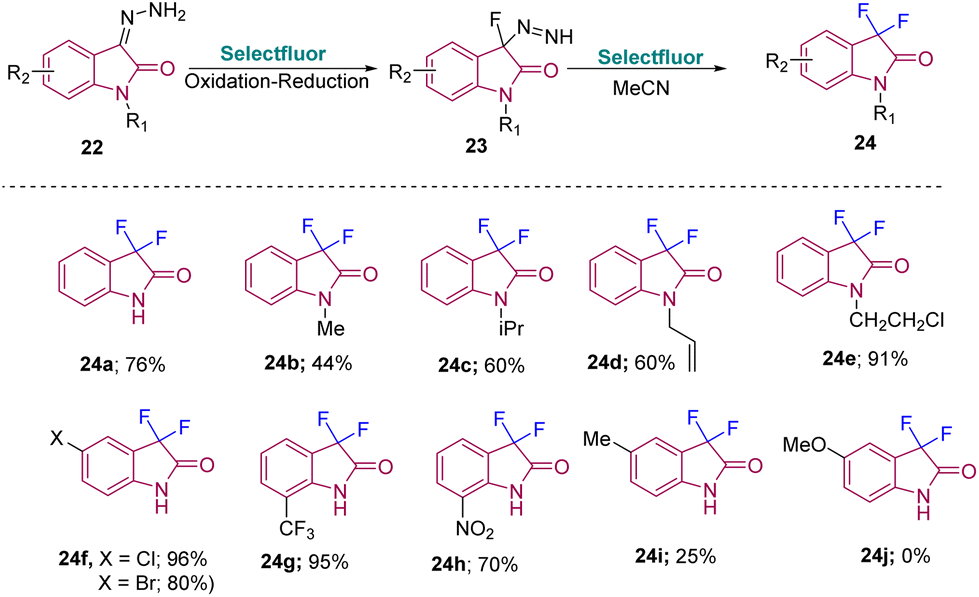 | ||
| Scheme 8 Synthesis of 3,3-difluorooxindole derivatives using isatin hydrazones and Selectfluor. | ||
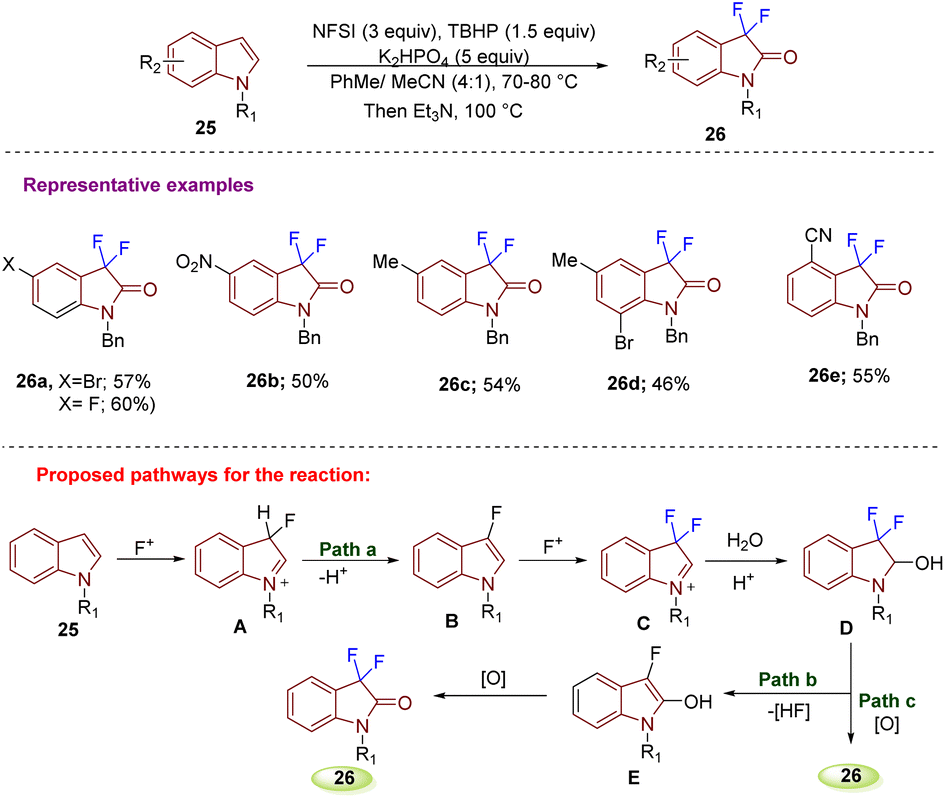 | ||
| Scheme 9 Synthesis of 3,3-difluoro-2-oxindoles through electrophilic fluorination of indoles. | ||
In 2017, Wu et al. constructed 3,3-difluoro-2-oxindoles 28 using N-substituted indoles 27via C-3 difluorination and C-2 oxidation simultaneously. The strategy was designed under metal-free conditions and was highly regioselective. Difluorination at C-3 was performed with PhI(OAc)2 and PhIF2 as a fluorinating agent (Scheme 10).20 Substrate scope was well explored with different substituents at different positions on the indole ring that afforded the desired products 28 in good yield. Further different substitutions on the N-benzyl ring gave products in moderate yields. According to the mechanism proposed, electrophilic fluorination (PhIF2) of 27 at C-3 gives A as an intermediate that is converted into an iodonium salt forming strained 3-membered ring B. Intermediate B is then attacked by a fluorine ion eliminated in the 1st step to generate C; thereafter, the intramolecular nucleophilic attack generates D. The moiety D gets attacked at the C-2 position by H2O and generates E, from which elimination of the fluoride ion leads to intermediate F, which is then converted into enolate G after rearomatization. Electrophilic fluorination of G then leads to intermediate H. Subsequent enolization and α-fluorination of H afford the desired compound 28. An H2O18 labelling experiment in anhydrous n-PrOH confirmed that the oxygen at the C-2 position of 29 comes from moisture in the solvent n-PrOH.
