
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFurin-targeting activity-based probes with phosphonate and phosphinate esters as warheads†
Shanping
Ji
a and
Steven H. L.
Verhelst
 *ab
*ab
aLaboratory of Chemical Biology, Department of Cellular and Molecular Medicine, KU Leuven – University of Leuven, 3000 Leuven, Belgium. E-mail: steven.verhelst@kuleuven.be
bAG Chemical Proteomics, Leibniz Institute for Analytical Sciences – ISAS, 44227 Dortmund, Germany
First published on 28th July 2023
Abstract
Activity-based probes (ABPs) are covalent chemical tools that are widely used to target proteases in chemical biology. Here, we report a series of novel ABPs for the serine protease furin with phosphonate and phosphinate esters as reactive electrophiles. We show that these probes covalently label furin and have nanomolar potencies, because of proposed interactions with the different recognition pockets around the active site of furin.
Introduction
Proprotein convertases (PCs) are serine proteases of the subtilisin/S8 family.1 They are type I transmembrane proteins that carry their protease domain extracellularly or in the lumen of the organelle. PCs occur in the secretory pathway and cleave their substrates in the Golgi, secretory granules, endosomes, at the cell surface or extracellularly. They are involved in the activation of proproteins and prohormones, and sometimes in the inactivation of proteins.The human genome codes for nine PCs, of which furin is probably the most well-known member. Furin participates in many physiological functions including the processing of cytokines, growth factors and receptors.2 Interestingly, it is involved in various aspects of carcinogenesis, including increased cell proliferation, angiogenesis, metastasis and immune evasion.3 Furthermore, furin contributes to entry of respiratory viruses such as SARS-CoV-2 by processing the viral spike protein, which binds to the human ACE2 receptor.4
The substrate specificity of furin (see Fig. 1a for general nomenclature) comprises Arg-Val-Arg/Lys-Arg in the P4–P1 position of its substrate. This has formed the basis for the design and synthesis of many peptide-based inhibitors, both covalent and non-covalent.5 Because furin inhibitors have potential as therapeutic drugs in the treatment of several human diseases,5 tools to monitor furin or PC activity in complex proteomes will aid characterization of cellular or in vivo inhibition, or may even find future use as diagnostic tools. Furthermore, it may contribute to our understanding of the complex trafficking of PCs from the secretory pathway to the cell surface and back. In order to detect active furin and potentially track its location, we started a research program to develop activity-based probes (ABPs) for covalent labelling of furin.
 | ||
| Fig. 1 Proteases and activity-based probes. (a) Proteases recognize substrates by using pockets around the scissile bond. The nomenclature of the pockets is S1, S2, etc. on the N-terminal side of the scissile bond, and S1′ etc. on the C-terminal side. The substrate side chains are named similarly: P1, P2 etc. on the N-terminal side and P1′ etc. on the C-terminal side. (b) The three elements that make up an ABP: a tag (lamp) that enables detection, a recognition element (peptide linker) for selectivity, and a warhead (fishhook) that covalently binds the target enzyme. (c) Different covalent inhibitors for furin: chloromethylketones react first with active site serine S368 and subsequently alkylate histidine H194. Diphenyl phosphonates react with the active site serine of serine proteases, giving rise to a phosphonate ester. Upon ‘ageing’ the second phenyl ester is hydrolysed. Phenyl phosphinates also react with the active site serine, but cannot undergo a second hydrolysis reaction. | ||
ABPs are mechanism-based small molecule probes that covalently react with the active site residue of enzymes. They have been particularly useful for the investigation of proteases.6,7 Generally, ABPs are assembled by the combination of three parts (Fig. 1b): (1) perhaps the most crucial element is the reactive electrophile, sometimes referred to as ‘warhead’. The warhead engages in a mechanism-based reaction with the active site nucleophile of a protease and results in covalent bond formation. The nature of the reactive electrophile determines whether a cysteine or serine protease is targeted. (2) A linker that acts as specificity element. This is usually a peptide derived from the substrate specificity of the target protease. (3) A tag, for example a fluorophore, a biotin, or biorthogonal group such as an alkyne amenable to click chemistry. It is used for detection of the covalent probe-protease complex.
