
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycosyl benzoates as novel substrates for glycosynthases†
Sabrina
de Lorenzo
,
Lauriane
Pillet
,
David
Lim
* and
Francesca
Paradisi
 *
*
Department of Chemistry, Biochemistry, and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, CH-3012, Bern, Switzerland. E-mail: francesca.paradisi@unibe.ch; david.lim@unibe.ch
First published on 19th July 2023
Abstract
The development of a procedure for the one-pot synthesis of glycosyl benzoates directly from unprotected sugars in aqueous media using 2-chloro-1,3-dimethylimidazolium chloride (DMC), thiobenzoic acid, and triethylamine is reported. These glycosyl donors are excellent substrates for wild-type and mutant glycosidases. β-Glucosyl benzoate was hydrolysed by the GH1 β-glucosidase derived from Halothermothrix orenii (HorGH1). Subsequent use of this substrate in thioligase-mediated glycosylation of p-nitrothiophenol demonstrated their superiority as donors compared to their p-nitrophenol counterparts with excellent conversions. Using a series of arene nucleophiles, we also demonstrate good to excellent conversions (up to 94%) of β-glucosyl benzoate to the corresponding p-nitrophenyl- and thioglycosides.
Introduction
Mimetics of carbohydrates that are resistant towards enzymatic hydrolysis have found value as therapeutics. For example, truncated variants of the natural MUC1 mucin are found overexpressed in many tumor tissues. Accordingly, when short epitopes bearing different tumor-associated carbohydrate antigens, such as the Tn determinant (α-O-GalNAc-Ser/Thr), are presented to the immune system, these molecules are recognized by anti-MUC1 antibodies.1 However, due to these structures possessing the natural O-glycosidic linkage, these molecules are susceptible to degradation by endogenous glycosidases, leading to shorter half-lives. To overcome this issue and similar drawbacks, strategies to incorporate unnatural linkages into these molecules has been implemented.2 Compañón et al. demonstrated that an O → S/Se replacement at the glycosidic linkage led to an increase in the carbohydrate-peptide distance and observed a change in both the dynamics and orientation of the glycosidic linkage.3 Consequently, the new glycopeptides demonstrated improved binding towards a representative anti-MUC1 antibody. A major drawback in the synthesis of these important molecules lies in the fact that complex protecting group strategies are required which are technically demanding and generate a lot of waste. Therefore, there has been a drive to develop methods for the synthesis of glycomimetics possessing similar unnatural linkages.Although stereoselective glycosylation has made significant progress within the past century, the chemical synthesis of glycoconjugates often still requires the use of protecting group strategies to introduce the desired linkages.4 Alternatively, biocatalytic methods are becoming increasingly attractive due to their excellent regio/stereoselectivity and their high efficiency while requiring much milder conditions.5,6 In particular, the use of mutant glycosidases, aptly termed glycosynthases, has allowed the stereoselective formation of glycosidic linkages. However, the use of glycosynthases necessitates the supplementation of activated glycosyl donors to compensate for the inability of the mutant to activate the glycosidic bond. Several classes of glycosyl donors have been reported for glycosynthases, including 4-nitrophenyl glycosides7 and glycosyl azides,8 fluorides,9 or oxazolines.10 The use of glycosyl acetates as substrates for glycosidases has been reported, albeit seldomly.11,12 Moreover, their use as substrates for glycosynthases has never, to-date, been reported. This may stem from the fact that this donor lacks a suitable chromophore for UV analyses of reaction mixtures.
Glycosyl esters have high potential as substrates for glycosidases and glycosynthases. For example, Zinin et al. demonstrated that the value of kcat for the hydrolysis of 1-O-acetyl-β-D-galactopyranose by β-galactosidase from Penicillium sp. was three orders of magnitude greater than for p-nitrophenyl β-D-galactopyranoside.11 This finding is hardly surprising due to the higher pKa of p-nitrophenol (7.15) compared to acetic acid (4.76), rendering the latter a better leaving group. These facts prompted us to investigate the possibility of glycosyl esters also being better substrates for glycosynthases.