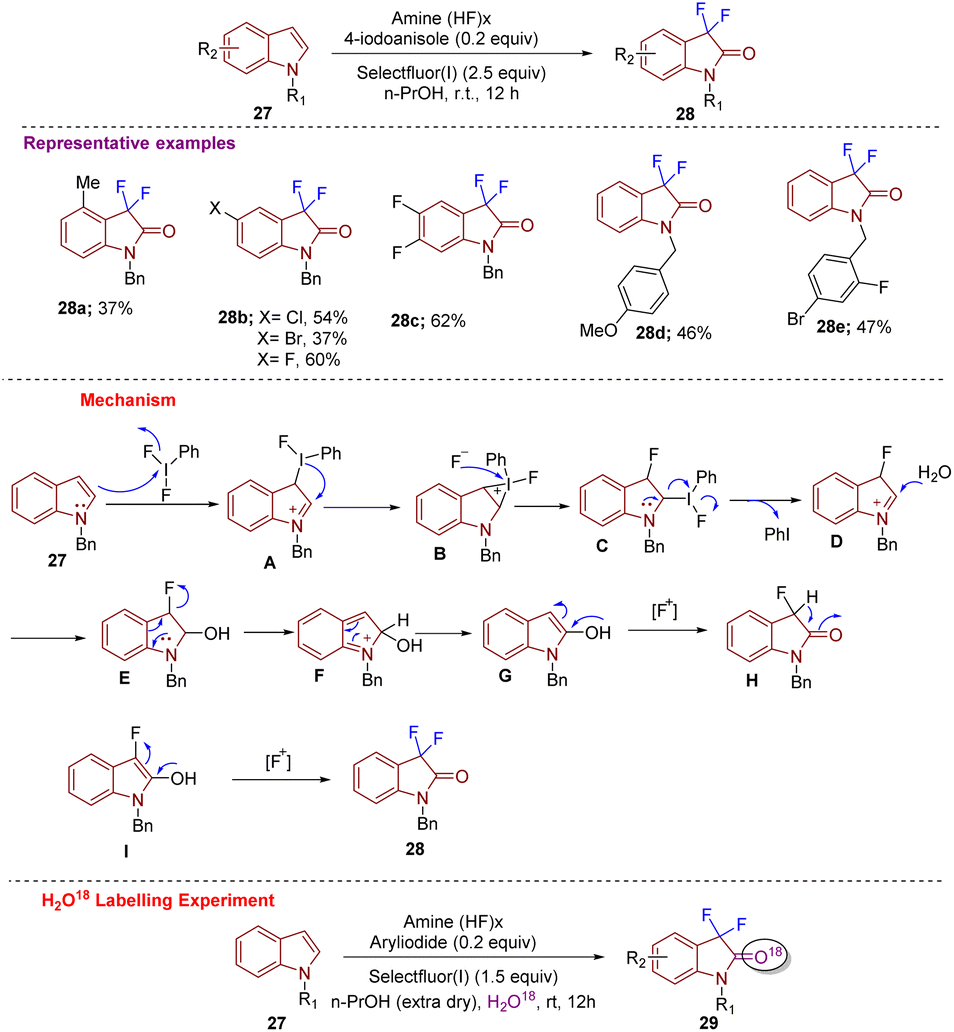 | ||
| Scheme 10 Synthesis of C3-difluorinated oxindole derivatives using N-substituted indoles. | ||
2.2 Intramolecular approach
 | ||
| Scheme 11 Synthesis of C3-difluorinated oxindoles through intramolecular cyclization of iododifluoroacetamides. | ||
In a further development, by using chlorodifluoroacetamides as starting materials, Buchwald and co-workers, in 2015, very efficiently constructed 3,3-difluorooxindoles with a broad substrate scope (35a–e) by C–H difluoro alkylation with a preheated catalyst solution of [Pd2dba3] and BrettPhos in anhydrous cyclopentylmethyl ether (CPME) (Scheme 12). Mechanistically, the process is initiated by the formation of a PdII enolate by oxidative addition of chlorodifluoro amide 34 to Pd0A. Later, six-membered palladacycle B is produced by electrophilic aromatic substitution of the arene, which is subsequently followed by deprotonation C and then, via reductive elimination, the noticed product 35 is synthesized and the Pd0 species is regenerated.22
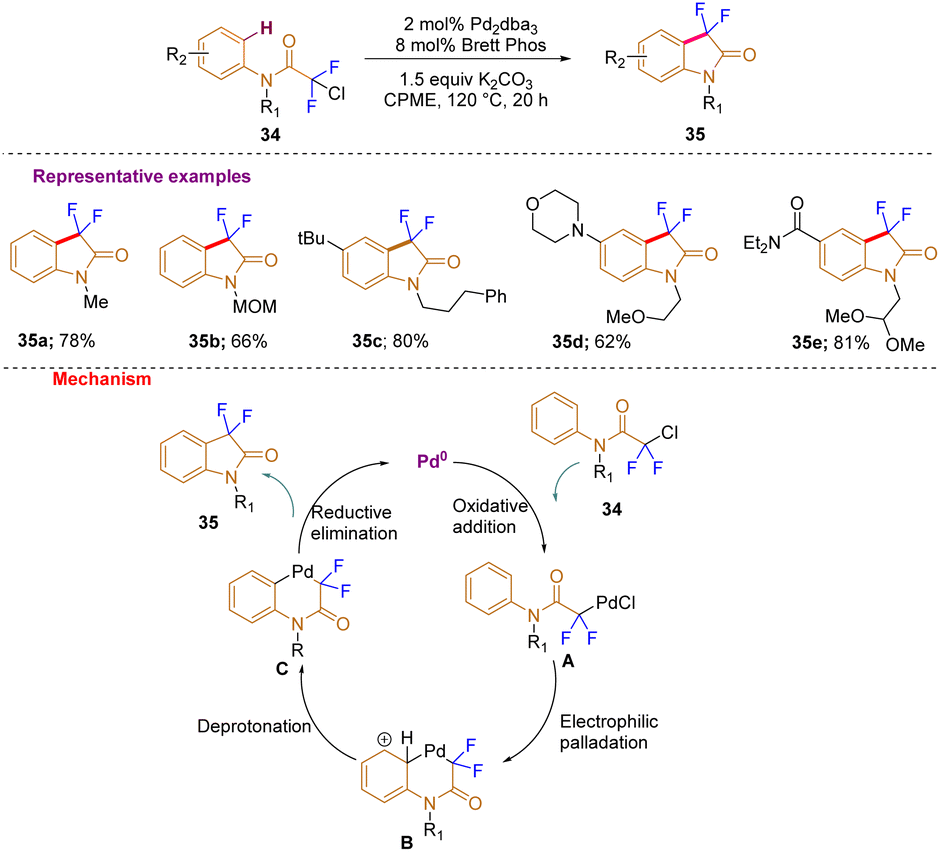 | ||
| Scheme 12 Synthesis of C3-difluorinated oxindoles via palladium catalyzed C–H difluoroalkylation. | ||
Furthermore, in a different approach, Liu's group reported a visible-light photocatalytic-mediated synthesis of 3,3-difluoro-2-oxindoles with wide substrate scope in good to excellent yields (37a–37e) through intramolecular C–H difluoroacetamidation of arenes 36 at room temperature using catalytic fac-Ir(ppy)3 and a 3 W blue LED light source. The substrates were mostly tertiary aryl bromodifluoroacetamides. This strategy was developed to avoid expensive fluorinating agents, complex ligands, and high temperatures (Scheme 13). According to the mechanism proposed, the visible-light illumination of the photoredox catalyst fac IrIII (ppy)3 generates an excited form of fac-IrIII(ppy)3* that reduces 2-bromo-2,2-difluorophenylacetamide with the help of SET (single electron transfer) and generates electron deficient radical (A) in addition to the generation of oxidized fac-IrIV(ppy)3. The electron-deficient radical undergoes intramolecular π-addition to form a cyclohexadiene radical (B) which after deprotonation with base K2HPO4 gives a radical anion (C). Further SET from C generates the desired difluorinated oxindole (37) and regenerates the photocatalyst fac-IrIII(ppy)3.23
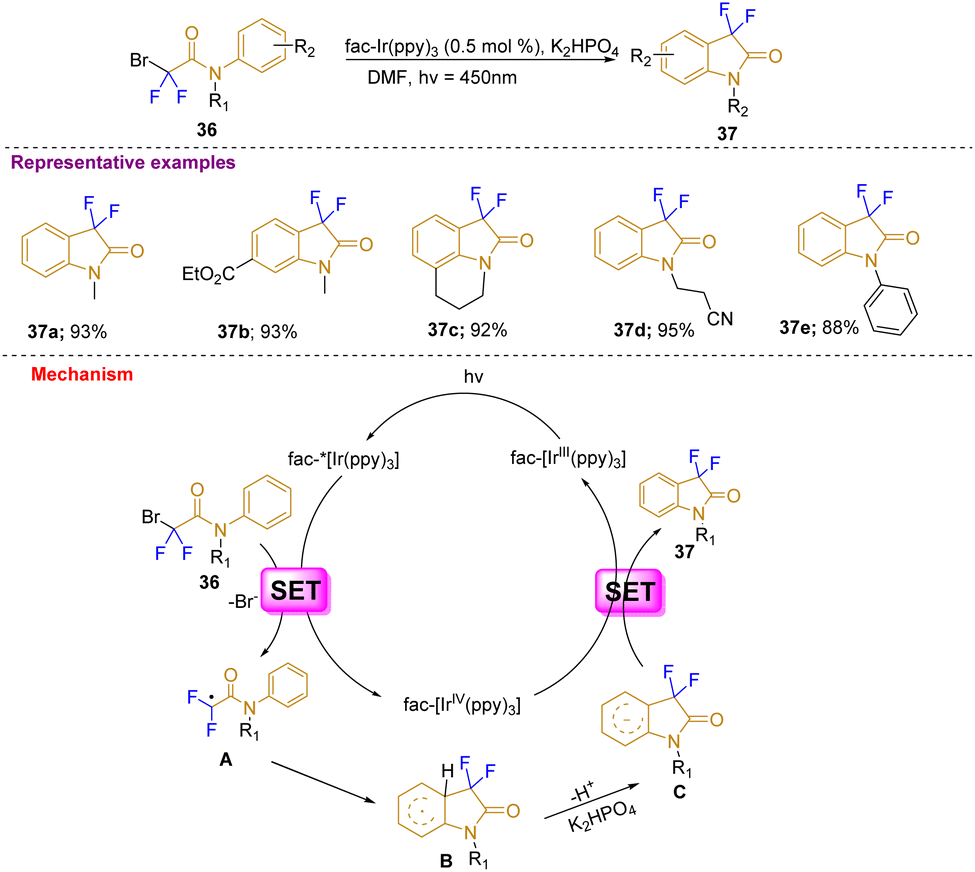 | ||
| Scheme 13 Synthesis of 3,3-difluorooxindoles via visible light photocatalysis. | ||
 | ||
| Scheme 14 Synthesis of 3,3-diflurooxindoles via ethoxycarbonyldifluoromethylation of anilines using BrCF2CO2Et. | ||