For furin, a limited number of irreversible electrophilic inhibitors have been reported from which ABPs may be derived. They include the chloromethylketone8,9 and diphenyl phosphonate10 electrophiles (Fig. 1c). To the best of our knowledge, in 2016 Ferguson et al. reported the only ABP for furin thus far, with a phosphonate ester as warhead.10 Recently, our group synthesized and applied a series of novel phosphinate ester ABPs for the S1 family of serine proteases, particularly the neutrophil serine proteases.11,12 Here, we explore whether phosphinate esters can be exploited as ABPs for the S8 family of serine proteases. Specifically, we report a series of new furin-targeting ABPs with different peptide elements and phosphinate or phosphonate esters as warhead.
Results and discussion
Probe design & synthesis
Furin prefers a basic amino acid side chain in the P1 position. In order to determine which side chain would be optimal in P1, we selected the reported diphenyl phosphonate building blocks 1–3 (Scheme 1a), which were synthesized as reported before (Scheme S1†).13–17 These building blocks were then coupled with the tripeptide hexynoyl-Arg-Val-Arg-OH to yield a first probe set (10–12; Scheme 2). Preliminary labelling of furin revealed that the Arg side chain was most optimal (Fig. S1†). We therefore chose this side chain for the synthesis of all subsequent phosphinate-based ABPs. To this end, we first synthesized phosphinate ester 9 (Scheme 1b). The synthesis of building block 9 started with the reported aldehyde 4, obtained from Swern oxidation of Phth-protected 4-amino-1-butanol.18 The phosphinate scaffold was made from compound 4 with benzyl carbamate and dichlorophenylphosphine. The latter reagent was chosen, because earlier research by our laboratory had indicated that a phenyl group attached to the phosphor atom was necessary for sufficient reactivity with the active site serine.12 This 3-component reaction led to formation of compound 5 in 58% yield. Compound 5 was subsequently esterified with phenol under influence of DIC and DMAP to give compound 6. Free amine 7, obtained after removal of the Phth protecting group was guanidinylated by using 1,3-di-Boc-2-methylisothiourea and HgCl2, yielding building block 8. We found that removal of the Cbz group under hydrogenation conditions with palladium on carbon led to hydrolysis of the phenyl ester. Therefore, the Cbz protecting group was removed by 33% HBr–AcOH19 to yield the phosphinate building block 9 (Scheme 1b).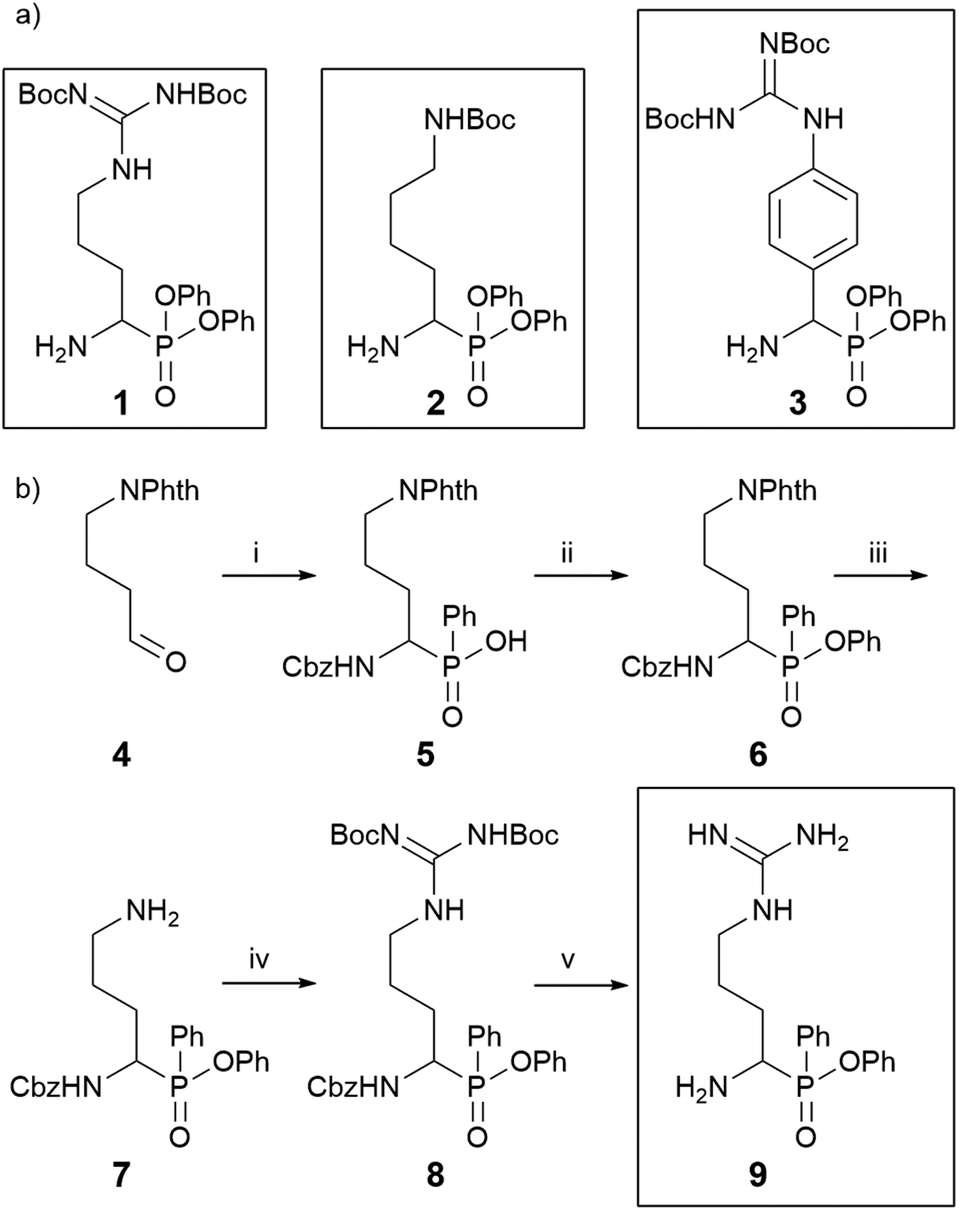 | ||