Herein, we report the synthesis of glycosyl benzoates using DMC. These substrates were then screened against glycosyl hydrolases and glycosynthases. The improved leaving group ability of benzoate was demonstrated by comparing the glycosylation of this substrate against common donors, such as p-nitrophenyl glycosides, thereby making glycosyl benzoates better sugar donors than their counterparts.
Results and discussion
Synthesis of glycosyl benzoates using DMC
We initially investigated the formation of glycosyl benzoates using the dehydrative reagent, 2-chloro-1,3-dimethylimidazolium chloride 1 (DMC).13 Prior reports suggested that free sugars could be transformed into the corresponding glycosyl acetate using thioacetic acid and DMC, affording the corresponding products in high yields and stereoselectivity.12 We envisaged that the use of commercially available thiobenzoic acid should yield a similar outcome. However, initial screens were quick to show that the decreased solubility of thiobenzoic acid in water necessitated the use of co-solvent in the reaction mixtures. In this vein, we treated N-acetyl-D-glucosamine 2 with thiobenzoic acid and DMC in a range of co-solvent mixtures (H2O/MeCN, H2O/dioxane, H2O/THF) and ratios (Scheme 1, Table S1, ESI†).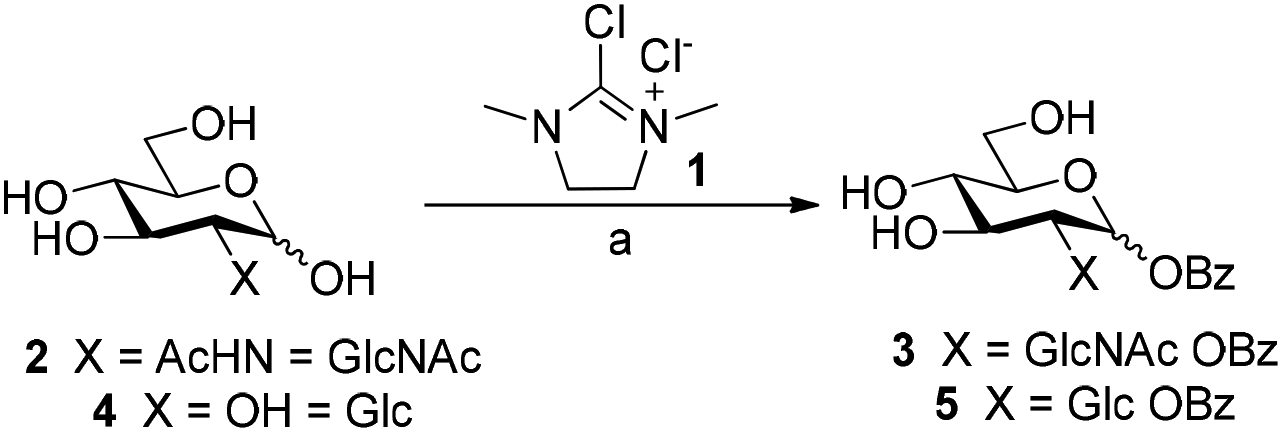 | ||
Scheme 1 Direct synthesis of glycosyl benzoates from unprotected sugars using DMC. Reaction conditions: 2 or 4, thiobenzoic acid (5 equiv.), Et3N (10 equiv.), D2O/CD3CN (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0 °C, then 1 (3 equiv.), 30 min. 3: 22%; 5: 17%. :1), 0 °C, then 1 (3 equiv.), 30 min. 3: 22%; 5: 17%. | ||
The use of MeCN as co-solvent, in a ratio of 5:1, gave the best yield of 22% and an anomeric mixture α:β of 4:1 (Table S1, entry 1, ESI†). We then moved to convert D-glucose 4 to the corresponding benzoate 5 using the same conditions (Table S2, ESI†). Similarly, we observed that the use of MeCN as the co-solvent, either as a ratio of 5:1 or 2:1, afforded 5 in a yield of 17%, exclusively as the β anomer. Further attempts to optimise the conversion by variation of reaction temperature, increasing reagents equivalents, iterative addition of reagents,12 or modifying the co-solvent ratios did not lead to any significant improvements (Fig. S1 and Tables S3, S4, ESI†). With our optimised procedure in hand, we subjected a series of unprotected mono-, di-, and trisaccharides to the reaction conditions. Subsequent isolation of the products by preparative RP-HPLC gave the corresponding glycosyl benzoates 7–10 in similar low yields (Fig. S5, ESI, entries 2–4, 3–11% yield†). While the galactosyl benzoate did form under our reaction conditions, we were unable to separate galactosyl benzoate 6 from formed by-products (Table S5, ESI, entry 1†). The rationale for the stereoselective formation of esters for sugars bearing a 2-OH was recently elucidated by Qiu et al.14 whereby any formed 1,2-cis-glycosyl acetates undergo migration of the ester from the anomeric position to 2-OH and, accordingly, we also isolated migration products by RP-HPLC. Moreover, the low yields observed in our DMC-mediated esterification, compared to that of the earlier work is likely due to the insolubility of thiobenzoic acid under our reaction conditions.