Using similar starting materials, Song's group developed the synthesis of 3,3-difluoro-2-oxindole and its derivatives in moderate to good yields (43a–e) from difluoro acetylation of anilines 42 catalyzed by copper/B2pin2via C–H activation that undergoes intramolecular amidation (Scheme 15). A plausible mechanism was proposed where, initially, there is the generation of LCu(I)-Bpin species I from LcuX (I) and B2pin2 in the presence of a base. Species I further activates BrCF2COOR to obtain a ˙CF2COOR radical. The amino group of aniline acts as a directing group to form a new C–CF2COOR bond to give intermediate A. SET led to intermediate B, which after deprotonation and intramolecular cycloamidation, gives the desired 43via intermediate C.25
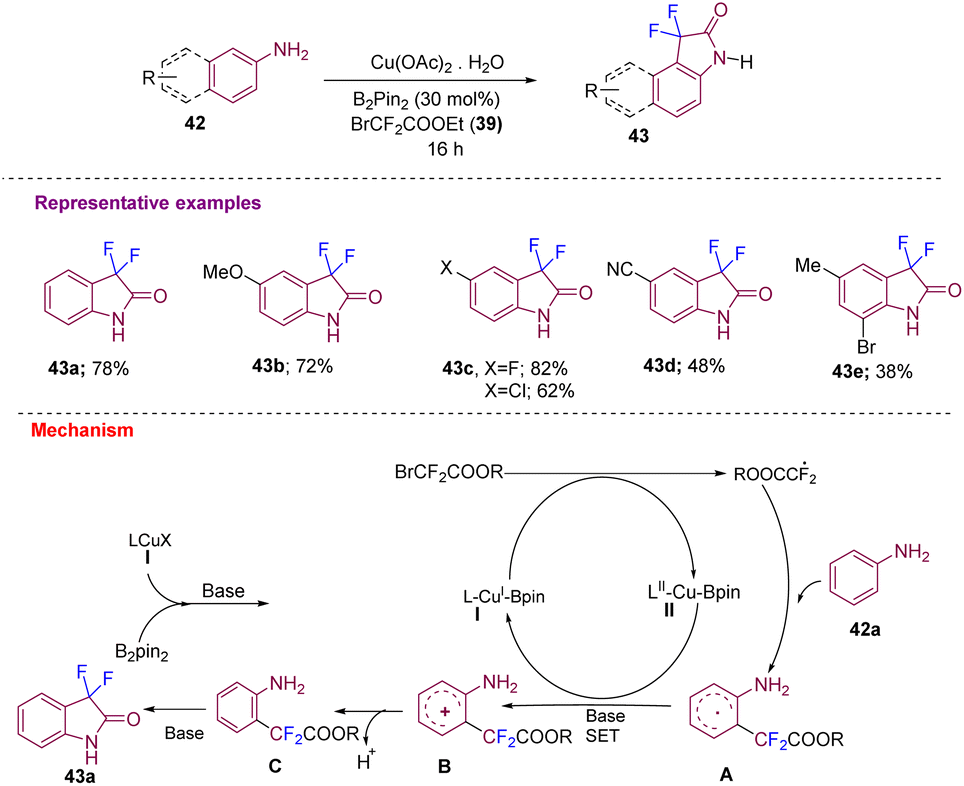 | ||
| Scheme 15 Synthesis of C3-difluorinated oxindoles via C–H difluoroacetylation–intramolecular amidation catalyzed by copper/B2pin2. | ||
Anilines (44) as starting materials were also utilized by Zhang's group who reported a strategy to construct 3,3-difluoro-2-oxindoles using BrCF2CO2Et (39) under a visible-light-induced photoredox difluoromethylation–amidation process (Scheme 16). Mechanistically, the excited state of the photocatalyst *[Ir(ppy)3] under visible light transfers a single electron to 39 to give a difluoroacetyl radical, which further reacts with aniline to afford the desired 45 in moderate to good yields via difluoroalkylation and successive intramolecular amidation following a similar mechanism to that mentioned in Scheme 13. The strategy showed broad functional group tolerance (45a–45e).26
 | ||
| Scheme 16 Synthesis of 3,3-difluorooxindoles using anilines via visible-light mediated difluoroalkylation–amidation. | ||
Along the same lines, Wang's group in 2017 reported the synthesis of 3,3-difluorooxindole 47 through difluoroalkylation and further intramolecular amidation using similar starting material to that discussed in Scheme 14, such as β-naphthylamine 46 and BrCF2CO2Et (39), but under different reaction conditions with CuI (10 mol%) and a ligand in CH3CN. The reaction conditions further involved basic PMDETA (pentamethyl-diethylenetriamine) ligand under an argon atmosphere at 80 °C (Scheme 17).27
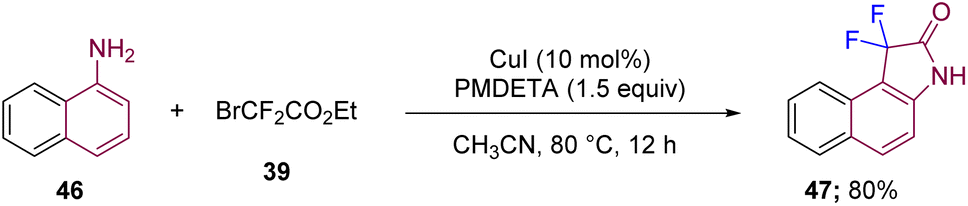 | ||
| Scheme 17 Synthesis of 3,3-difluorooxindoles with a Cu catalyst via difluoromethylation–amidation. | ||
Aniline derivatives 48 with BrCF2COOEt 39 as starting materials were further explored by Noel and co-workers in 2019 that afforded C3-difluorinated oxindole derivatives (49a–49c) in reasonable yields using visible light photoredox catalysis with 1 mol% photocatalyst A and TMEDA (N,N,N′,N′-tetramethyl ethane-1,2-diamine) in DMF at room temperature (Scheme 18).28
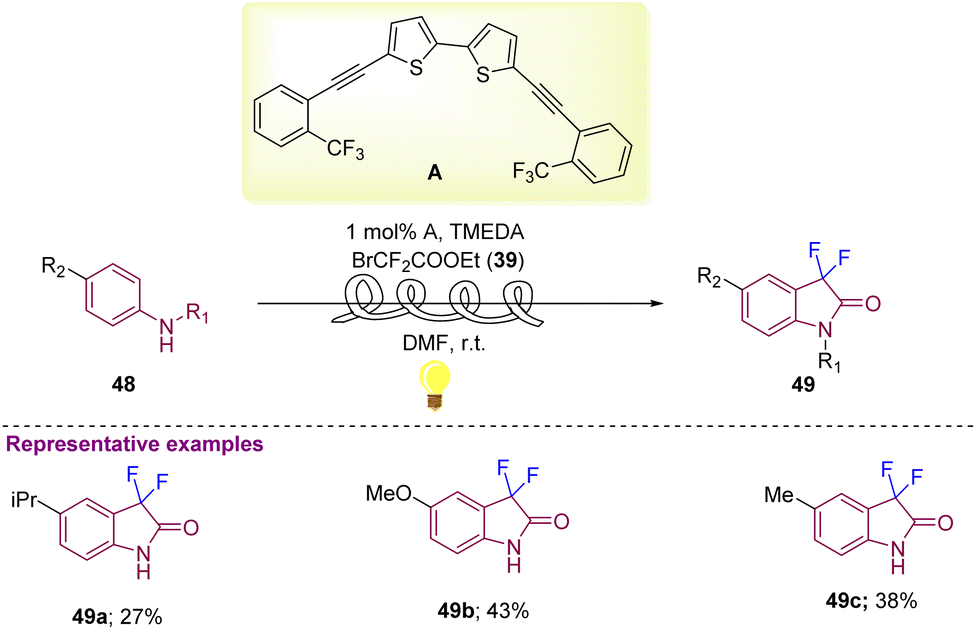 | ||
| Scheme 18 Synthesis of C3-difluorinated oxindoles using difluoroalkylation–amidation of anilines via visible light photoredox catalysis. | ||