| Scheme 1 Phosphonate and phosphinate reactive electrophile warheads. (a) Chemical structures of the used diphenyl phosphonate building blocks of Arg (1), Lys (2), and p-guanidino-phenylglycine (3), an Arg mimic, synthesized as reported before.18–26 (b) Synthesis of the Arg phenyl phosphinate building block 9, (i), CbzNH2, dichlorophenylphosphine, acetonitrile, 58%; (ii), DIC, DMAP, PhOH, toluene, 84%; (iii), H2NNH2·H2O, in i-PrOH, 69%; (iv) 1,3-di-Boc-2-methylisothiourea, Et3N, HgCl2, yield 68%; (v) 33% HBr/AcOH. | ||
 | ||
| Scheme 2 ABP synthesis. N-Hexynoyl-capped tri- or tetrapeptides were synthesized by standard Fmoc solid phase peptide synthesis. These peptides were coupled in solution to building blocks 1, 2, 3 or 9. Reagents and conditions: (i) HATU, collidine, DMF. (ii) 95% TFA, 2.5% TIS, 2.5% H2O. The structures of the final probes with their sequences in P5–P1 and the substituent on the phosphor atom are summarized below the general probe structure. | ||
Next, we coupled the Arg phosphonate and phosphinate building blocks to various hexynoyl-labeled peptides in order to complete our furin probes. We made use of the known furin substrate specificity, which is reported as Arg-Val-Arg-Arg (P4–P1) as well as information on previously reported furin inhibitors: an additional basic residue in the P5 position boosts potency of non-covalent peptide-based inhibitors.20 Moreover, replacement of the of the P2 Arg by Lys decreases in vivo toxicity in mice.21 For our final probe set 13–19, we therefore used a peptide with Arg or Lys in the P2 position and an optional P5 element consisting of an Arg or a 4-aminomethyl-phenylalanine non-natural amino acid (Scheme 2). The probes were created by coupling the warhead building blocks to the hexynoyl-capped tri- or tetrapeptides, which were generated by general solid phase peptide synthesis on chlorotrityl resin (Scheme 2a). With this probe set in hand, we proceeded to labeling experiments with recombinantly expressed furin.