Glycosyl benzoates as substrates for glycosyl hydrolases
We first probed the use of glycosyl benzoates to act as substrates for glycosyl hydrolases (Scheme 2). Earlier work demonstrated that β-glucosyl acetate was successfully hydrolysed and transglycosylated with MeOH using β-glucosidase from almonds.12 When we incubated the same substrate with β-glucosidase from Halothermothrix orenii (HorGH1, EC 3.2.1.21), we observed rapid hydrolysis of 5 of 74% after 1 h. These values also consider the auto-hydrolysis of 5, (2% after 1 h), which reached a maximum value of 8% after 24 h. With these promising results in hand, we moved on to using 5 as a substrate for glycosynthases.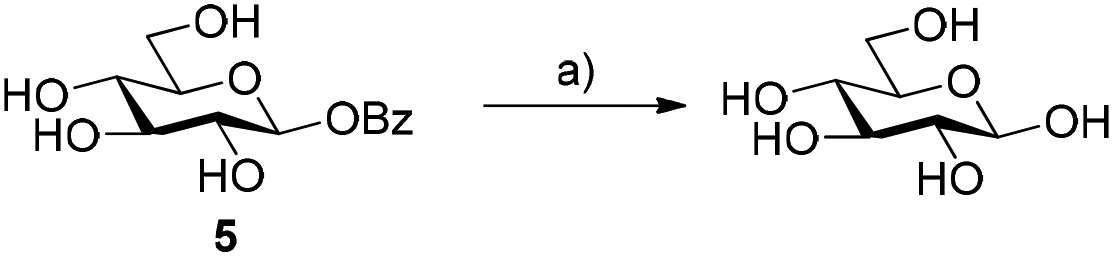 | ||
| Scheme 2 Use of β-glucosyl benzoate as a substrate for β-glucosidase from H. orenii. Reagents and conditions: (a) 1 mM β-Bz-Glc 5, HorGH1 (5.13 U), 50 mM HEPES buffer, 150 mM NaCl, pH 7.4, 25 °C, 74% after 1 h. Conversions were determined by monitoring the formation of benzoic acid over time by RP-HPLC. | ||
β-Glucosyl benzoate as a substrate for HorGH1 mutants
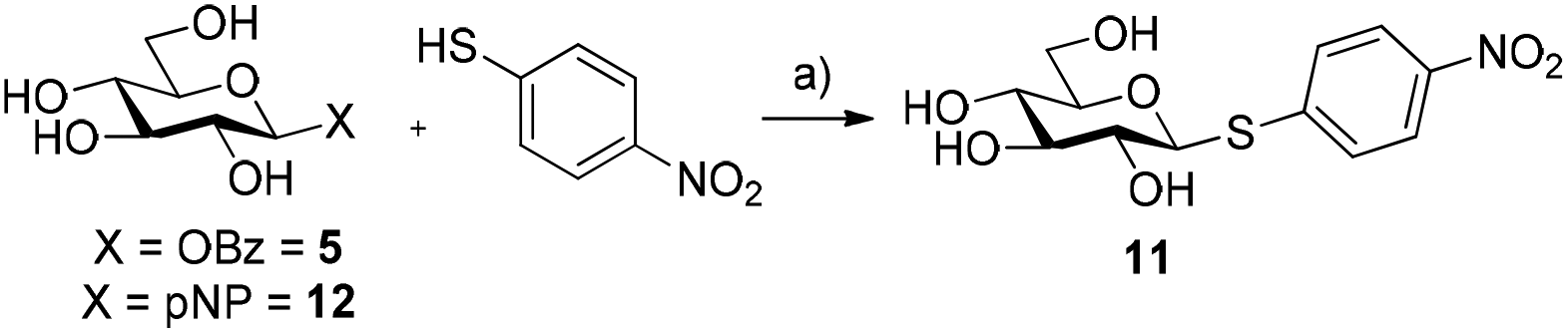
The ability of glycosyl benzoates to act as substrates for glycosynthases was then investigated. Recently, Pillet et al. demonstrated that several mutants of HorGH1 accepted aromatic thiols as substrates for thioglycoside synthesis.15 In their work, the classic p-nitrophenyl glycoside and glycosyl fluoride were used as sugar donors for the thioglycosynthase. We surmised that glycosyl benzoates would be better substrates for these mutants due to the fact that while glycosyl fluorides are good donors for glycosynthases (HF pKa = 3.8), these substrates lack a suitable chromophore for UV-spectroscopic monitoring, while p-nitrophenyl glycosides (pNP pKa = 7.15) have a poorer leaving group ability compared to that of a carboxylic acid (pKa = ∼5). In this vein, we incubated 5 with HorGH1 E166A and E166A/M299R mutants in the presence of p-nitrothiophenol as the acceptor. Pleasingly, we observed rapid formation of β-pNT-Glc 11 reaching a maximum conversion of 94% and 92% with HorGH1 E166A and HorGH1 E166A/M299R mutants, respectively, after 4 h (Table 1, entries 1 and 2; Fig. S3, ESI†). Comparatively, and in accordance with the results previously obtained by Pillet et al., when p-nitrophenyl-β-D-glucopyranoside 12 was incubated with the same enzymes under the same conditions, a low 21% and 15% conversion was observed with HorGH1 E166A and HorGH1 E166A/M299R mutants, respectively, after 24 h (Table 1, entries 3 and 4; Fig. S3, ESI†). These results confirm the superior leaving group ability of benzoate over