Generation of diclofenac (DCF) analogs, processed from target C3-difluorinated oxindole compounds 52, which were accessed from esters 51 using NFSI with LiHMDS in THF at 78 °C for 1–2 hours was reported by Ohe's group in 2020. The esters 51 were obtained from their corresponding acids 50, under standard reaction conditions (H2SO4, C2H5OH under reflux for 0.5–2 hours) (Scheme 19).29
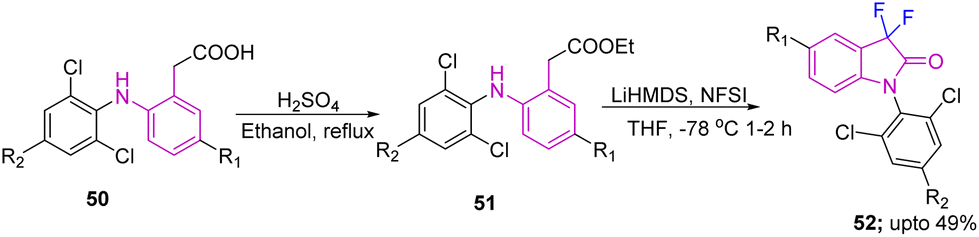 | ||
| Scheme 19 Synthesis of C3-difluorinated oxindoles as intermediates while forming DCF analogs. | ||
Furthermore, a [3 + 2] annulation strategy to synthesize 3,3-difluorooxindole 54 was developed by Li's group from the reaction between N-substituted anilines 53 and α-carbonyl alkyl bromides 39 catalyzed by copper. The method undergoes a C–H cycloamidation process under a radical mechanism, as confirmed by inhibiting the reaction using radical scavengers like TEMPO. The authors also demonstrated a broad substrate scope (Scheme 20).30
 | ||
| Scheme 20 Synthesis of C3-difluorinated oxindoles by [3 + 2] annulation of N,N-disubstituted anilines with α-carbonyl alkyl bromides catalyzed by copper. | ||
Zhang's group in 2020 produced 3,3-difluoroindolin-2-ones 57a–b in good yields from anilines 55 and difluoroiodoacetates 56 using P(4-CF3Ph)3 as the catalyst and K2CO3 as a base in degassed MeCN, which were irradiated with a 16 W blue LED at room temperature for 24 h under photocatalyst-free conditions (Scheme 21).31 Later, a mechanism was proposed, wherein initially, donor–acceptor (EDA) complex II is formed that under visible light generates a difluoroalkyl radical (˙CF2CO2R) that reacts with aniline 56a and generates a radical intermediate A that undergoes SET in the presence of a base to generate an intermediate B, which after deprotonation and subsequent cycloamidation gives the desired compound 57avia intermediate C.
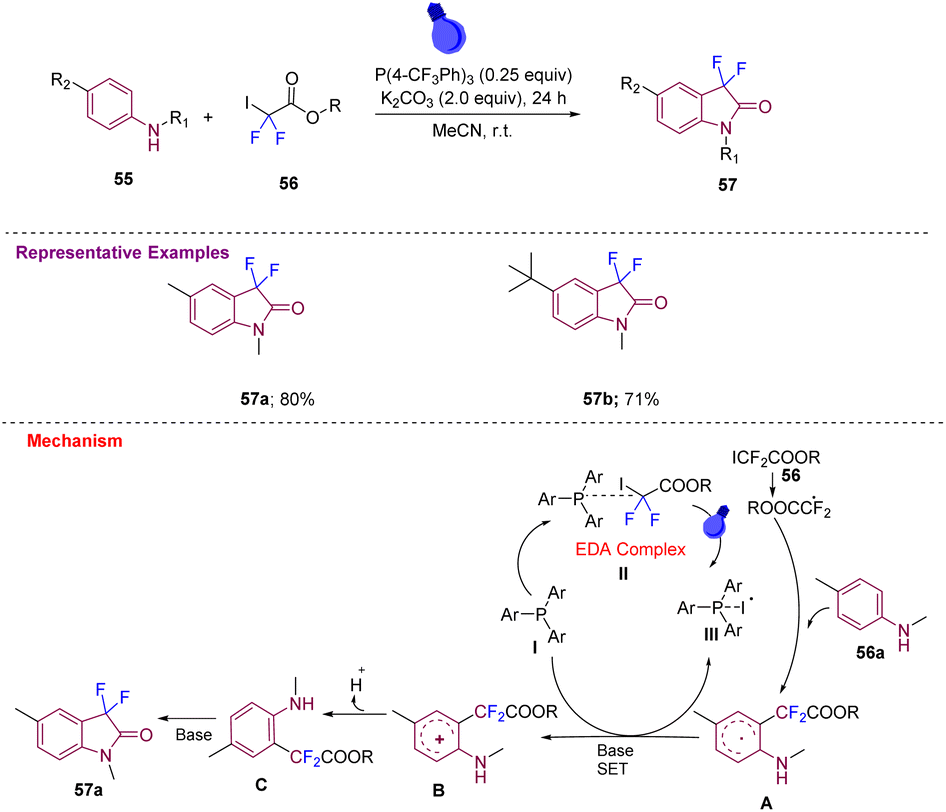 | ||
| Scheme 21 Synthesis of 3,3-difluoroindolin-2-one derivatives using a 16 W blue LED. | ||
Gou's group, in 2022, reported difluoromethylation at the ortho or para positions of different aromatic amines (58) with difluorobromoacetate 39 to yield 59 with a nickel metal catalyst and bidentate phosphine ligand L (Scheme 22).32 The reaction showed vast substrate scope with good to excellent yields of 59a and 59b. The application of the method was demonstrated by the efficient synthesis of a Baclofen derivative 59c in good yield. The authors demonstrated further application of the method by synthesizing a potent HDAC6 inhibitor starting from commercially available 58d, which under the standard reaction conditions gave 59d, which was then converted into the bioactive moiety 59d′.