Probe evaluation
To test labeling of furin by the obtained phosphonate and phosphinate ABPs 10–19, we transfected HEK293T cells with a plasmid coding for recombinant, flag-tagged furin.22 Because furin is partially shed from the membrane, we concentrated the serum-free conditioned medium and used this for our labelling experiments. Reactions were performed in an optimized buffer containing 2 mM CaCl2 (Fig. S2†). Treatment with 10 μM of probes 10–19 followed by click chemistry with a TAMRA-azide fluorophore showed clear fluorescent bands at the molecular weight of furin (Fig. 2a). This band was absent in the dmso control, indicating no click chemistry background. Importantly, the bands diminished upon pretreatment with Furin I inhibitor, a widely used active-site directed chloromethyl ketone based inhibitor (see Fig. S3† for structure), confirming the specificity of the probes for the active site. | ||
| Fig. 2 Probe evaluation experiments. (a) Labeling of furin with probes 10–19. Pretreatment with Furin I inhibitor completely blocked labeling. (b) ICApp50 determined by a serial dilution of probes 10–19 with measurement of residual activity by a fluorogenic furin substrate. (c) Probe labelling by a titration series of probes 10–19. (d) Furin labelling in live HEK293T cells expressing FLAG-tagged furin. Furin labelling appears as a band at approximately 100 kDa. Note that the anti-FLAG western blot reveals two furin bands, the upper being the proenzyme. Ponceau stain as loading control is provided as Fig. S5.† | ||
To gain insight into the differences in potency of these probes, we determined the apparent IC50 values using the commercially available fluorescent Furin substrate Pyr-Arg-Thr-Lys-Arg-AMC. In the phosphonate series, the pentapeptide probes 13 and 14 were most potent, with ICApp50 values of 65 and 23 nM, respectively (Fig. 2b and Fig. S4†). Of all phosphinates, the pentapeptide probe 18 was the most potent inhibitor with an ICApp50 value of 99 nM (Fig. 2b). When comparing phosphonate probes and phosphinate probes carrying the same peptide recognition element, the phosphonates display an overall somewhat better inhibitory capacity than the phosphinates.
Labeling of furin with a serial dilution of the probes followed by click chemistry detection of the covalent complex also revealed differences in the probe potencies (Fig. 2c). The general trend matches with the IC50 differences, with ABP 14 being the phosphonate that most sensitively detects furin and ABP 18 as the best phosphinate. Both were able to visualize furin on gel at 16 nM probe concentration (Fig. 2c).
Before performing experiments on live cells, we evaluated the probes for cytotoxicity. Fortunately, ABPs displayed no substantial negative effects on cell viability (Fig. S6†). We next performed labelling of furin in live cells. To this end, HEK293T cells, transfected to express recombinant furin, were incubated with the different probes at 20 μM concentration. After cell lysis using a buffer compatible with click chemistry, probe-labeled furin was tagged with a TAMRA-azide fluorophore under influence of copper(II)sulfate, sodium ascorbate and a Cu+ ligand. All probes, except probe 11, showed labelling of furin, which appeared at a MW of approximately 100 kDa, in contrast to shed furin, which misses the transmembrane domain and has a lower molecular weight. Labeling intensity shows a similar trend as on shed furin, with the most efficient labelling for probes 13, 14 and 18 (Fig. 2d).
To obtain insight into the binding interactions of the new phosphinate probes with the furin active site, we performed covalent molecular docking with the most potent phosphinate probe, 18. To this end, we used a reported crystal structure of human furin in complex with a peptidomimetic inhibitor.23 To minimize the amount of rotatable bonds during docking and to maximize possible overlap with the co-crystallized inhibitor, the P5 element and detection tag were removed from probe 18 prior to docking. The phosphinate building blocks have two chiral centers: one at the α-carbon and one at the phosphor center. We here only used the R-configuration at the α-carbon, as this corresponds to the natural L-configuration of the amino acid in the P1 position and constitutes the active isomer as shown in previous studies.24 We only obtained a good docking pose from an S-chirality at the phosphine center (Fig. S7†). We therefore propose that the RS isomer is likely the (most) active species. Interestingly, a similar preference for the RS isomer was suggested for S1 family serine proteases.12 Overall, probe 18, covalently bound to active site Ser368, fits well into the substrate binding cleft (Fig. 3a) It displays very good overlap with a crystal structure of a previously reported active-site directed furin inhibitor (Fig. 3b).23 The side chains of the inhibitor make various interactions with the protein. For example, the P1 Arg interacts with Asp258 and Asp306 as well as various backbone atoms in the S1 pocket (Fig. 3c) and the P2 Lys hydrogen bonds to Asp154 as well as a backbone carbonyl (Fig. 3d). The phenyl group of the phosphinate extends towards the primed site. Future extension of this substituent may lead to increased potency through interactions with proximal side chains Arg298, Trp328 or Thr365 (Fig. 3e). In conclusion, the docking reveals interactions with the furin active site similar to those of other peptide-like inhibitors.