pNP. Incubation of 5 with the acid/base and nucleophilic mutant (HorGH1 E166A/E354G) gave 11 solely as the β-anomer (Table 1, entry 5). Given that HorGH1 E166A/E354G has lost both its acid/base and nucleophilic residues, we would expect that an activated β-glycosyl donor should not be accepted as a substrate. The observation that we see the β-linked product 12 suggests that the enzyme catalyses the reaction following an SN1-type pathway, whereby the scaffold of HorGH1 E166A/E354G promotes a ping-pong like mechanism, assisting the departure of the benzoyl group, followed by subsequent thioglycosylation. Interestingly, incubation of 5 with the triple mutant (HorGH1 E166A/E354G/M299R) yielded no thioglycoside 11 (Table 1, entry 6).|
|
|||
|---|---|---|---|
| Entry | Substrate | Mutant | Conversion [%] |
| Reaction conditions: 1 mM glycosyl donor, 10 mM p-nitrothiophenol, 50 mM HEPES buffer pH 7.4, 150 mM NaCl, 30% DMSO, 20 mM β-mercaptoethanol, enzyme (0.3 mg mL−1), 25 °C. Conversions were determined by HPLC based on product formation.a Only β-linked thioglycoside was observed. | |||
| 1 | β-Bz-Glc 5 | E166A | 94 |
| 2 | β-Bz-Glc 5 | E166A/M299R | 92 |
| 3 | β-pNP-Glc 12 | E166A | 21 |
| 4 | β-pNP-Glc 12 | E166A/M299R | 15 |
| 5 | β-Bz-Glc 5 | E166A/E354G | 8a |
| 6 | β-Bz-Glc 5 | E166A/E354G/M299R | 0 |
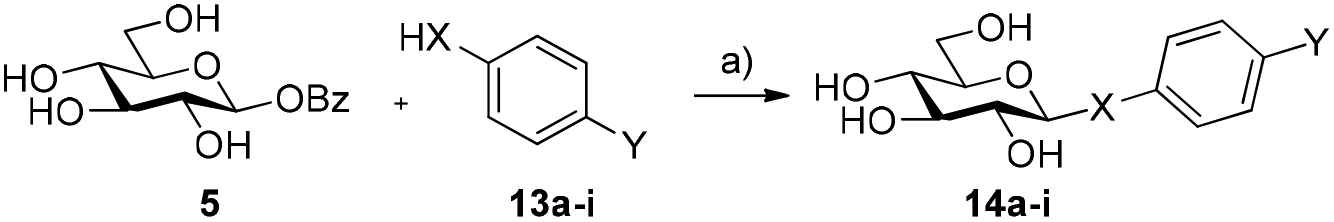
Encouraged by these findings, we then decided to screen the scope of nucleophiles capable of being accepted by the acid/base mutants under these conditions. The use of alternative aryl thiols 13a–c as acceptors yielded the corresponding thioglycosides in good to excellent conversions (72–89%, Table 2, entries 1–3; Fig. S4, ESI†), while no conversion was observed with p-aminothiophenol 13d or N,N′-dibenzoylcysteine 13e (Table 2, entries 4 and 5, respectively). Interestingly, amongst the aryl alcohols, pNP 13f gave the corresponding β-pNP-Glc 12 in 45% conversion after 1 h (Table 2, entry 6). We found that after this time, 12 gradually hydrolyses to D-glucose under the reaction conditions, reaching 22% of 12 after 24 h. This result suggests that 12 is a substrate for HorGH1 E166A and is hydrolysed by the enzyme over time. This is further supported by the fact that 12, in the absence of enzyme, was not observed to hydrolyse under the reaction conditions (data not shown). Moreover, doubling the concentration of pNP (10 mM to 20 mM) led to a slower rate of hydrolysis of the corresponding product (38% after 24 h, Fig. S5, ESI†). Interestingly, the reaction of 5 with 13f gave the corresponding product with β-stereochemistry. The more electron-rich p-methoxyphenol 13g and phenol 13h did not yield their corresponding products (Table 2, entries 7 and 8).