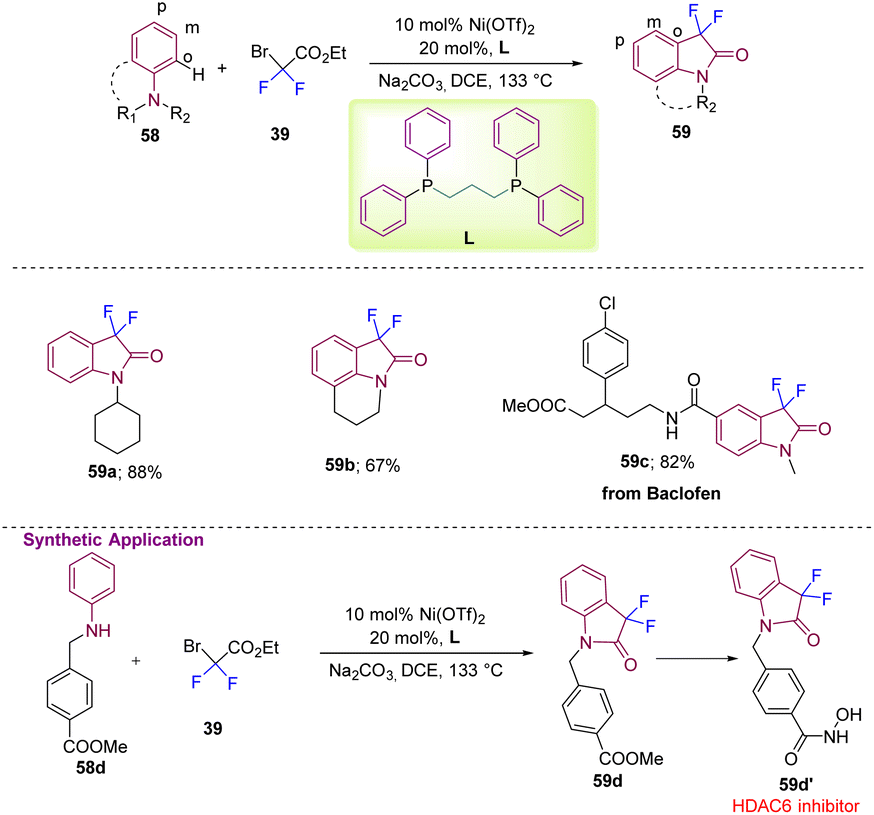 | ||
| Scheme 22 Synthesis of 3,3-difluoroindolin-2-ones using a nickel metal catalyst and a bidentate phosphine ligand. | ||
3. C-3 monofluorinated oxindoles
3.1 Intermolecular approach
 | ||
| Scheme 23 Synthesis of C3-monofluorinated oxindoles through enantioselective catalysis of oxindole derivatives and synthesis of MaxiPost. | ||
Following the same strategy, Feng's group synthesised a biologically important molecule, MaxiPost (BMS-204352), an anti-stroke drug, by starting from trifluorinated methyl derivative 60a using NFSI as the fluorinating agent and a Sc(III)/N,N′-dioxide complex as the catalyst. The reaction conditions required Na2CO3 as the base in DCM as the solvent at −30 °C for three days. However, the requirement of a transition metal and long-duration reaction may make this approach non-viable for pilot-plant production and hence unsuitable for industrial purposes (Scheme 23).34
Tang et al. in 2018, produced 3-fluorooxindoles (63a–e) from hydrazonoindolin-2-ones 62 using Selectfluor as fluorinating reagent at 80 °C in DCE. Mechanistically, intermediate A was generated from 62 that, after deprotonation with a weak base, such as LiOAc, was converted into intermediate B, which finally afforded the desired 63 after elimination of nitrogen gas and protonation of B (Scheme 24).17
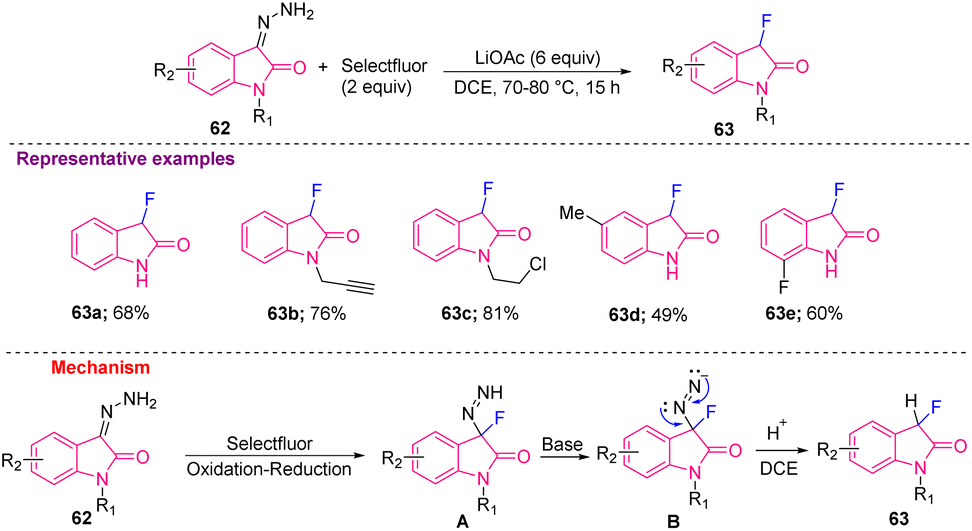 | ||
| Scheme 24 Synthesis of C3-monofluorinated oxindoles using hydrazonoindolin-2-ones. | ||
Later, in 2019, Gnanaprakasam's group synthesized C3-monofluorinated oxindoles 65 with Selectfluor from Z-3-(aminobenzylidene/aminoalkylidene) indolin-2-ones 66, which were synthesized from 2-oxindoles 64 using LiOtBu and 2,2′-BPY via a metal-free approach. The reaction was further explored with different substituents at R1 and R2 affording the desired 66 in moderate yields (Scheme 25).35
 | ||
| Scheme 25 Synthesis of C3-monofluorinated oxindoles from 2-oxindoles under metal-free reaction conditions. | ||
Hazra's group, in 2018, achieved asymmetric nucleophilic fluorination for the first time at the sp3-tertiary carbon to produce 3-fluoro-3-substituted oxindoles (68a–e) having excellent enantioselectivity. They used tetrabutylammonium fluoride (TBAF) as the fluorinating agent in the absence of metal catalysts (Scheme 26).36
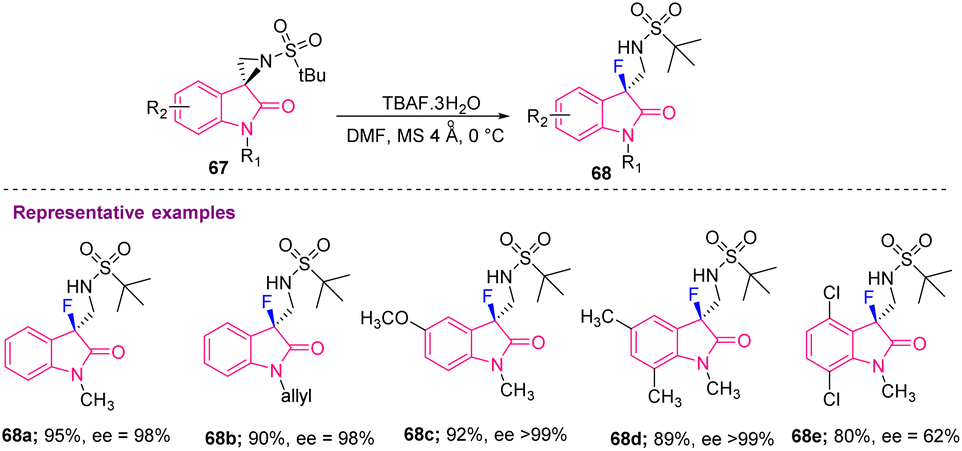 | ||
| Scheme 26 Synthesis of C3-monofluorinated oxindoles using asymmetric nucleophilic fluorination. | ||
Furthermore, to provide a sustainable method, Najera's group, in 2019, reported Mg-catalyzed synthesis of C3-monofluorinated oxindole derivatives (71a–c). In this report, 3-acetyl-2-oxindoles 69 were fluorinated using 1 equiv. of NFSI (N-fluorobenzenesulfonimide) with a Lewis acid catalyst Mg(ClO4)2 to provide 3-acetyl-2-oxindoles 70, which were later converted into 71 in moderate yields via deacylation, after the addition of benzyltrimethyl ammonium hydroxide (Triton B) in a degassed reaction mixture (Scheme 27).37
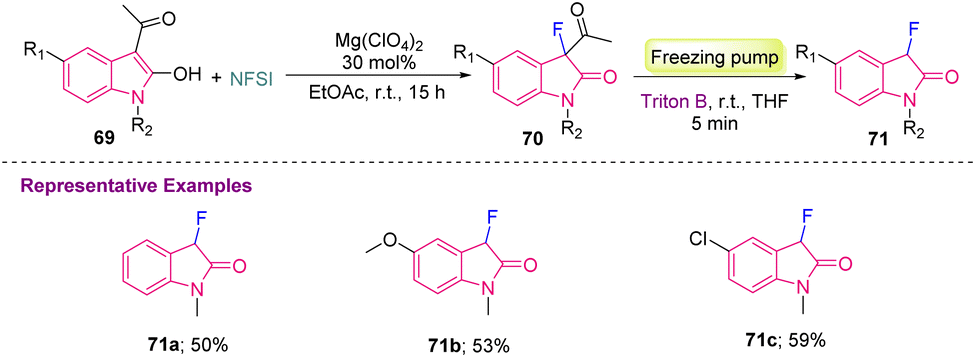 | ||
| Scheme 27 Synthesis of monofluorinated oxindoles using 3-acetyl-2-oxindoles. | ||
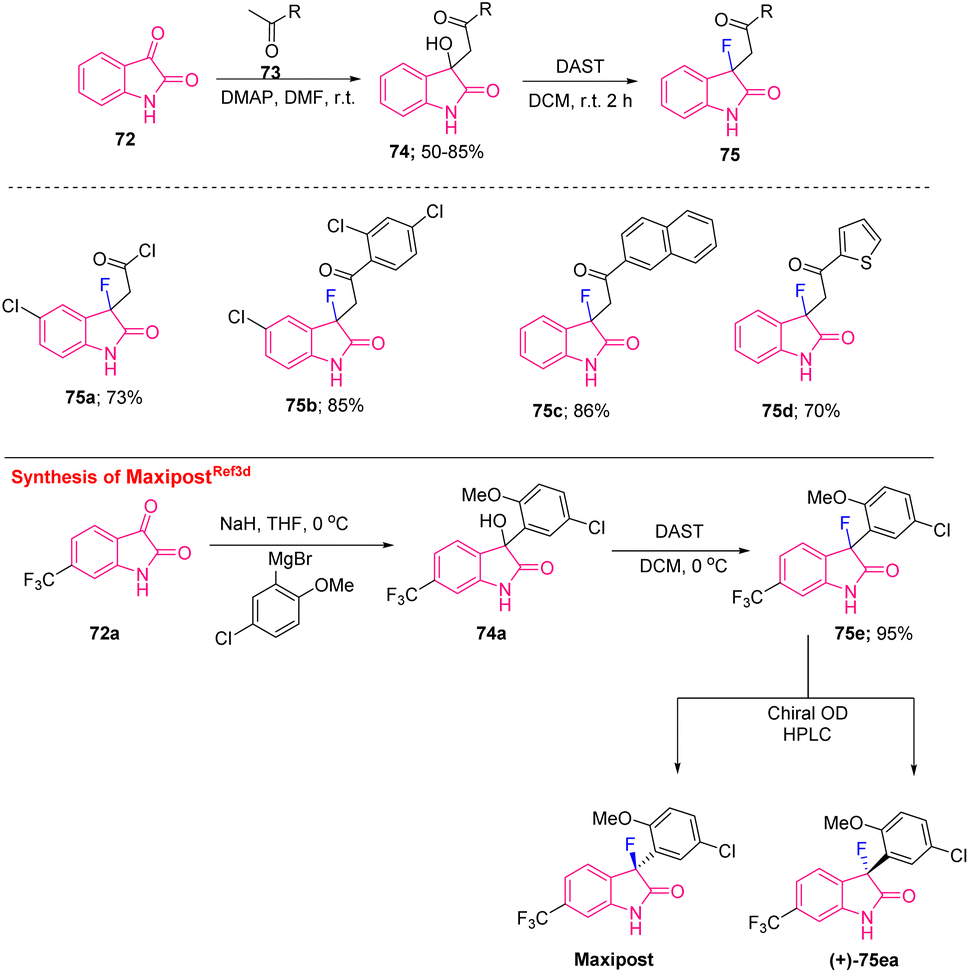 | ||
| Scheme 28 Synthesis of C3-monofluorinated oxindoles from isatin and synthesis of the MaxiPost drug. | ||
The abovementioned approach was utilized by Starrett's group to synthesise MaxiPost (BMS-204352) as openers of Maxi-K potassium channels used for neuroprotective therapy (protects against the risk of stroke). The synthetic route started with the reaction of a Grignard reagent to the sodium salt of isatin 72a in THF that provided the intermediate alcohol 74a, which, after treatment with DAST in DCM, furnished the racemic drug 75e. Further resolution of the racemic compound using chiral HPLC gave an industrially suitable mild method for the synthesis of MaxiPost and its isomer (+)-75ea (Scheme 28).3d