 | ||
| Fig. 3 Covalent docking results of phosphinate probe 18 in the active site of furin (PDB code: 5JXH). The protein is displayed in cartoon mode, probe and a few selected residues of the furin active site in stick model. (a) Probe 18 (in cyan) with the furin protein as semi-transparent surface reveals a good fit with the substrate binding cleft. (b) Overlay of the docked structure with the original peptidomimetic inhibitor. Note the good overlay in the P1, P2 and P3 site. (c) Close-up of the S1 pocket with interactions of the arginine P1 with Asp258 and Asp306. (d) Close-up of the S2 pocket shows interaction of the lysine P1 with Asp 154. (e) Close-up of the primed site with residues indicated for potential future interactions if the primed site is extended. | ||
Lastly, we checked whether these probes also show activity against other PCs. To this end, we determined ICApp50 values against PC1, a PC whose expression mainly occurs in neurons and endocrine tissues. As for furin, we found that most of our probes also display nanomolar potencies against PC1 (Fig. S8 and Table S1†), which is unsurprising in view of the similar substrate specificities amongst the PCs.25 Hence, the here reported probes may also be used for labeling other PCs, and future work may be directed towards obtaining higher probe selectivity.
Conclusions
Here, we have reported the first pentapeptide phosphonate ABPs and the first phosphinate-derived inhibitors and ABPs for furin. Activity assays revealed that attaching a basic residue in the P5 position increases the potency of the probes towards human furin. Although in general the phosphonates had higher potency, the best phosphinate, compound 18, still displayed an ICApp50 value of 99 nM, and could sensitively detect furin by gel-based ABPP. Molecular docking experiments suggest a good fit into the substrate binding cleft and possibilities to extend the phenyl substituent on the phosphinate in order to increase interactions with the furin primed site. With the ability of these ABPs to covalently label furin at low concentration and in live cells, we think that attaching fluorophores to these molecules will result in probes compatible with fluorescent microscopy. Conveniently, the phosphinates contain one single leaving group to which potentially a fluorescent quencher may be added, resulting in quenched fluorescent ABPs,26,27 which may be amenable to live cell imaging. We therefore expect that the probes reported here hold promise as powerful new tools for detection and imaging of active furin in disease models.Author contributions
SJ performed all experiments. SV conceptualized the study. SJ and SV wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank J. Creemers for the kind gift of vectors for expression of recombinant furin. SJ acknowledges funding of the Chinese Scholarship Council (fellowship no.: 202008440293). This work was further funded by the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin–inkl. Wissenschaft und Forschung, and the Bundesministerium für Bildung und Forschung.References
- N. D. Rawlings, M. Waller, A. J. Barrett and A. Bateman, Nucleic Acids Res., 2013, 42, D503–D509 CrossRef PubMed.
- G. Thomas, Nat. Rev. Mol. Cell Biol., 2002, 3, 753–766 CrossRef CAS PubMed.
- Z. He, A.-M. Khatib and J. W. M. Creemers, Oncogene, 2022, 41, 1252–1262 CrossRef CAS PubMed.
- M. Takeda, Microbiol. Immunol., 2022, 66, 15–23 CrossRef CAS PubMed.
- F. Couture, A. Kwiatkowska, Y. L. Dory and Y. R. Da, Expert Opin. Ther. Pat., 2015, 25, 1–18 CrossRef.
- S. Serim, U. Haedke and S. H. L. Verhelst, ChemMedChem, 2012, 7, 1146–1159 CrossRef CAS PubMed.
- S. Chakrabarty, J. P. Kahler, M. A. T. van de Plassche, R. Vanhoutte and S. H. L. Verhelst, Curr. Top. Microbiol. Immunol., 2018, 420, 253–281 Search PubMed.
- A. Stieneke-Gröber, M. Vey, H. Angliker, E. Shaw, G. Thomas, C. Roberts, H. D. Klenk and W. Garten, EMBO J., 1992, 11, 2407–2414 CrossRef PubMed.
- W. Garten, S. Hallenberger, D. Ortmann, W. Schäfer, M. Vey, H. Angliker, E. Shaw and H. D. Klenk, Biochimie, 1994, 76, 217–225 CrossRef CAS PubMed.