|
|
||
|---|---|---|
| Entry | Acceptor | Conversion [%] |
| Reaction conditions: 1 mM β-Bz-Glc 5, 10 mM acceptor, 50 mM HEPES buffer pH 7.4, 150 mM NaCl, 30% DMSO, 20 mM β-mercaptoethanol, HorGH1 E166A (0.3 mg mL−1), 25 °C. Conversions were determined by HPLC based on product formation. The stereochemistry of the products was confirmed by 1H NMR (Fig. S6, ESI†). | ||
| 1 | p-Thiotoluene (X = S; Y = CH3), 13a | 72 |
| 2 | p-Bromothiophenol (X = S; Y = Br) 13b | 89 |
| 3 | 2-Naphthalenethiol, 13c | 78 |
| 4 | p-Aminothiophenol (X = S; Y = NH2), 13d | 0 |
| 5 | N,N′-Dibenzoyl-L-cystine, 13e | 0 |
| 6 | p-Nitrophenol (X = O; Y = NO2), 13f | 45 |
| 7 | p-Methoxyphenol (X = O, Y = OCH3), 13g | 0 |
| 8 | Phenol (X = O, Y = H), 13h | 0 |
| 9 | Diphenyl diselenide, 13i | 0 |
Conclusions
In conclusion, we have developed a protecting-group-free synthesis of glycosyl benzoates using DMC and thiobenzoic acid. Albeit being low yielding reactions due to unwanted migratory and degradative processes, these substrates were shown to be suitable substrates for glycosidases and glycosynthases. We demonstrated that glycosyl benzoates perform much better than classic p-nitrophenyl donors in the thioglycosylation reaction using our single acid/base mutant HorGH1 E166A and our double mutant E166A/M299R. Therefore, we provide critical insight on how to overcome the leaving group ability limitation in the thioglycoligase strategy for the enzymatic synthesis of thioglycosides. Moreover, by mitigating the use of protecting groups, our strategy complies with the 8th principle of Green Chemistry.16 By expanding the repertoire of sugar donors available for glycosynthase-catalysed glycoside formation, we pave the way to more efficient synthetic routes for the selective and sustainable synthesis of these stable glycosidic analogues of glycosides present in many commercial products and pharmaceuticals.17,18Author contributions
F. P. supervised the project. D. L. conceptualized the idea, wrote the initial draft, and guided the synthesis of the glycosyl benzoates as well as the biotransformations. S.d.L. performed most of the experimental work. L. P. expressed and purified the enzyme variants and helped in the design of biotransformations. All authors discussed and agreed to the final version of the manuscript.Conflicts of interest
There are no conflict of interest to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation (SNSF) (grant number 200021_192274). Open access funding was provided by Universität Bern.References
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed.