3.2 Intramolecular synthesis
 | ||
| Scheme 29 Synthesis of C3-monofluorinated oxindoles from methyl esters using a reductive cyclization strategy. | ||
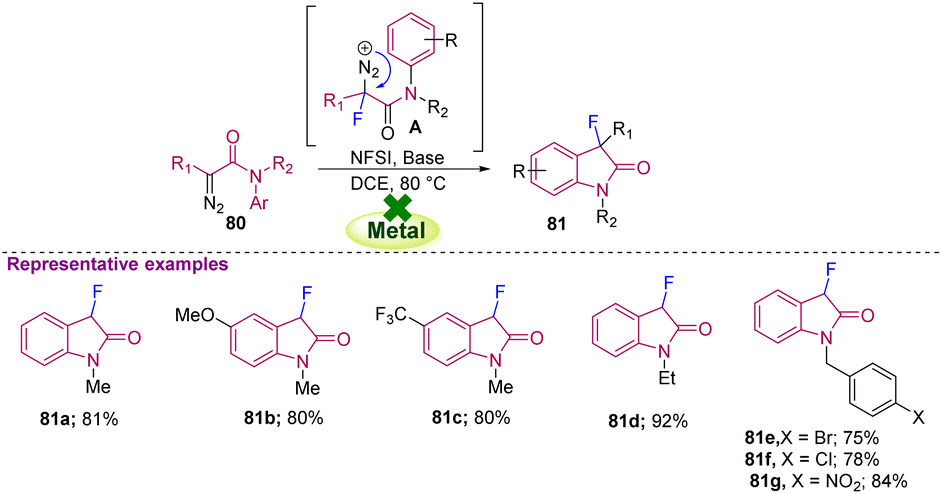 | ||
| Scheme 30 Synthesis of C3-monofluorinated oxindoles from N-aryl diazoacetamides. | ||
Furthermore, Xu and co-workers in 2017 reported a novel method for producing C3-monofluorinated oxindoles (83a–g) via electrochemical synthesis, where the oxidation of Cp2Fe at the anode and the reduction of MeOH at the cathode are triggered by the generation of a potential difference across the electrolysis cell. Generated MeO− ions then abstract the malonyl proton from 82 and form the conjugate base A, which is in equilibrium with 82. The generated conjugate base A transfers its electron to Cp2Fe, which later helps in the formation of key radical intermediate B that undergoes radical cyclization and rearomatization to furnish the desired C3-monofluorinated oxindole 83. This electrochemical approach showed satisfactory results with different functional groups, including the full range of halogens 83a–d, pyridines 83e, redox-sensitive pyrrole 83f, and amino esters 83g (Scheme 31).41
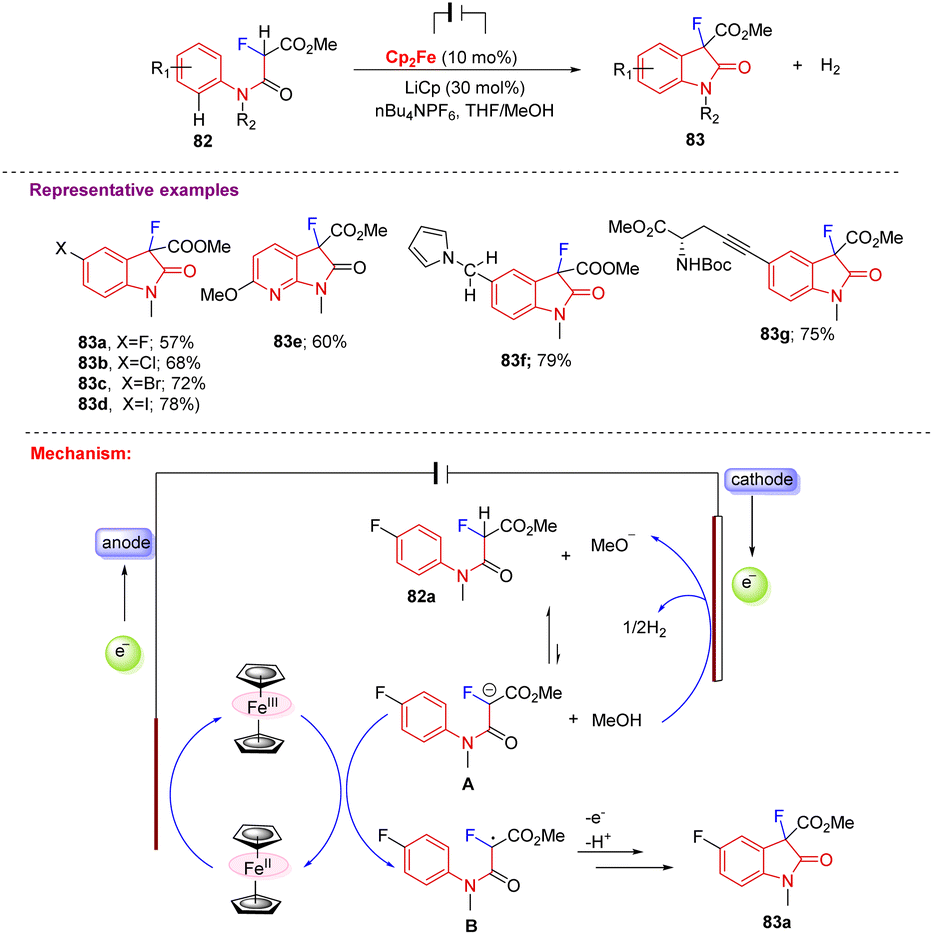 | ||
| Scheme 31 Synthesis of C3-monofluorinated oxindoles from malonate amides using Cp2Fe. | ||
4. Conclusions
C3-fluorinated oxindoles have been a topic of interest in recent years due to their myriad biological properties. Along with their significance in medicinal chemistry, these scaffolds are also utilized for the synthesis of complex architectures bearing fluorine atoms under appropriate reaction conditions.42 Furthermore, C3-difluorinated oxindoles are also used as nucleophilic catalysts for regenerative glycosylation.43 All these properties have thus stimulated the development of new methods to access C3-fluorinated oxindoles. Herein, we overviewed the different strategies for constructing C3-di and monofluorinated oxindoles via intermolecular and intramolecular approaches. The C3-difluorinated oxindoles were synthesised via intermolecular approaches that mainly used nucleophilic and electrophilic fluorination of isatin and indoles, respectively, with appropriate reagents (DAST, Selectfluor, and Deoxo-Fluor). In contrast, intramolecular approaches hinged on the difluoroalkylation of halodifluoroacetamides besides difluoroalkylation with its concomitant amidation of anilines. Furthermore, owing to the immense biological significance of C3-monofluorinated oxindoles, different approaches for their synthesis have been covered with emphasis on the approach and in comparison with other known literature processes for the synthesis of the anti-stroke drug MaxiPost. Among other state-of-the-art methods, electrochemical synthesis removes the requirement of transition metals and oxidants and is quite promising. Although some decent progress has been achieved toward the development of various processes via intramolecular strategies, the intermolecular approaches are limited to the utilization of isatin, oxindoles, and indoles, which provide ample scope for further discovery and development of new intermolecular methods to access such C-3 fluorinated oxindoles with emphasis on asymmetric versions of C3-monofluorinated oxindoles.Author contributions
This review was written by Deeksha and Bittu under the supervision of RS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RS and Deeksha are grateful to the DST-SERB (CRG/2020/001633) for its financial support and the Central University of Rajasthan for infrastructure.References