- T. E. G. Ferguson, J. A. Reihill, B. Walker, R. A. Hamilton and S. L. Martin, PLoS One, 2016, 11, 1–9 Search PubMed.
- J. P. Kahler and S. H. Verhelst, RSC Chem. Biol., 2021, 2, 1285–1290 RSC.
- J. P. Kahler, S. Lenders, M. A. T. van de Plassche and S. H. L. Verhelst, ACS Med. Chem. Lett., 2020, 11, 1739–1744 CrossRef CAS PubMed.
- D. S. Jackson, S. A. Fraser, L.-M. Ni, C.-M. Kam, U. Winkler, D. A. Johnson, C. J. Froelich, D. Hudig and J. C. Powers, J. Med. Chem., 1998, 41, 2289–2301 CrossRef CAS PubMed.
- J. Oleksyszyn, B. Boduszek, C.-M. Kam and J. C. Powers, J. Med. Chem., 1994, 37, 226–231 CrossRef CAS PubMed.
- T. L. T. C.-L. J. Wang, A. J. Mical, S. Spitz and T. M. Reilly, Tetrahedron Lett., 1992, 7667–7670 CrossRef.
- M. Skorenski, A. Milewska, K. Pyrc, M. Sienczyk and J. Oleksyszyn, J. Enzyme Inhib. Med. Chem., 2019, 34, 8–14 CrossRef CAS PubMed.
- M. Sienczyk and J. Oleksyszyn, Tetrahedron Lett., 2004, 45, 7251–7254 CrossRef CAS.
- M. Spallarossa, Q. Wang, R. Riva and J. Zhu, Org. Lett., 2016, 18, 1622–1625 CrossRef CAS PubMed.
- T. Kryza, T. Khan, S. Lovell, B. S. Harrington, J. Yin, S. Porazinski, M. Pajic, H. Koistinen, J. K. Rantala, T. Dreyer, V. Magdolen, U. Reuning, Y. W. He, E. W. Tate and J. D. Hooper, Nat. Chem. Biol., 2021, 17, 776–783 CrossRef CAS PubMed.
- G. L. Becker, Y. Lu, K. Hardes, B. Strehlow, C. Levesque, I. Lindberg, K. Sandvig, U. Bakowsky, R. Day, W. Garten and T. Steinmetzer, J. Biol. Chem., 2012, 287, 21992–22003 CrossRef CAS PubMed.
- T. Ivanova, K. Hardes, S. Kallis, S. O. Dahms, M. E. Than, S. Kunzel, E. Bottcher-Friebertshauser, I. Lindberg, G. S. Jiao, R. Bartenschlager and T. Steinmetzer, ChemMedChem, 2017, 12, 1953–1968 CrossRef CAS PubMed.
- J. W. M. Creemers, L. E. Pritchard, A. Gyte, P. Le Rouzic, S. Meulemans, S. L. Wardlaw, X. Zhu, D. F. Steiner, N. Davies, D. Armstrong, C. B. Lawrence, S. M. Luckman, C. A. Schmitz, R. A. Davies, J. C. Brennand and A. White, Endocrinology, 2006, 147, 1621–1631 CrossRef CAS PubMed.
- S. O. Dahms, M. Arciniega, T. Steinmetzer, R. Huber and M. E. Than, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11196–11201 CrossRef CAS PubMed.
- B. Walker, S. Wharry, R. J. Hamilton, S. L. Martin, A. Healy and B. J. Walker, Biochem. Biophys. Res. Commun., 2000, 276, 1235–1239 CrossRef CAS PubMed.
- A. G. Remacle, S. A. Shiryaev, E. S. Oh, P. Cieplak, A. Srinivasan, G. Wei, R. C. Liddington, B. I. Ratnikov, A. Parent, R. Desjardins, R. Day, J. W. Smith, M. Lebl and A. Y. Strongin, J. Biol. Chem., 2008, 283, 20897–20906 CrossRef CAS PubMed.
- S. Serim, P. Baer and S. H. L. Verhelst, Org. Biomol. Chem., 2015, 13, 2293–2299 RSC.
- E. Bisyris, E. Zingkou, G. G. Kordopati, M. Matsoukas, P. A. Magriotis, G. Pampalakis and G. Sotiropoulou, Org. Biomol. Chem., 2021, 19, 6834–6841 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00948c |
| This journal is © The Royal Society of Chemistry 2023 |