- X. Wu, H. McFall-Boegeman, Z. Rashidijahanabad, K. Liu, C. Pett, J. Yu, M. Schorlemer, S. Ramadan, S. Behren, U. Westerlind and X. Huang, Org. Biomol. Chem., 2021, 19, 2448–2455 RSC.
- I. Compañón, A. Guerreiro, V. Mangini, J. Castro-Lopez, M. Escudero-Casao, A. Avenoza, J. H. Busto, S. Castillon, J. Jimenez-Barbero, J. L. Asensio, G. Jimenez-Oses, O. Boutureira, J. M. Peregrina, R. Hurtado-Guerrero, R. Fiammengo, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2019, 141, 4063–4072 CrossRef PubMed.
- L. Q. Wan, X. Zhang, Y. Zou, R. Shi, J. G. Cao, S. Y. Xu, L. F. Deng, L. Zhou, Y. Gong, X. Shu, G. Y. Lee, H. Ren, L. Dai, S. Qi, K. N. Houk and D. Niu, J. Am. Chem. Soc., 2021, 143, 11919–11926 CrossRef CAS PubMed.
- D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1, 18–23 CrossRef PubMed.
- R. A. Rocha, R. E. Speight and C. Scott, Org. Process Res. Dev., 2022, 26, 1914–1924 CrossRef CAS.
- Z. Meszaros, P. Nekvasilova, P. Bojarova, V. Kren and K. Slamova, Biotechnol. Adv., 2021, 51, 107820 CrossRef PubMed.
- B. Cobucci-Ponzano, F. Conte, E. Bedini, M. M. Corsaro, M. Parrilli, G. Sulzenbacher, A. Lipski, F. Dal Piaz, L. Lepore, M. Rossi and M. Moracci, Chem. Biol., 2009, 16, 1097–1108 CrossRef CAS PubMed.
- S. J. Williams and S. G. Withers, Carbohydr. Res., 2000, 327, 27–46 CrossRef CAS PubMed.
- C. D. Heidecke, T. B. Parsons and A. J. Fairbanks, Carbohydr. Res., 2009, 344, 2433–2438 CrossRef CAS PubMed.
- A. I. Zinin, E. V. Eneyskaya, K. A. Shabalin, A. A. Kulminskaya, S. M. Shishlyannikov and K. N. Neustroev, Carbohydr. Res., 2002, 337, 635–642 CrossRef CAS PubMed.
- D. Lim and A. J. Fairbanks, Chem. Sci., 2017, 8, 1896–1900 RSC.
- T. Isobe and T. Ishikawa, J. Org. Chem., 1999, 64, 6984–6988 CrossRef CAS.
- X. Qiu, D. Chong and A. J. Fairbanks, Org. Lett., 2023, 25, 1989–1993 CrossRef CAS PubMed.
- L. Pillet, D. Lim, N. Almulhim, A. I. Benitez-Mateos and F. Paradisi, Chem. Commun., 2022, 58, 12118–12121 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106(1), 160–187 CrossRef CAS PubMed.
- M. Qiao, L. Zhang, R. Jiao, S. Zhang, B. Li and X. Zhang, Tetrahedron, 2021, 81, 131920 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00979c |
| This journal is © The Royal Society of Chemistry 2023 |