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- (a) M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS PubMed; (b) E. T. Newcomb, P. C. Knutson, B. A. Pedersenand and E. M. Ferreira, J. Am. Chem. Soc., 2016, 138, 108–111 CrossRef CAS PubMed; (c) G. O. Fonseca and J. M. Cook, Org. Chem. Insights, 2016, 6, 1–55 CrossRef; (d) A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979–4982 CrossRef CAS PubMed; (e) W. C. Sumpter, Chem. Rev., 1945, 37, 443–479 CrossRef CAS PubMed.
- (a) A. K. Podichetty, A. Faust, K. Kopka, S. Wagner, O. Schober, M. Schäfers and G. Haufe, Bioorg. Med. Chem., 2009, 17, 2680–2688 CrossRef CAS PubMed; (b) N. Zhou, A. M. Polozov, M. O'Connell, J. Burgeson, P. Yu, W. Zeller, J. Zhang, E. Onua, J. Ramirez, G. A. Palsdottir, G. V. Halldorsdottir, T. Andresson, A. S. Kiselyov, M. Gurney and J. Singh, Bioorg. Med. Chem. Lett., 2010, 20, 2658–2664 CrossRef CAS PubMed; (c) J. C. Kath and M. J. Luzzio, Pyrimidine Derivatives for the Treatment of Abnormal Cell Growth. PatentW004/056807A1, 2004 Search PubMed; (d) P. Hewawasam, V. K. Gribkoff, Y. Pendri, S. I. Dworetzky, N. A. Meanwell, E. Martinez, C. G. Boissard, D. J. Post-Munson, J. T. Trojnacki, K. Yeleswaram, L. M. Pajor, J. Knipe, Q. Gao, R. Perrone and J. E. Starrett, Bioorg. Med. Chem. Lett., 2002, 12, 1023–1026 CrossRef CAS PubMed; (e) P. J. Dollings, A. F. Donnell, A. M. Gilbert, M. Zhang, B. L. Harrison, C. J. Stanton, S. V. O'Neil, L. M. Havran and D. C. Chong, Substituted Oxindol Cb2 Agonists for Pain Treatment, WO2010077839A1, 2010; (f) A. Bort, S. Quesada, Á. Ramos-Torres, M. Gargantilla, E. M. Priego, S. Raynal, F. Lepifre, J. M. Gasalla, N. Rodriguez-Henche, A. Castro and I. Díaz-Laviada, Sci. Rep., 2018, 8, 4370 CrossRef PubMed; (g) Y.-L. Liu, X.-P. Wang, J. Wei and Y. Li, Org. Biomol. Chem., 2022, 20, 538–552 RSC.
- J. W. Middleton and M. E. Bingham, J. Org. Chem., 1980, 45, 2883–2887 CrossRef.
- J. C. Torres, S. J. Garden, A. C. Pinto, F. S. Q. da Silva and N. Boechat, Tetrahedron, 1999, 55, 1881–1892 CrossRef CAS.
- (a) G. D. Zhu, V. B. Gandhi, J. Gong, Y. Luo, X. Liu, Y. Shi, R. Guan, S. R. Magnone, V. Klinghofer, E. F. Johnson, J. Bouska, A. Shoemaker, A. Oleksijew, K. Jarvi, C. Park, R. D. Jong, T. Oltersdorf, Q. Li, S. H. Rosenberg and V. L. Giranda, Bioorg. Med. Chem. Lett., 2006, 16, 3424–3429 CrossRef CAS PubMed; (b) S. Y. Lin, T. K. Yeh, C. C. Kuo, J. S. Song, M. F. Cheng, F. Y. Liao, M. W. Chao, H. L. Huang, Y. L. Chen, C. Y. Yang, M. H. Wu, C. L. Hsieh, W. Hsiao, Y. H. Peng, J. S. Wu, L. M. Lin, M. Sun, Y. S. Chao, C. Shih, S. Y. Wu, S. L. Pan, M. S. Hung and S. H. Ueng, J. Med. Chem., 2016, 59, 419–430 CrossRef CAS PubMed; (c) T. S. Chundawat, P. Kumari, N. Sharma and S. Bhagat, Med. Chem. Res., 2016, 25, 2335–2348 CrossRef CAS; (d) L. D. Azevedo, M. M. Bastos, F. C. Vasconcelos, L. V. B. Hoelz, F. P. S. Junior, R. F. Dantas, A. C. M. de Almeida, A. P. de Oliveira, L. C. Gomes, R. C. Maia and N. Boechat, Med. Chem. Res., 2017, 26, 2929–2941 CrossRef CAS; (e) F. S. C. Branco, E. C. de Lima, J. L. O. Domingos, A. C. Pinto, M. C. S. Lourenco, K. M. Gomes, M. M. Costa-Lima, C. F. Arauja-Lima, C. A. F. Aiub, I. Felzenszwalb, T. E. M. M. Costa, C. Penido, M. G. Henriques and N. Boechat, Chem. – Eur. J., 2018, 146, 529–540 CrossRef PubMed; (f) G. Luo, X. J. Jiang, L. Chen, C. M. Conway, M. Gulianello, W. Kostich, D. Keavy, L. J. Signor, P. Chen, C. Davis, V. J. Whiterock, R. Schartman, K. A. Widmann, J. E. Macor and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2021, 43, 128077 CrossRef CAS PubMed; (g) A. Oliveira, S. Moura, L. Pimental, J. Neto, R. Dantas, F. Sliva-Jr, M. Bastos and N. Boechat, Molecules, 2022, 27, 750 CrossRef CAS PubMed.
- G. D. Zhu, V. B. Gandhi, J. Gong, Y. Luo, X. Liu, Y. Shi, R. Guan, S. R. Magnone, V. Klinghofer, E. F. Johnson, J. Bouska, A. Shoemaker, A. Oleksijew, K. Jarvis, C. Park, R. D. Jong, T. Oltersdorf, Q. Li, S. H. Rosenberg and V. L. Giranda, Bioorg. Med. Chem. Lett., 2006, 16, 3424–3429 CrossRef CAS PubMed.
- E. Crizanto de Lima, F. S. Castelo-Branco, C. C. Maquiaveli, A. B. Farias, M. N. Renno, N. Boechat and E. R. Silva, Bioorg. Med. Chem., 2019, 27, 3853–3859 CrossRef CAS PubMed.
- (a) H. Sun, Y. Zhang, W. Ding, X. Zhao, X. Song, D. Wang, Y. Li, K. Han, Y. Yang, Y. Ma, R. Wang, D. Wang and P. Yu, Eur. J. Med. Chem., 2016, 123, 365–378 CrossRef CAS PubMed; (b) Q. Zhang, Y. Tang, Y. Yuan, T. Ruan, Q. Wang, X. Gao, Y. Zhou, K. Han, P. Yu and K. Lu, Eur. J. Med. Chem., 2018, 156, 800–814 CrossRef CAS PubMed; (c) A. Hurtevent, M. le Naour, V. Leclerc, P. Carato, P. Melnyk, N. Hennuyer, B. Staels, M. Beucher-Gaudin, D. H. Carignard, C. Dacquet and N. Lebegue, J. Enzyme Inhib. Med. Chem., 2020, 35, 524–538 CrossRef CAS PubMed.
- L. A. McAllister, R. A. McCormick, K. M. James, S. Brand, N. Willets and D. J. Procter, Chem. – Eur. J., 2007, 13, 1032–1046 CrossRef CAS PubMed.
- N. Boechat, W. B. Kover, M. M. Bastos, A. C. Pinto, A. C. Maciel, L. M. U. Mayer, F. S. Q. da Silva, P. M. Sá, J. S. Mendonca, S. M. S. V. Wardell and M. S. L. Arruda, J. Braz. Chem. Soc., 2008, 19(3), 445–457 CrossRef CAS.
- M. Baumann, I. R. Baxendale and S. V. Ley, Synlett, 2008, 2111–2114 CAS.
- M. Baumann, I. R. Baxendale, L. J. Martin and S. V. Ley, Tetrahedron, 2009, 65, 6611–6625 CrossRef CAS.
- A. K. Podichetty, A. Faust, K. Kopka, S. Wagner, O. Schober, M. Schäfers and G. Haufe, Bioorg. Med. Chem., 2009, 17, 2680–2688 CrossRef CAS PubMed.
- N. Zhou, A. M. Polozov, M. O'Connell, J. Burgeson, P. Yu, W. Zeller, J. Zhang, E. Onua, J. Ramirez, G. A. Palsdottir, G. V. Halldorsdottir, T. Andresson, A. S. Kiselyov, M. Gurney and J. Singh, Bioorg. Med. Chem. Lett., 2010, 20, 2658–2664 CrossRef CAS PubMed.
- R. P. Singh, U. Majumderu and J. M. Shreeve, J. Org. Chem., 2001, 66, 6263–6267 CrossRef CAS PubMed.
- Q. Yang, G. L. Dai, Y. M. Yang, Z. Luo and Z. Y. Tang, J. Org. Chem., 2018, 83, 6762–6768 CrossRef CAS PubMed.
- Y. H. Lim, Q. Ong, H. A. Duong, T. M. Nguyen and C. W. Johannes, Org. Lett., 2012, 14(22), 5676–5679 CrossRef CAS PubMed.
- B. J. Melancon, M. S. Poslusney, P. R. Gentry, J. C. Tarr, D. J. Sheffler, M. E. Mattmann, T. M. Bridges, T. J. Utley, J. S. Daniels, C. M. Niswender, P. J. Conn, C. W. Lindsley and M. R. Wood, Bioorg. Med. Chem. Lett., 2013, 23, 412–416 CrossRef CAS PubMed.
- X. Sun, X. J. Zhao and B. Wu, Asian J. Org. Chem., 2017, 6, 690–693 CrossRef CAS.
- J. Zhu, W. Zhang, L. Zhang, J. Liu, J. Zheng and J. Hu, J. Org. Chem., 2010, 75, 5505–5512 CrossRef CAS PubMed.
- S. L. Shi and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 1646–1650 CrossRef CAS PubMed.
- X. J. Wei, L. Wang, S. F. Du, L. Z. Wu and Q. Liu, Org. Biomol. Chem., 2016, 14, 2195–2199 RSC.
- Y. Ohtsuka and T. Yamakawa, Tetrahedron, 2011, 67, 2323–2331 CrossRef CAS.
- M. Ke and Q. Song, Chem. Commun., 2017, 53, 2222–2225 RSC.
- L. C. Yu, J. W. Gu, S. Zhang and X. Zhang, J. Org. Chem., 2017, 82, 3943–3949 CrossRef CAS PubMed.
- X. Wang, S. Zhao, J. Liu, D. Zhu, M. Guo, X. Tang and G. Wang, Org. Lett., 2017, 19(16), 4187–4190 CrossRef CAS PubMed.
- C. Bottecchia, R. Martin, I. Abdiaj, E. Crovini, J. Alcazar, J. Orduna, M. J. Blesa, J. R. Carillo, P. Prieto and T. Noel, Adv. Synth. Catal., 2019, 361, 945–950 CrossRef CAS.
- Y. Tateishi, T. Ohe, M. Ogawa, K. Takahashi, S. Nakamura and T. Mashino, ACS Omega, 2020, 5(50), 32608–32616 CrossRef CAS PubMed.
- A. Z. Cao, Y. T. Xiao, Y. C. Wu, R. J. Song, Y. X. Xie and J. H. Li, Org. Biomol. Chem., 2020, 18, 2170–2174 RSC.
- H. Lu, D. Y. Wang and A. Zhang, J. Org. Chem., 2020, 85, 942–951 CrossRef CAS PubMed.
- Q. Gou, Q. Chen, Q. Tan, M. Zhu, H. Huang, M. Deng, W. Yi and S. He, Org. Lett., 2022, 24, 3549–3554 CrossRef CAS PubMed.
- Y. Hamashima, T. Suzuki, H. Takamo, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164–10165 CrossRef CAS PubMed.
- J. Li, Y. Cai, W. Chen, X. Liu, L. Lin and X. Feng, J. Org. Chem., 2012, 77, 9148–9155 CrossRef CAS PubMed.
- G. S. Bisht and B. Gnanaprakasam, J. Org. Chem., 2019, 84, 13516–13527 CrossRef CAS PubMed.
- S. Hajra, A. Hazra and P. Mandal, Org. Lett., 2018, 20, 6471–6475 CrossRef CAS PubMed.
- C. Molina, A. O. Martin, J. M. Sansano and C. Najera, Org. Biomol. Chem., 2019, 17, 482–489 RSC.
- P. L. N. Ranganath and A. V. Narsaiah, J. Fluor. Chem., 2023, 268, 110134 CrossRef CAS.
- A. Harsanyi, G. Sandford, D. S. Yufit and J. A. K. Howard, J. Org. Chem., 2014, 10, 1213–1219 Search PubMed.
- K. Dong, B. Yan, S. Chang, Y. Chi, L. Qiu and X. Xu, J. Org. Chem., 2016, 81, 6887–6892 CrossRef CAS PubMed.
- Z. J. Wu and H. C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed.
- H. Uno, K. Kawai, M. Shiro and N. Shibata, ACS Catal., 2020, 10, 14117–14126 CrossRef CAS.
- S. S. Nigudkar, K. J. Stine and A. V. Demchenko, J. Am. Chem. Soc., 2014, 136, 921–923 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |