
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Peptides incorporating polyhydroxylated cyclohexane β-amino acid: synthesis and conformational study†‡
David
Reza
 a,
Rosalino
Balo
ac,
José M.
Otero
a,
Ai M.
Fletcher
c,
Rebeca
García-Fandino
ab,
Víctor M.
Sánchez-Pedregal
b,
Stephen G.
Davies
c,
Ramón J.
Estévez
ab and
Juan C.
Estévez
*ab
a,
Rosalino
Balo
ac,
José M.
Otero
a,
Ai M.
Fletcher
c,
Rebeca
García-Fandino
ab,
Víctor M.
Sánchez-Pedregal
b,
Stephen G.
Davies
c,
Ramón J.
Estévez
ab and
Juan C.
Estévez
*ab
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica. Universidade de Santiago de Compostela, c/Jenaro de la Fuente s/n, 15782 Santiago de Compostela, Spain. E-mail: juancarlos.estevez@usc.es
bDepartamento de Química Orgánica, Universidade de Santiago de Compostela, Avda. das Ciencias s/n, 15782 Santiago de Compostela, Spain
cChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
First published on 6th October 2023
Abstract
We describe the synthesis of trihydroxylated cyclohexane β-amino acids from (−)-shikimic acid, in their cis and trans configuration, and the incorporation of the trans isomer into a trans-2-aminocyclohexanecarboxylic acid peptide chain. Subsequently, the hydroxyl groups were partially or totally deprotected. The structural study of the new peptides by FTIR, CD, solution NMR and DFT calculations revealed that they all fold into a 14-helix secondary structure, similarly to the homooligomer of trans-2-aminocyclohexanecarboxylic acid. This means that the high degree of substitution of the cyclohexane ring of the new residue is compatible with the adoption of a stable helical secondary structure and opens opportunities for the design of more elaborate peptidic foldamers with oriented polar substituents at selected positions of the cycloalkane residues.
Introduction
β-Amino acids are attractive building blocks for the synthesis of peptidomimetics because of their propensity to fold into diverse secondary structures (such as helices, turns, sheets, and hairpins) in short peptide sequences.1–6 In addition, their resistance to protease degradation7,8 is beneficial for therapeutic applications.4,9–14Carbocyclic β-amino acids are of special interest due to the limited flexibility derived from their constrained cyclic structures and have been demonstrated to act as particularly strong helix or turn inducers.5,15–17 This has led, for instance, to the design of bioactive β-peptides,18–20 self-assembling nanomaterials21–24 and catalysts.25,26
The folding properties of carbocyclic β-peptidic foldamers have been widely studied. Pioneering work demonstrated that homooligomers of trans-2-aminocyclohexanecarboxylic acid (trans-ACHC) fold as a robust 14-helix in short peptide sequences15,27 while homooligomers of trans-2-aminocyclopentanecarboxylic acid (trans-ACPC) fold as a 12-helix16,28,29 in the crystal and in solution. Other secondary structures can be accessed by modification of the backbone stereochemical configuration30,31 or the side chains32,33 of homooligomers. Another strategy to tune the peptide secondary structure has been the combination of two or more β-amino acids into heterooligomers.6,34–37 Peptides containing the cis-ACHC diastereoisomer have also been studied. Homochiral oligomers of cis-ACHC do not form helical structures, although it has been shown to participate in helical folds of heterooligomers and hybrid peptides.33,38,39
Most of these studies have been carried out with highly hydrophobic compounds because the number of foldamers containing cyclic β-amino acid monomers functionalized with polar substituents is still limited.6 The most frequent strategy to obtain polar or water soluble foldamers based on cyclic β-amino acids has been the synthesis of heterooligomers of apolar trans-ACPC or trans-ACHC residues with structurally simple polar acyclic β-amino acids holding a polar group in their side chain (e.g. β-serine or β-lysine).19,35,40–44 Other examples have the polar group in an otherwise structurally simple cyclic amino acid, as the trans-4-aminopyrrolidine-3-carboxylic acid (trans-APC),18,20,34trans-DCHC,45,46 or an aza-ACPC analogue.47
Polyhydroxylated cycloalkane β-amino acids are particularly interesting targets, because their rich functionality makes them useful scaffolds for the access of a variety of lipo- or hydrosoluble peptides, by protection or deprotection of their hydroxy substituents. In addition, they can bear pharmacophoric groups in well defined spatial orientations, a property that may facilitate their interaction with biological receptors, or with substrates if the goal is to use them as peptidic catalysts. Some previous work on the stereoselective synthesis of mono-, di-, tri- and tetrahydroxylated cycloalkane β-amino acids has been reported but, to our knowledge, their incorporation into peptides has not yet been addressed17,36,48–50 except in one example.51
In connection with our interest in polyhydroxylated cycloalkane amino acids and their peptides,52,53 we present here an efficient stereoselective synthesis of two new polyhydroxylated cyclohexane β-amino acids with cis- and trans- configurations, starting from commercially available (−)-shikimic acid. The newly synthesized trans-β-amino acid was incorporated into a short heterooligomeric peptide also containing trans-ACHC and its structure in solution has been studied.54
Results and discussion
Synthesis of polyhydroxylated cyclohexane β-amino acids 5 and 8
(−)-Shikimic acid is an attractive starting material on account of its convenient structural properties: a preformed cyclohexane ring bearing an α,β-unsaturated carboxylic acid moiety and three hydroxy substituents with well-defined spatial orientations.55,56 The synthetic plan involved a stereoselective Michael addition of dibenzylamines to α,β-unsaturated esters, in this case methyl shikimates.57Addition of the known shikimate ester 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 53 to a cooled (−78 °C) solution of lithium amide (R)-2 in THF provided, after 2 h reaction followed by work-up with saturated aq. ammonium chloride solution, a mixture of cis-β-amino acid 3 (major component, 61% yield) and trans-β-amino acid 6 (minor component, 5% yield). In addition, reaction of compound 3 with a 2 M solution of NaOMe in MeOH resulted in its isomerization to compound 6 (89% yield). Compounds 3 and 6 were easily identified from their analytical and spectroscopic data. The structure of compound 3 was unambiguously established by X-ray crystallography (Scheme 1).
53 to a cooled (−78 °C) solution of lithium amide (R)-2 in THF provided, after 2 h reaction followed by work-up with saturated aq. ammonium chloride solution, a mixture of cis-β-amino acid 3 (major component, 61% yield) and trans-β-amino acid 6 (minor component, 5% yield). In addition, reaction of compound 3 with a 2 M solution of NaOMe in MeOH resulted in its isomerization to compound 6 (89% yield). Compounds 3 and 6 were easily identified from their analytical and spectroscopic data. The structure of compound 3 was unambiguously established by X-ray crystallography (Scheme 1).
 | ||
| Scheme 1 | ||
Catalytic hydrogenation of compound 3, using Pd(OH)2, provided amino acid ester 4, which upon reaction with 6 M HCl resulted in the formation of the new trihydroxylated cis-2-aminocyclohexanecarboxylic acid 5 (76% yield over the two steps). Similarly, compound 6 provided the new trihydroxylated trans-2-aminocyclohexanecarboxylic acid 8 (77% yield over the three steps), via compound 7.
The favoured Michael addition of the chiral amide (R)-2 to the si-face of the α,β-unsaturated carboxylic acid ester moiety of 1 can be justified as a case of double chiral induction by the reagent and the substrate, leading to the stereospecific formation of the intermediate enolate shown in Scheme 2 by the complexation of the litium ion with the carbonyl oxygen and the amino atoms. Then, protonation of this enolate can occur according either to path a (more favoured) or path b (less favoured), the result being the formation of a mixture of compound 3 (kinetic isomer, major component) and compound 6 (thermodynamic isomer, minor component) (Scheme 2).58,59
 | ||
| Scheme 2 | ||
We chose to introduce the nitrogen substituent with amide (R)-2 because it highly favours the formation of the cis-β-amino ester 3 (kinetic product), which can then be quantitatively transformed into the trans-β-amino ester 6 (thermodynamic product). Although dibenzylamine could also be tried, our previous experience with similar compounds indicated that it would likely lead to a mixture of both isomers in uncertain proportions.53
Synthesis of pentamers 14, 15 and 16, containing trans-polyhydroxylated cyclohexane β-amino acid 8
The newly synthesized beta amino acids 5 and 8 can be used as components of peptidomimetics. Oligomers of trans-ACHC are known to adopt the 14-helix secondary structure.15,16,27 while the conformational properties of peptides containing cis-ACHC and derivatives are more uncertain33,38,39 and depend on the specific composition in heterooligomers and/or hybrid peptides. With the aim of obtaining a predictable structure, in this work we focused on the trans-β-amino acid 8. Therefore, we synthesized pentamer 14 that has the functionalized β-amino acid 8 in the central position, flanked by trans-ACHC residues (Scheme 3). Sequential deprotection of the hydroxyls of 14 led to pentamers 15 and 16, which allowed us to study the influence of their degree of polarity and steric hindrance on their propensity to adopt the 14-helix secondary structure. | ||
| Scheme 3 | ||
The synthesis of the target pentamers 14–16 was performed in solution starting from compound 7, an orthogonally protected equivalent of β-amino acid 8, that was coupled with dimer 10 (Boc-trans-(ACHC)2), obtained from dimer 9 (Boc-trans-(ACHC)2-OBn),27 to give trimer 11 in 93% yield characterized from their spectroscopic and spectrometric properties (see ESI‡). Dimer 13 (trans-(ACHC)2-OBn) was also obtained from (Boc-trans-(ACHC)2-OBn) (see the ESI‡). Treatment of trimer 11 with LiOH to give the acid derivative 12 (92%), followed by coupling with dimer 13, gave the expected pentamer 14 (Scheme 3).
Pentamer 14 was transformed into the monohydroxylated pentamer 15 in 76% yield by treatment with TBAF. Finally, pentamer 15 was transformed into the trihydroxylated pentamer 16 when submitted to a hydrolysis reaction catalyzed by acetic acid. All three pentamers 14–16 were purified and then subjected to structural studies in solution.
Infrared spectroscopy
We measured the ATR-FTIR spectra of dimer 9, trimer 11 and the three pentamers 14–16 in the solid state to assess the formation of hydrogen bonds. Significant differences were observed for the vibration of the Amide A band, with clearly lower wavenumber values for pentamers 14 (3281 cm−1) and 16 (3269 cm−1) than for dimer 9 (3297 cm−1) and trimer 11 (3327 cm−1) as shown in Fig. 1A. The vibration Amide II band shifts to higher wavenumbers from dimer 9 (1533 cm−1) and trimer 11 (1528 cm−1) to pentamers 14 (1536 cm−1), 15 (1547 cm−1) and 16 (1547 cm−1) (Fig. 1B). Differences in the Amide I band of the five compounds were negligible. | ||
Fig. 1
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) max of the ATR-FTIR spectra of peptides 9, 11, 14, 15 and 16 in the Amide A (A) and Amide II (B) regions.
max of the ATR-FTIR spectra of peptides 9, 11, 14, 15 and 16 in the Amide A (A) and Amide II (B) regions. | ||
The signals of Amide A and Amide II are compatible with the increase of hydrogen bonding as the polyamide chain gets longer. Such behaviour is the expected for a higher level of secondary structure formation as oligomer length increases. Upon formation of H-bonds, vibration frequencies of Amide A shift to lower wavenumbers, while vibrations of Amide II shift to higher wavenumbers. However, this information is not sufficient to determine what fold (e.g. what helix type) is adopted by the peptide.60,61
CD spectroscopy
Circular dichroism (CD) spectroscopy can be used to establish the secondary structure of polyamide chains, since each type of secondary structure gives rise to a characteristic CD spectrum in the far ultraviolet region (190–240 nm).62 In fact, it has been used routinely to establish the secondary structure of peptide α-amino acids and even establish the proportion of each of these secondary structures in natural proteins.63 For the case of polyamides constituted by other than α-amino acids, there is no simple way to correlate far-ultraviolet CD spectra with their specific conformations,64 but comparison of CD data from new and existing polyamides can provide useful information if structures are similar.The CD spectra of dimer 9, trimer 11 and pentamer 14, at 1 mM concentration in methanol (Fig. 2A), present an ellipticity maximum in the far ultraviolet region around 220 nm, that arises from the backbone amide groups. The intensity of this maximum increases with β-peptide length.
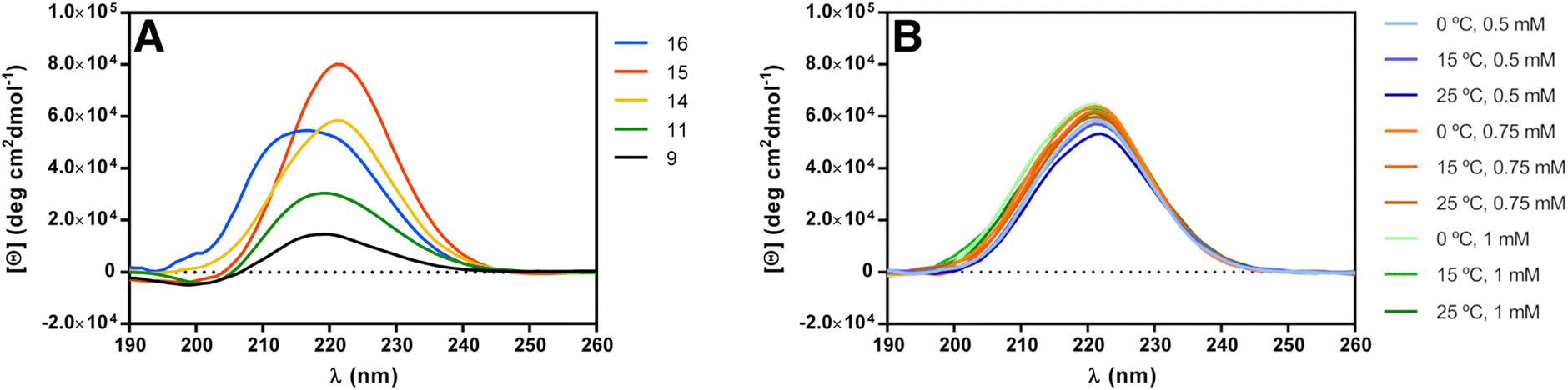 | ||
| Fig. 2 (A) CD spectra of peptides 9, 11, 14, 15 and 16 in methanol (c = 1 mM). (B) Concentration and temperature dependence of the CD spectra of peptide 14 in methanol. | ||
We then compared the CD spectra of the three differently protected pentamers 14–16, at 1 mM concentration in methanol solution (Fig. 2A). The three pentamers present an absorption maximum in the far ultraviolet region of about 220 nm. The slight shift of θmax (221 nm for 14, 221 nm for 15 and 217 nm for 16) can be justified assuming that the conformation of the helices are slightly different when the substituted cyclohexane ring is strained by the acetonide group (14 and 15) to when this acetonide is not present (16). Indeed, the value of θmax of 16 is identical to the reported value for the unsubstituted trans-ACHC oligomers (217 nm). Slight shifts of the CD maxima have also been attributed to different populations of the 14-helix averaging with other conformers existing in solution.27,43
Additionally, we performed CD experiments with pentamer 14 by changing the concentration and temperature (Fig. 2B). Increasing the concentration resulted in higher intensity but no shift of the θmax, consistent with the molecule not aggregating in this concentration range. More interestingly, negligible changes occurred upon increasing temperature from 0 °C to 25 °C, demonstrating the overall stability of the structure.
NMR spectroscopy
We analyzed the structure of pentamers 14–16 by NMR spectroscopy in CDCl3, DMSO-d6, and methanol-d3 solutions. 1H NMR spectra of peptides 14 and 15 in CDCl3 solution had multiple amide HN peaks and broad lines, indicating more than a single conformation and aggregation. Peptide 16 formed a gel in CDCl3 and no further NMR studies were done in this solvent.Dispersion of chemical shifts in DMSO-d6 solution was sufficient to analyze the NMR spectra of compounds 14–16.65 Amide HN resonances were well dispersed in the 1D proton spectra, suggesting a high population of a single well-defined conformation of each compound. Residue-specific assignments of the backbone HN, Hα, and Hβ protons were made based on a combination of COSY, TOCSY and ROESY spectra. The other side-chain resonances were typically not assigned due to extensive overlap (see tables of assignments in ESI‡).
The backbone fold of the peptides was determined from NOE contacts characteristic of the 14-helix secondary structure (Fig. 3), like the strong long-range Hα(i)/Hβ(i + 3) and sequential HN(i)/Hα(i − 1) NOEs, that correspond to H–H distances of ≈2.2–2.6 Å in a 14-helix.15,37,42,65 Further support to the 14-helix fold comes from the HN(i)/Hβ(i + 2) and the HN(i)/Hβ(i + 3) NOE peaks of medium intensity, that correspond to distances in the range of 3.0–3.5 Å (see the ESI for tables of NOEs of the three peptides‡). Some NOEs from residue 3 to the terminal t-Bu and OBn groups are also compatible with the 14-helix conformation. The uniform amide three-bond coupling values 3J(HNi–Hβi) in the range of 7–9 Hz (Table 1) correspond to dihedral angles of the HN(i)–CHβ(i) fragments close to 160°,66,67 that is also characteristic of the 14-helix secondary structure.15,65
 | ||
| Fig. 3 Summary of detected ROESY cross-peaks of peptides 14–16 in DMSO-d6 solution. Arrows representing the strong intraresidual HN(i)/Hα(i) cross-peaks have been omitted for clarity. Intensities: HN(i)/Hα(i – 1) and Hα(i)/Hb(i + 3), strong; HN(i)/Hβ(i), medium; HN(i)/Hβ(i + 2), medium or unknown due to peak overlap. The complete tables of ROESY cross-peak intensities are included in the ESI.‡ | ||
| 3 J HN–Hβ (Hz) | T-coef (–ppb K−1) | |||||
|---|---|---|---|---|---|---|
| 14 | 15 | 16 | 14 | 15 | 16 | |
| HN1 | 8.3 | 8.5 | 8.8 | 7.3 | 7.5 | 5.2 |
| HN2 | 7.9 | 8.3 | 8.6 | 5.6 | 4.9 | 4.3 |
| HN3 | 8.3 | 9.2 | 7.3 | 0.9 | 1.7 | 3.4 |
| HN4 | 8.4 | 8.6 | 8.6 | 9.0 | 9.2 | 8.2 |
| HN5 | 7.9 | 8.6 | 8.9 | 5.2 | 5.4 | 4.9 |
Restrained molecular dynamics calculated with XPLOR-NIH68,69 using the NOE and scalar coupling data led to structures with the 14-helical fold for peptides 14–16. These models were further optimized using density functional theory (DFT) calculations that employed the hybrid density functional M052X70 with the 6-31G(d) basis set. DFT calculations were performed using Gaussian 09.71
To analyze the alignment of the atoms intervening in hydrogen bonding, we considered the distances and angles listed in Table 2. In principle, as it is stated in the IUPAC definition for hydrogen bonds, the closer the X–H⋯Y angles angle is to 180°, the stronger is the hydrogen bond and the shorter is the H⋯Y distance.72 The structure of 16 resembled very closely the geometry of the crystallographic structure of the trans-ACHC homohexamer (RMSD = 0.236 Å for backbone atoms Cα, Cβ, N and C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of residues 1–5; Fig. 4),15 presenting the shortest O⋯N and O⋯H distances and the largest CO⋯H angles. In contrast, the geometry of the hydrogen bonds of peptides 14 and 15 in the DFT optimized models was a bit distorted, while keeping the 14-helix fold, due to the steric constraints imposed by the substituents on positions γ, δ and ε of residue 3 (Fig. 5). This agrees well with the CD data recorded in methanol, as the maximum ellipticity of peptides 14 and 15 deviates a few nm from the values of the trans-ACHC homohexamer and peptide 16. Further support to the 14-helix fold derives from the NMR amide proton temperature coefficients (Table 1). The usual interpretation is that intramolecularly hydrogen-bonded HN groups give small values, while solvent-exposed HN groups give larger values.65 In peptides 14–16, the HN of residues 1–3 participate in intramolecular hydrogen bonds and, accordingly, the HN of residues 2 and 3 give the smallest coefficients. The larger value of the HN(1) temperature coefficient is consistent with fraying at the terminus. The solvent exposed HN(4) gives the largest value (≈−9 ppb K−1) in the three peptides.
O) of residues 1–5; Fig. 4),15 presenting the shortest O⋯N and O⋯H distances and the largest CO⋯H angles. In contrast, the geometry of the hydrogen bonds of peptides 14 and 15 in the DFT optimized models was a bit distorted, while keeping the 14-helix fold, due to the steric constraints imposed by the substituents on positions γ, δ and ε of residue 3 (Fig. 5). This agrees well with the CD data recorded in methanol, as the maximum ellipticity of peptides 14 and 15 deviates a few nm from the values of the trans-ACHC homohexamer and peptide 16. Further support to the 14-helix fold derives from the NMR amide proton temperature coefficients (Table 1). The usual interpretation is that intramolecularly hydrogen-bonded HN groups give small values, while solvent-exposed HN groups give larger values.65 In peptides 14–16, the HN of residues 1–3 participate in intramolecular hydrogen bonds and, accordingly, the HN of residues 2 and 3 give the smallest coefficients. The larger value of the HN(1) temperature coefficient is consistent with fraying at the terminus. The solvent exposed HN(4) gives the largest value (≈−9 ppb K−1) in the three peptides.
 | ||
| Fig. 4 Alignment of peptide 16 and the crystallographic structure of the ACHC homohexamer reported by Gellman et al.15 Backbone Cα, Cβ, N and C(O) atoms of residues 1–5 were selected for the alignment of the two molecules (rmsd = 0.236 Å). Colours: C of peptide 16 green, C of ACHC homohexamer grey, O red, N blue, amide HN and hydroxyl HO white. Hydrogen atoms bound to carbons have been omitted for clarity. | ||
 | ||
| Fig. 5 Geometry of peptides 14–16 optimized using DFT calculations at M052X/6-31G(d) level. Colours: C green, O red, N blue, Si black, amide HN and hydroxyl HO white. Hydrogen atoms bound to carbons have been omitted for clarity. Hydrogen bonds are shown as dotted purple lines. | ||
| O⋯N | O⋯H | O⋯H–N | CO⋯H |
|
|---|---|---|---|---|
| 14 | 2.91 | 1.94 | 160.2 | 138.6 |
| 3.06 | 2.20 | 142.9 | 144.4 | |
| 3.04 | 2.18 | 142.3 | 143.0 | |
| 15 | 2.91 | 1.93 | 161.8 | 138.4 |
| 3.22 | 2.36 | 141.6 | 140.2 | |
| 3.00 | 2.18 | 138.1 | 148.0 | |
| 16 | 2.92 | 1.96 | 156.6 | 154.3 |
| 2.95 | 2.01 | 153.5 | 174.9 | |
| 2.96 | 2.03 | 153.1 | 162.8 |
We also studied peptide 16 in CD3-OH solution at 500 and 750 MHz at temperatures 273 and 298 K. The amide HN peaks in the 1D 1H spectrum were consistent with the existence of a major conformer and small amounts of other species. There is some extent of chemical exchange in the amide region of the NOESY and some peaks of the minor species exchange with the solvent HO proton at δH ≈ 5.1 ppm. Although chemical shift dispersion of the Hα(i) peaks was poor, assignment of backbone resonances of the major conformer was possible with spectra recorded at 750 MHz. Amide HN three-bond scalar couplings, temperature coefficients and NOESY peak pattern were similar to those observed for the same peptide in DMSO-d6 solution and hence compatible with the 14-helix conformation. Amide three-bond couplings 3J(HNi−Hβi) were in the range of 8.2–9.2 Hz, that correspond to dihedral angles φi ≈ 160° according to the Karplus relationship.66,67 The small amide temperature coefficients of residues 1–3 (values −5.2, −4.0 and −4.2 ppb K−1, respectively) are consistent with the participation in intramolecular hydrogen bonds. Most of the characteristic NOEs of the 14-helix15,65 were detected in the NOESY spectrum (tmix = 500 ms, T = 273 K, 750 MHz), like the strong intraresidue HN(i)/Hα(i) and sequential HN(i)/Hα(i – 1) peaks. All of the expected intra-residue HN(i)/Hβ(i) and inter-residue HN(i)/Hβ(i + 2) [i = 1, 2, 3] peaks also appear in the NOESY with reasonable intensities for the H–H distances (≈3.0 and ≈3.4 Å, respectively) in the canonical 14-helix conformation. Finally, two important strong long-range contacts are expected for Hα(i)/Hβ(i + 3) [i = 1 or 2, distance <2.5 Å]; those peaks do appear in the NOESY spectrum, although assignment is only tentative due to overlap of several Hα protons with very close chemical shifts. All these observations are in agreement with the major species being in the 14-helix conformation in methanol.
In summary, the FTIR, CD, NMR and DFT calculations data support that pentamers 14–16 adopt the expected 14-helical fold, similarly to homooligomers of trans-ACHC, regardless the functionalization of residue 3. The strain of the cyclohexane ring due to the fused five membered acetonide ring (in pentamers 14–15) or the bulky hydrophobic TBS group protecting the Oε(3) atom (in pentamer 14) does not impede the adoption of a backbone conformation compatible with the 14-helix (i.e. that placing the carbonyl and the amino groups in equatorial orientation), at least in these short peptides. Although CD spectra cannot be measured in DMSO, but only in methanol, the solution NMR analysis of peptide 16 in DMSO-d6 and methanol-d3 revealed that it adopts essentially the same fold in both solvents.
Conclusions
In conclusion, we present here a stereocontrolled synthesis of highly functionalized cyclohexane β-amino acids from (−)-shikimic acid that has allowed the synthesis, on the gram scale, of the first trihydroxylated cyclohexane β-amino acids described having cis- or trans- relative configurations in their free form (5 and 8) or orthogonally protected for their incorporation into peptides (4 and 7, respectively). The availability of highly functionalized β-amino acids is useful for obtaining cycloalkane β-peptides with polar side chains, including the previously unreported polyhydroxylated cycloalkane β-amino acids, of potential interest in chemical biology and material sciences. As an example, herein we present the synthesis of a pentameric β-peptide containing the orthogonally protected trans β-amino acid 7 at an internal position, flanked by apolar trans-ACHC residues. We also demonstrate the chemical stability of this class of peptides (by sequentially deprotecting the hydroxyl groups of pentamer 14 to give pentamers 15 and 16), and their conformational stability, as the structural studies revealed that they adopt a 14-helix fold despite the bulky substituents of the cyclohexane ring. Work to construct more elaborate peptides containing combinations of this new polar residue with apolar ones is in progress to obtain functionalized and potentially amphiphilic peptides.Experimental section
General information
All non-aqueous reactions were carried out under a positive atmosphere of argon in flame-dried glassware unless otherwise stated. Air- and moisture-sensitive liquid reagents were added by dry syringe or cannula. Anhydrous tetrahydrofuran (THF) was freshly distilled from sodium/benzophenone under argon and all other solvents and reagents were used as obtained from commercial sources without further purification unless stated. Flash chromatography was performed using 60 Merck 230–400 mesh (flash, 0.04–0.063) silica. Thin layer chromatography (tlc) was carried out on aluminium backed sheets coated with 60 GF254 silica. Plates were developed using a spray of 0.2% w/v cerium(IV) sulfate and 5% ammonium molybdate in 2 M sulfuric acid, or in 5% w/v ninhydrin in methanol. 1H- and 13C-NMR spectra were recorded on Varian Mercury 300 (300 MHz for 1H and 75 MHz for 13C) spectrometers at room temperature unless otherwise stated. All chemical shifts are quoted on the δ scale using residual solvent as internal standard; s, d, t, q, m, and br designate singlet, doublet, triplet, quadruplet, multiplet, and broad, respectively. Coupling constants (J) are measured in Hz. Mass spectra were recorded on a Micromass VG-Autospec spectrometer [by chemical ionisation (NH3, CI) or electrospray techniques, as stated]. Infrared spectra were recorded on a FT-IR Mattson Cygnus-100 spectrometer. Only the characteristic peaks are quoted (in units of cm−1); st, m, and br designate strong, medium, and broad, respectively. All spectra were measured in KBr unless otherwise stated. Optical rotations were measured on a Jasco DIP-370 polarimeter with a path length of 0.5 dm and Na (589 nm) lamp. Concentrations are given in g per 100 mL.Methyl (3aR,4S,5S,7R,7aR)-4-(benzyl((R)-1-phenylethyl)amino)-7-((tert-butyldimethylsilyl)oxy)-2,2-dimethylhexahydrobenzo[d] [1,3]dioxole-5-carboxylate (3) and methyl (3aR,4S,5R,7R,7aR)-4-(benzyl((R)-1-phenylethyl)amino)-7-((tert-butyldimethylsilyl)oxy)-2,2-dimethylhexahydrobenzo[d][1,3]dioxole-5-carboxylate (6)
n-Butyllithium (1.6 M hexanes solution, 4.6 mL, 2.0 eq.) was added to a solution of (R)-N-benzyl(1-phenylethyl)amine (1.57 mL, 7.36 mmol, 2.0 eq.) in dry THF (46 mL) at −78 °C and stirred for 30 min at the same temperature. To this mixture was added a solution of 1 (1.26 g, 3.68 mmol) in dry THF (12.5 mL) at −78 °C. After stirring the mixture for 2 h at the same temperature, a saturated solution of NH4Cl was added and the mixture was allowed to warm to room temperature over 30 min and extracted with Et2O (3 × 75 mL). The organic layers combined were washed with a 10% solution of citric acid (75 mL), a saturated solution of NaHCO3 (75 mL), and brine (75 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (Et2O/hexanes 1:20) to give 3 (1.24 g, 61%) and 6 (0.10 g, 5%), both as a colorless oils.
O). HRMS (ESI+): calculated for C32H48NO5Si [M + H]+, 554.3296; found, 554.3296. IR (cm−1): 1738 (CO).
O). HRMS (ESI+): calculated for C32H48NO5Si [M + H]+, 554.3296; found, 554.3295. IR (cm−1): 1738 (CO).
Methyl (3aR,4S,5R,7R,7aR)-4-(benzyl((R)-1-phenylethyl)amino)-7-((tert-butyldimethylsilyl)oxy)-2,2-dimethylhexahydrobenzo[d] [1,3]dioxole-5-carboxylate (6)
Compound 3 (0.960 g, 1.73 mmol) was stirred in a solution of 2 M NaOMe in MeOH, freshly prepared (2.55 g of Na in 54 mL of dry MeOH at 0 °C), for 40 h at room temperature. The solvent was evaporated at reduced pressure and the residue was redissolved in DCM (50 mL) and washed with brine (2 × 50 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography (AcOEt/Hex 1:20) to give 6 (0.85 g, 1.54 mmol, 89%) as a colorless oil.
Methyl (3aR,4S,5S,7R,7aR)-4-amino-7-((tert-butyldimethylsilyl)oxy)-2,2-dimethylhexahydrobenzo[d][1,3]dioxole-5-carboxylate (4)
Pd(OH)2/C at 20% (0.075 g (50 wt%)) was added over a deoxygenated solution of 3 (0.150 g, 0.27 mmol) in AcOEt (6 mL). This mixture was stirred under hydrogen over 15 h at rt. Then, the solution was filtered through Celite and washed with AcOEt. The solvent was dried under vacuum and the residue was purified by flash chromatography (AcOEt/Hex 3:1) to give 4 (0.086 g, 0.24 mmol, 88%) as a colorless oil. [α]23D: −34.9 (c 0.4, CHCl3). 1H-NMR (CDCl3, 300 MHz, ppm): 0.06 (m, 6H, 2× CH3), 0.87 (s, 9H, C(CH3)3), 1.33 (s, 3H, CH3), 1.45 (s, 3H, CH3), 1.86–2.02 (m, 2H, CH2), 2.84 (ddd, J = 9.2, 6.0, 4.0 Hz, 1H, CH–CO), 3.36 (t, J = 4.2 Hz, 1H, CH–N), 3.68 (s, 3H, CH3–O), 3.86 (dt, J = 7.6, 5.5 Hz, 1H, CH-OTBS), 3.99 (t, J = 5.5 Hz, 1H, CH–O), 4.19 (dd, J = 5.6, 4.3 Hz, 1H, CH–O). 13C-NMR (CDCl3, 75 MHz, ppm): −4.9 (CH3), −4.7 (CH3), 18.1 (C), 25.8 (3× CH3), 26.0 (CH3), 28.1 (CH3), 29.2 (CH2), 41.4 (CH), 50.2 (CH), 51.7 (CH3), 70.9 (CH), 79.1 (CH), 79.3 (CH), 108.4 (C), 174.0 (CO). HRMS (ESI+): calculated for C17H34NO5Si [M + H]+, 360.2201; found, 360.2200. IR (ν, cm−1): 3388 (NH), 1735 (CO).
Methyl (3aR,4S,5R,7R,7aR)-4-amino-7-((tert-butyldimethylsilyl)oxy)-2,2-dimethylhexahydrobenzo[d][1,3]dioxole-5-carboxylate (7)
Pd(OH)2/C at 20% (0.075 g (50 wt%)) was added over a deoxygenated solution of 6 (0.150 g, 0.27 mmol) in AcOEt (6 mL). This mixture was stirred under hydrogen for 15 h at rt. Then, the solution was filtered through Celite and washed with AcOEt. The solvent was dried under vacuum and the residue was purified by flash chromatography (AcOEt/Hex 3:1) to give 7 (0.089 g, 0.24 mmol, 92%) as a colorless oil. [α]23D: +66.9 (c 0.5, CHCl3). 1H-NMR (CDCl3, 300 MHz, ppm): 0.06 (s, 6H, 2× CH3), 0.86 (s, 9H, 3× CH3), 1.33 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.77 (dtd, J = 13.6, 3.3, 0.8 Hz, 1H, CHH), 1.88 (ddd, J = 13.6, 11.3, 2.7 Hz, 1H, CHH), 2.60 (td, J = 11.2, 3.6 Hz, 1H, CH–CO), 3.01 (dd, J = 11.2, 8.4 Hz, 1H, CH–N), 3.68 (s, 3H, CH3–O), 3.82 (dd, J = 8.4, 5.1 Hz, 1H, CH–O), 3.91–4.00 (m, 1H, CH–O), 4.19 (q, J = 3.0 Hz, CH-OTBS). 13C-NMR (CDCl3, 75 MHz, ppm): −5.1 (CH3), −5.0 (CH3), 17.9 (C), 25.6 (3× CH3), 26.4 (CH3), 28.3 (CH3), 32.0 (CH2), 42.3 (CH), 51.7 (CH), 54.1 (CH3), 67.2 (CH), 77.8 (CH), 80.4 (CH), 109.1 (C), 175.0 (CO). HRMS (ESI+): calculated for C17H34NO5Si [M + H]+, 360.2201; found, 360.2200. IR (ν, cm−1): 3382 (NH), 1737 (CO).
(1S,2S,3R,4S,5R)-2-Amino-3,4,5-trihydroxycyclohexane-1-carboxylic acid (5)
Compound 4 (0.039 g, 0.11 mmol) in 6 M HCl aqueous solution (5 mL) was refluxed for 15 h. The solvent was concentrated in vacuum, and the residue was redissolved in H2O (1 mL). Activated Dowex 50WX4-50 was added, and the mixture was stirred for 1 h. Then, the Dowex was washed with water and MeOH. The compound was released from the resin with a 10% aqueous solution of NH3. The combined aqueous layers were concentrated in vacuum to give 5 (0.018 g, 0.09 mmol, 86%) as a pale-yellow oil. [α]23D: +33.7 (c 1.5, H2O). 1H-NMR (D2O, 300 MHz, ppm): 2.01 (m, 1H, CHH), 2.24 (m, 1H, CHH), 2.80 (q, J = 4.4 Hz, 1H, CH–CO), 3.49 (dd, J = 10.1, 5.0 Hz, 1H, CH–N), 3.77–3.91 (m, 2H, 2× CH–O), 4.06 (dd, J = 10.1, 2.3 Hz, 1H, CH–O). 13C-NMR (D2O, 75 MHz, ppm): 29.4 (CH2), 40.0 (CH), 49.5 (CH), 51.8 (CH), 69.7 (CH), 72.3 (CH), 179.7 (CO). HRMS (ESI+): calculated for C7H14NO5 [M + H]+, 192.0866; found, 192.0866. IR (ν, cm−1): 3340 (NH and OH), 1740 (CO).
(1R,2S,3R,4S,5R)-2-Amino-3,4,5-trihydroxycyclohexane-1-carboxylic acid (8)
Compound 7 (0.070 g, 0.19 mmol) in 6 M HCl aqueous solution (8 mL) is refluxed for 15 h. The solvent was concentrated in vacuum, and the residue was redissolved in H2O (1 mL). Activated Dowex 50WX4-50 was added, and the mixture was stirred for 1 h. Then, the Dowex was washed with water and MeOH. The compound was released of the resin with a 10% aqueous solution of NH3. The combined aqueous layers were concentrated in vacuum to give 8 (0.031 g, 0.16 mmol, 84%) as pale-yellow oil. [α]23D: −26.9 (c 2.0, H2O). 1H-NMR (D2O, 300 MHz, ppm): 1.78–2.06 (m, 2H, CH2), 2.68 (td, J = 12.5, 2.8 Hz, 1H, CH–CO), 3.45 (t, J = 10.9 Hz, 1H, CH–N), 3.88–4.03 (m, 2H, 2× CH–O), 4.10 (m, 1H, CH–O). 13C-NMR (D2O, 75 MHz, ppm): 30.2 (CH2), 41.7 (CH), 53.3 (CH), 68.8 (CH), 69.2 (CH), 71.9 (CH), 179.9 (CO). HRMS (ESI+): calculated for C7H14NO5 [M + H]+, 192.0866, found, 192.0866. IR (ν, cm−1): 3246 (NH and OH), 1744 (CO).
Dipeptide 10
Pd(OH)2 at 10% (75 mg, 50 wt%) was added to a deoxygenated solution of dipeptide 9 (Boc-trans-(ACHC)2-OBn) (150 mg, 0.327 mmol) in MeOH (7 mL). The mixture was stirred under hydrogen atmosphere for 16 h, then was filtered over Celite and washed with MeOH. The solvent was removed under vacuum to afford dipeptide 10 (135 mg) as a white solid that was used in the next reaction without further purification.Tripeptide 11
EDCI·HCl (222 mg, 1.159 mmol) was added over a solution of dipeptide 10 (194 mg, 0.527 mmol) in dry DMF (3.8 mL) and the mixture was stirred for 30 min. Compound 7 (189 mg, 0.527 mmol) and DMAP (193 mg, 1.581 mmol) were then added to the former mixture and the stirring followed for 17 h under an argon atmosphere. The solvent was then removed under vacuum, the residue was redissolved in AcOEt (5 mL) and washed with 5 mL portions of HCl (1 M), NaCl (sat.) y NaHCO3 (sat.) and NaCl (sat.). The organic layer was dried over Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography (AcOEt/Hex 1:1) to afford tripeptide 11 (347 mg, 93%) as a white solid. [α]20D: +33.1 (c 10, CHCl3). 1H-NMR (CDCl3, 300 MHz, ppm): 0.07 (s, 3H, SiCH3), 0.08 (s, 3H, SiCH3), 0.89 (s, 9H, SiC(CH3)3), 1.06–1.37 (m, 14H, 2× CH3 + 4× CH2), 1.40 (s, 9H, C(CH3)3), 1.62–2.15 (m, 10H, 5× CH2), 2.62–2.79 (m, 2H, 2× CH–CO), 2.99 (m, 1H, CH–CO), 3.59 (m, 4H, OCH3 + NCH), 3.87 (q, J = 9.5 Hz, 1H, NCH), 3.99–4.10 (m, 2H, NCH + OCH), 4.13–4.28 (m, 2H, 2× OCH), 5.83 (d, J = 9.3 Hz, 1H, HN), 6.13 (d, J = 7.1 Hz, 1H, HN), 7.01 (d, J = 8.2 Hz, 1H, HN). 13C-NMR (CDCl3, 75 MHz, ppm): −4.81 (SiCH3), −4.78 (SiCH3), 18.22 (C), 22.79 (CH2), 24.72 (CH2), 25.19 (CH2), 25.38 (CH2), 25.60 (3× CH3), 25.96 (CH3), 26.59 (CH3), 28.12 (3× CH3), 29.01 (CH2), 29.79 (CH2), 31.85 (2× CH2), 33.79 (CH2), 41.83 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H–CO), 48.34 (H–CO), 49.57 (H–CO), 51.60 (CH3), 52.08 (CH–N), 52.64 (CH–N), 53.47 (CH–N), 67.12 (CH–O), 76.92 (CH–O), 78.30 (CH–O), 79.02 (C, Boc), 109.24 (C), 156.18 (CO, Boc), 173.26 (2× CO), 174.99 (CO). HRMS (ESI+): calculated for C36H64N3O9Si [M + H]+, 710.4405; found 710.4406. IR (cm−1): 3327.38 (NH), 1736.29 and 1679.77 (CO).
H–CO), 48.34 (H–CO), 49.57 (H–CO), 51.60 (CH3), 52.08 (CH–N), 52.64 (CH–N), 53.47 (CH–N), 67.12 (CH–O), 76.92 (CH–O), 78.30 (CH–O), 79.02 (C, Boc), 109.24 (C), 156.18 (CO, Boc), 173.26 (2× CO), 174.99 (CO). HRMS (ESI+): calculated for C36H64N3O9Si [M + H]+, 710.4405; found 710.4406. IR (cm−1): 3327.38 (NH), 1736.29 and 1679.77 (CO).
Tripeptide 12
LiOH·H2O (50 mg, 1.180 mmol) was added to a solution of dipeptide 11 (336 mg, 0.472 mmol) in THF/MeOH/H2O (1:1:1) (8 mL) at 0 °C. The mixture was stirred for 24 h at room temperature. The solvent was removed under reduced pressure, the mixture was diluted with a 10% citric acid solution (5 mL) and extracted with Et2O (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated to afford tripeptide 12 (301 mg, 92%) as a white solid that was used in the next reaction without further purification.
Dipeptide 13
Dipeptide 9 (Boc-trans-(ACHC)2-OBn) (216 mg, 0.472 mmol) was stirred in a mixture of TFA/H2O (1:1) (2.4 mL) for 1 h. Then, the solvent was evaporated under vacuum to afford dipeptide 13 (295 mg) as a white solid, which was used in the next reaction without further purification.
Pentapeptide 14
EDCI·HCl (54 mg, 0.282 mmol) was added to a solution of dipeptide 12 (89 mg, 0.128 mmol) in dry DMF (0.91 mL) and the mixture was stirred over 30 min. Then, tripeptide 13 (46 mg, 0.128 mmol) in dry DMF (0.91 mL) and DMAP (47 mg, 0.384 mmol), were added to the mixture and the reaction was further stirred for 21 h under argon atmosphere. Then, the solvent was removed in vacuum, the residue was redissolved in DCM (5 mL) and washed with 5 mL portions of HCl (1 M), NaCl (sat.), NaHCO3 (sat.) and NaCl (sat.). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by flash chromatography (DCM/MeOH 1:20) to afford 14 (60 mg, 45%) as a white solid. [α]23D: +48.3 (c 1, CH2Cl2). For NMR data see the ESI.‡HRMS (ESI+): calculated for C56H89N5O11NaSi [M + Na]+, 1058.6218; found 1058.6220. IR: 3281.07 (NH), 1721.22 and 1644.93 (CO).
Pentapeptide 15
TBAF (1 M THF solution, 0.7 mL) was added dropwise to a solution of pentapeptide 14 (50 mg, 0.048 mmol) in dry THF (0.78 mL) and the mixture was stirred for 4 days. The solvent was them removed under reduced pressure, the residue was redissolved in DCM (10 mL) and washed with 1 M HCl (3 × 10 mL) and NaCl (10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (MeOH/DCM 1:15) to afford pentapeptide 15 (34 mg, 76%) as a white solid. [α]23D: +34.2 (c 1, CH2Cl2). For NMR data see the ESI.‡HRMS (ESI+): calculated for C50H75N5NaO11 [M + Na]+, 944.5359; found, 944.5355. IR: 3369.60 (NH), 1721.09 and 1649.42 (CO).
Pentapeptide 16
Pentapetide 15 (18 mg, 0.02 mmol) was stirred in a mixture of AcOH/THF/H2O (0.176 mL) for 29 h at rt. Then, the solvent was removed under reduced pressure and the residue was purified by flash chromatography (MeOH/DCM 1:10) to give pentapeptide 16 (8.5 mg, 50%) as a white solid. [α]23D: +44.0 (c 1, CH2Cl2). For NMR data see the ESI.‡HRMS (ESI+): calculated for C47H71N5NaO11 [M + Na]+, 904.5048; found 904.5045. IR: 3269.40 (NH), 1723.10 and 1649.50 (CO).
Infrared spectroscopy
Fourier-transform infrared (FTIR) spectra were acquired using a MIDAC Prospect FT-IR PerkinElmer Spectrum Two spectrometer. Samples were deposited on an ATR diamond accessory as dry solids.Circular dichroism spectroscopy
Circular dichroism (CD) spectra were acquired with a JASCO DIP-370 optical polarimeter using a 1 mm path length quartz cell (Hellma Analytics, Germany). Wavelength scans were collected between 260 and 195 nm with a 1 nm bandwidth, 1 nm data interval and 100 nm min−1 scanning speed.Samples were prepared by dissolving dry peptide in HPLC-grade methanol (Fisher Scientific) to give 1 mM solutions. Samples of 700, 500, and 100 μM concentration were prepared by serial dilution of the 1 mM stock.
Three scans of each peptide solution were taken and averaged. Solvent blanks were subtracted from the raw spectra and smoothened over 17 data points prior to normalization to molar ellipticity. The CD signal was converted into molar ellipticity ([Θ], deg cm2 dmol−1) using the equation:
| [Θ] = 100 × Ψ/(l × c) |
70 with the 6-31+G(d) basis set. DFT calculations were performed using Gaussian 09.71
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has received financial support from the Xunta de Galicia and the European Regional Development Fund - ERDF (ED431C 2018/30, ED431F 2020/05 and Centro singular de investigación de Galicia accreditation 2019-2022, ED431G 2019/03). Funding from the Agencia Estatal de Investigación (AEI) and the European Regional Development Fund - ERDF (RTI2018-098795-A-I00 and PDC2022-133402-I00) is also acknowledged. R. G.-F. thanks Ministerio de Ciencia, Innovación y Universidades for a “Ramón y Cajal” contract (RYC-2016-20335). L. B. thanks Ministerio de Educación, Cultura y Deporte for a FPU fellowship (FPU13/01243). D. R. thanks Xunta de Galicia for a predoctoral fellowship. All calculations were carried out at the Centro de Supercomputación de Galicia (CESGA). Funding for open access charge: Universidade de Santiago de Compostela/CRUE.References
- S. H. Gellman, Foldamers: A Manifesto, Acc. Chem. Res., 1998, 31, 173–180, DOI:10.1021/ar960298r.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, β-Peptides: From Structure to Function, Chem. Rev., 2001, 101, 3219–3232, DOI:10.1021/cr000045i.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, A Field Guide to Foldamers, Chem. Rev., 2001, 101, 3893–4012, DOI:10.1021/cr990120t.
- Y.-D. Wu, W. Han, D.-P. Wang, Y. Gao and Y.-L. Zhao, Theoretical Analysis of Secondary Structures of β-Peptides, Acc. Chem. Res., 2008, 41, 1418–1427, DOI:10.1021/ar800070b.
- P. G. Vasudev, S. Chatterjee, N. Shamala and P. Balaram, Structural Chemistry of Peptides Containing Backbone Expanded Amino Acid Residues: Conformational Features of β, γ, and Hybrid Peptides, Chem. Rev., 2011, 111, 657–687, DOI:10.1021/cr100100x.
- T. A. Martinek and F. Fülöp, Peptidic foldamers: ramping up diversity, Chem. Soc. Rev., 2012, 41, 687–702, DOI:10.1016/j.jasms.2007.08.005.
- D. Seebach, S. Abele, J. V. Schreiber, B. Martinoni, A. K. Nussbaum, H. Schild, H. Schulz, H. Hennecke, R. Woessner and F. Bitsch, Biological and Pharmacokinetic Studies with β-Peptides, Chimia, 1998, 52, 734, DOI:10.2533/chimia.1998.734.
- J. Frackenpohl, P. I. Arvidsson, J. V. Schreiber and D. Seebach, The Outstanding Biological Stability ofβ- andγ-Peptides toward Proteolytic Enzymes: An In Vitro Investigation with Fifteen Peptidases, ChemBioChem, 2001, 2, 445–455, DOI:10.1002/1439-7633(20010601)2:6<445::AID-CBIC445>3.0.CO;2-R.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756, DOI:10.1073/pnas.0902663106.
- L. M. Johnson, W. S. Horne and S. H. Gellman, Broad Distribution of Energetically Important Contacts across an Extended Protein Interface, J. Am. Chem. Soc., 2011, 133, 10038–10041, DOI:10.1021/ja203358t.
- L. M. Johnson, D. E. Mortenson, H. G. Yun, W. S. Horne, T. J. Ketas, M. Lu, J. P. Moore and S. H. Gellman, Enhancement of α-Helix Mimicry by an α/β-Peptide Foldamer via Incorporation of a Dense Ionic Side-Chain Array, J. Am. Chem. Soc., 2012, 134, 7317–7320, DOI:10.1021/ja302428d.
- L. Diao and B. Meibohm, Pharmacokinetics and Pharmacokinetic–Pharmacodynamic Correlations of Therapeutic Peptides, Clin. Pharmacokinet., 2013, 52, 855–868, DOI:10.1007/s40262-013-0079-0.
- J. A. Kritzer, O. M. Stephens, D. A. Guarracino, S. K. Reznik and A. Schepartz, β-Peptides as inhibitors of protein–protein interactions, Bioorg. Med. Chem., 2005, 13, 11–16, DOI:10.1016/j.bmc.2004.09.009.
- V. K. Outlaw, R. W. Cheloha, E. M. Jurgens, F. T. Bovier, Y. Zhu, D. F. Kreitler, O. Harder, S. Niewiesk, M. Porotto, S. H. Gellman and A. Moscona, Engineering Protease-Resistant Peptides to Inhibit Human Parainfluenza Viral Respiratory Infection, J. Am. Chem. Soc., 2021, 143, 5958–5966, DOI:10.1021/jacs.1c01565.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, β-Peptide Foldamers: Robust Helix Formation in a New Family of β-Amino Acid Oligomers, J. Am. Chem. Soc., 1996, 118, 13071–13072, DOI:10.1021/ja963290l.
- D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi and S. H. Gellman, Residue-based control of helix shape in beta-peptide oligomers., Nature, 1997, 387, 381–384, DOI:10.1038/387381a0.
- L. Kiss and F. Fülöp, Synthesis of Carbocyclic and Heterocyclic β-Aminocarboxylic Acids, Chem. Rev., 2014, 114, 1116–1169, DOI:10.1021/cr300454h.
- E. A. Porter, X. Wang, H. S. Lee, B. Weisblum and S. H. Gellman, Non-haemolytic beta-amino-acid oligomers, Nature, 2000, 404, 565, DOI:10.1038/35007145.
- T. L. Raguse, E. A. Porter, B. Weisblum and S. H. Gellman, Structure−Activity Studies of 14-Helical Antimicrobial β-Peptides: Probing the Relationship between Conformational Stability and Antimicrobial Potency, J. Am. Chem. Soc., 2002, 124, 12774–12785, DOI:10.1021/ja0270423.
- E. P. English, R. S. Chumanov, S. H. Gellman and T. Compton, Rational development of beta-peptide inhibitors of human cytomegalovirus entry., J. Biol. Chem., 2006, 281, 2661–2667, DOI:10.1074/jbc.M508485200.
- T. A. Martinek, A. Hetényi, L. Fülöp, I. M. Mándity, G. K. Tóth, I. Dékány and F. Fülöp, Secondary Structure Dependent Self-Assembly of β-Peptides into Nanosized Fibrils and Membranes, Angew. Chem., Int. Ed., 2006, 45, 2396–2400, DOI:10.1002/anie.200504158.
- S. Kwon, A. Jeon, S. H. Yoo, I. S. Chung and H.-S. Lee, Unprecedented Molecular Architectures by the Controlled Self-Assembly of a β-Peptide Foldamer, Angew. Chem., Int. Ed., 2010, 49, 8232–8236, DOI:10.1002/anie.201003302.
- J. Kim, S. Kwon, S. H. Kim, C.-K. Lee, J.-H. Lee, S. J. Cho, H.-S. Lee and H. Ihee, Microtubes with Rectangular Cross-Section by Self-Assembly of a Short β-Peptide Foldamer, J. Am. Chem. Soc., 2012, 134, 20573–20576, DOI:10.1021/ja3088482.
- K. Kulkarni, N. Habila, M. P. Del Borgo and M.-I. Aguilar, Novel Materials From the Supramolecular Self-Assembly of Short Helical β3-Peptide Foldamers, Front. Chem., 2019, 7, 70, DOI:10.3389/fchem.2019.00070.
- Z. C. Girvin and S. H. Gellman, Foldamer Catalysis, J. Am. Chem. Soc., 2020, 142, 17211–17223, DOI:10.1021/jacs.0c07347.
- P. Milbeo, J. Martinez, M. Amblard, M. Calmès and B. Legrand, 1-Aminobicyclo[2.2.2]octane-2-carboxylic Acid and Derivatives As Chiral Constrained Bridged Scaffolds for Foldamers and Chiral Catalysts, Acc. Chem. Res., 2021, 54, 685–696, DOI:10.1021/acs.accounts.0c00680.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, Synthesis and Characterization of trans-2-Aminocyclohexanecarboxylic Acid Oligomers: An Unnatural Helical Secondary Structure and Implications for β-Peptide Tertiary Structure, J. Am. Chem. Soc., 1999, 121, 6206–6212, DOI:10.1021/ja990748l.
- J. Applequist, K. A. Bode, D. H. Appella, L. A. Christianson and S. H. Gellman, Theoretical and Experimental Circular Dichroic Spectra of the Novel Helical Foldamer Poly[(1 R, 2 R)- trans -2-aminocyclopentanecarboxylic acid], J. Am. Chem. Soc., 1998, 120, 4891–4892, DOI:10.1021/ja9742186.
- D. H. Appella, L. A. Christianson, D. A. Klein, M. R. Richards, D. R. Powell and S. H. Gellman, Synthesis and Structural Characterization of Helix-Forming β-Peptides: trans-2-Aminocyclopentanecarboxylic Acid Oligomers, J. Am. Chem. Soc., 1999, 121, 7574–7581, DOI:10.1021/ja991185g.
- T. A. Martinek, I. M. Mándity, L. Fülöp, G. K. Tóth, E. Vass, M. Hollósi, E. Forró and F. Fülöp, Effects of the Alternating Backbone Configuration on the Secondary Structure and Self-Assembly of β-Peptides, J. Am. Chem. Soc., 2006, 128, 13539–13544, DOI:10.1021/ja063890c.
- I. M. Mándity, E. Wéber, T. A. Martinek, G. Olajos, G. K. Tóth, E. Vass and F. Fülöp, Design of Peptidic Foldamer Helices: A Stereochemical Patterning Approach, Angew. Chem., Int. Ed., 2009, 48, 2171–2175, DOI:10.1111/j.1399-3011.1994.tb01023.x.
- A. Hetényi, Z. Szakonyi, I. M. Mándity, É. Szolnoki, G. K. Tóth, T. A. Martinek and F. Fülöp, Sculpting the β-peptide foldamer H12 helix via a designed side-chain shape, Chem. Commun., 2009, 177–179, 10.1039/B812114A.
- I. M. Mándity, L. Fülöp, E. Vass, G. K. Tóth, T. A. Martinek and F. Fülöp, Building β-Peptide H10/12 Foldamer Helices with Six-Membered Cyclic Side-Chains: Fine-Tuning of Folding and Self-Assembly, Org. Lett., 2010, 12, 5584–5587, DOI:10.1021/ol102494m.
- X. Wang, J. F. Espinosa and S. H. Gellman, 12-Helix Formation in Aqueous Solution with Short β-Peptides Containing Pyrrolidine-Based Residues, J. Am. Chem. Soc., 2000, 122, 4821–4822, DOI:10.1021/ja000093k.
- É. Szolnoki, A. Hetényi, T. A. Martinek, Z. Szakonyi and F. Fülöp, Self-association-driven transition of the β-peptidic H12 helix to the H18 helix, Org. Biomol. Chem., 2012, 10, 255–259, 10.1039/C1OB06627G.
- L. Kiss, I. M. Mándity and F. Fülöp, Highly functionalized cyclic β-amino acid moieties as promising scaffolds in peptide research and drug design, Amino Acids, 2017, 49, 1441–1455, DOI:10.1007/s00726-017-2439-9.
- F. Sussman, V. M. Sánchez-Pedregal, J. C. Estévez, R. Balo, J. Jiménez-Barbero, A. Ardá, A. Gimeno, M. Royo, M. C. Villaverde and R. J. Estévez, Environmental Effects Determine the Structure of Potential β-Amino Acid Based Foldamers, Chem. – Eur. J., 2018, 24, 10625–10629, DOI:10.1002/chem.201801953.
- M. Lee, J. Shim, P. Kang, M.-G. Choi and S. H. Choi, Stabilization of 11/9-helical α/β-peptide foldamers in protic solvents, Chem. Commun., 2016, 52, 5950–5952, 10.1039/C6CC01189F.
- S. H. Choi, M. Ivancic, I. A. Guzei and S. H. Gellman, Structural Characterization of Peptide Oligomers Containing (1 R, 2 S)-2-Aminocyclohexanecarboxylic Acid (cis -ACHC): Structural Characterization of Peptide Oligomers, Eur. J. Org. Chem., 2013, 3464–3469, DOI:10.1002/ejoc.201300118.
- T. L. Raguse, J. R. Lai, P. R. LePlae and S. H. Gellman, Toward β-Peptide Tertiary Structure: Self-Association of an Amphiphilic 14-Helix in Aqueous Solution, Org. Lett., 2001, 3, 3963–3966, DOI:10.1021/ol016868r.
- P. R. LePlae, J. D. Fisk, E. A. Porter, B. Weisblum and S. H. Gellman, Tolerance of Acyclic Residues in the β-Peptide 12-Helix: Access to Diverse Side-Chain Arrays for Biological Applications, J. Am. Chem. Soc., 2002, 124, 6820–6821, DOI:10.1021/ja017869h.
- T. L. Raguse, J. R. Lai and S. H. Gellman, Environment-Independent 14-Helix Formation in Short β-Peptides: Striking a Balance between Shape Control and Functional Diversity, J. Am. Chem. Soc., 2003, 125, 5592–5593, DOI:10.1021/ja0341485.
- W. C. Pomerantz, T. L. R. Grygiel, J. R. Lai and S. H. Gellman, Distinctive Circular Dichroism Signature for 14-Helix-Bundle Formation by β-Peptides, Org. Lett., 2008, 10, 1799–1802, DOI:10.1021/ol800622e.
- C. Wang, N. A. Biok, K. Nayani, X. Wang, H. Yeon, C.-K. Derek Ma, S. H. Gellman and N. L. Abbott, Cationic Side Chain Identity Directs the Hydrophobically Driven Self-Assembly of Amphiphilic β-Peptides in Aqueous Solution, Langmuir, 2021, 37, 3288–3298, DOI:10.1021/acs.langmuir.0c03255.
- D. H. Appella, J. J. Barchi, S. R. Durell and S. H. Gellman, Formation of Short, Stable Helices in Aqueous Solution by β-Amino Acid Hexamers, J. Am. Chem. Soc., 1999, 121, 2309–2310, DOI:10.1021/ja983918n.
- J. A. Kritzer, J. Tirado-Rives, S. A. Hart, J. D. Lear, W. L. Jorgensen and A. Schepartz, Relationship between Side Chain Structure and 14-Helix Stability of β 3 -Peptides in Water, J. Am. Chem. Soc., 2005, 127, 167–178, DOI:10.1021/ja0459375.
- A. Hetényi, G. K. Tóth, C. Somlai, E. Vass, T. A. Martinek and F. Fülöp, Stabilisation of Peptide Foldamers in an Aqueous Medium by Incorporation of Azapeptide Building Blocks, Chem. – Eur. J., 2009, 15, 10736–10741, DOI:10.1002/chem.200900724.
- L. Kiss, E. Forró, T. A. Martinek, G. Bernáth, N. De Kimpe and F. Fülöp, Stereoselective synthesis of hydroxylated β-aminocyclohexanecarboxylic acids, Tetrahedron, 2008, 64, 5036–5043, DOI:10.1016/j.tet.2008.03.068.
- E. Forró, L. Schönstein, L. Kiss, A. Vega-Peñaloza, E. Juaristi and F. Fülöp, Direct Enzymatic Route for the Preparation of Novel Enantiomerically Enriched Hydroxylated β-Amino Ester Stereoisomers, Molecules, 2010, 15, 3998–4010, DOI:10.3390/molecules15063998.
- M. Risseeuw, M. Overhand, G. W. J. Fleet and M. I. Simone, A compendium of cyclic sugar amino acids and their carbocyclic and heterocyclic nitrogen analogues, Amino Acids, 2013, 45, 613–689, DOI:10.1007/s00726-013-1521-1.
- M. Lee, T. L. Raguse, M. Schinnerl, W. C. Pomerantz, X. Wang, P. Wipf and S. H. Gellman, Origins of the High 14-Helix Propensity of Cyclohexyl-Rigidified Residues in β-Peptides, Org. Lett., 2007, 9, 1801–1804, DOI:10.1021/ol070511r.
- F. Fernández, A. G. Fernández, R. Balo, V. M. Sánchez-Pedregal, M. Royo, R. G. Soengas, R. J. Estévez and J. C. Estévez, Polyhydroxylated Cyclopentane β-Amino Acids Derived from d -Mannose and d -Galactose: Synthesis and Protocol for Incorporation into Peptides, ACS Omega, 2022, 7, 2002–2014, DOI:10.1021/acsomega.1c05468.
- M. A. González, A. M. Estévez, M. Campos, J. C. Estévez and R. J. Estévez, Protocol for the Incorporation of γ-Amino Acids into Peptides: Application to (−)-Shikimic Acid Based 2-Amino-Methylcyclohexanecarboxylic Acids, J. Org. Chem., 2018, 83, 1543–1550, DOI:10.1021/acs.joc.7b02671.
- An initial version of this work was deposited in ChemRxiv on December 29, 2022, reference: D. Reza, R. Balo, J. M. Otero, A. M. Fletcher, R. García-Fandino, V. M. Sanchez-Pedregal, S. G. Davies, R. J. Estevez and J. C. Estevez, β-Peptides incorporating polyhydroxylated cyclohexane β-amino acids: synthesis and conformational study, ChemRxiv DOI:10.26434/chemrxiv-2022-zdb5j.
- J. C. Borah, Shikimic acid: a highly prospective molecule in pharmaceutical industry, Curr. Sci., 2015, 109, 8, DOI:10.18520/v109/i9/1672-1679.
- M. A. González-Castro, D. L. Poole, J. C. Estévez, G. W. J. Fleet and R. J. Estévez, A stereoselective transformation of (−)-shikimic acid into (3R,4S,5R,7R)-7-(hydroxymethyl)azepane-3,4,5-triol, a potential glycosidase inhibitor, Tetrahedron: Asymmetry, 2015, 26, 320–323, DOI:10.1016/j.tetasy.2015.02.005.
- S. G. Davies and O. Ichihara, Asymmetric synthesis of R-β-amino butanoic acid and S-β-tyrosine: Homochiral lithium amide equivalents for Michael additions to α,β-unsaturated esters., Tetrahedron: Asymmetry, 1991, 2, 183–186, DOI:10.1016/S0957-4166(00)82354-X.
- J. F. Costello, S. G. Davies and O. Ichihara, Origins of the high stereoselectivity in the conjugate addition of lithium(α-methylbenzyl)benzylamide to t-butyl cinnamate, Tetrahedron: Asymmetry, 1994, 5, 1999–2008, DOI:10.1016/S0957-4166(00)86275-8.
- S. G. Davies, O. Ichihara, I. Lenoir and I. A. S. Walters, Asymmetric synthesis of (–)-(1R,2S)-cispentacin and related cis- and trans-2-amino cyclopentane- and cyclohexane-1-carboxylic acids, J. Chem. Soc., Perkin Trans. 1, 1994, 1411–1415, 10.1039/P19940001411.
- G. Montalvo, M. M. Waegele, S. Shandler, F. Gai and W. F. DeGrado, Infrared Signature and Folding Dynamics of a Helical β-Peptide, J. Am. Chem. Soc., 2010, 132, 5616–5618, DOI:10.1021/ja100459a.
- L. Zhai, M. Nara, Y. Otani and T. Ohwada, Unexpectedly rigid short peptide foldamers in which NH–π and CH–π interactions are preserved in solution, Chem. Commun., 2021, 57, 8344–8347, 10.1039/D1CC02998C.
- R. W. Woody, Circular Dichroism: Principles and Applications, VCH Publishers, New York, 1994 Search PubMed.
- S. Yu. Venyaminov and J. T. Yang, in Circular Dichroism and the Conformational Analysis of Biomolecules, ed. G. D. Fasman, Springer US, Boston, MA, 1996, pp. 69–107 Search PubMed.
- A. Glättli, X. Daura, D. Seebach and W. F. van Gunsteren, Can One Derive the Conformational Preference of a β-Peptide from Its CD Spectrum?, J. Am. Chem. Soc., 2002, 124, 12972–12978, DOI:10.1021/ja020758d.
- J. J. Barchi, X. Huang, D. H. Appella, L. A. Christianson, S. R. Durell and S. H. Gellman, Solution Conformations of Helix-Forming β-Amino Acid Homooligomers, J. Am. Chem. Soc., 2000, 122, 2711–2718, DOI:10.1021/ja9930014.
- A. Pardi, M. Billeter and K. Wüthrich, Calibration of the angular dependence of the amide proton-Cα proton coupling constants, 3JHNα, in a globular protein, J. Mol. Biol., 1984, 180, 741–751, DOI:10.1016/0022-2836(84)90035-4.
- B. Vögeli, J. Ying, A. Grishaev and A. Bax, Limits on variations in protein backbone dynamics from precise measurements of scalar couplings., J. Am. Chem. Soc., 2007, 129, 9377–9385, DOI:10.1021/ja070324o.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, The Xplor-NIH NMR molecular structure determination package, J. Magn. Reson., 2003, 160, 65–73, DOI:10.1016/S1090-7807(02)00014-9.
- C. Schwieters, J. Kuszewski and G. M. Clore, Using Xplor–NIH for NMR molecular structure determination, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62, DOI:10.1016/j.pnmrs.2005.10.001.
- Y. Zhao and D. G. Truhlar, Density Functionals for Noncovalent Interaction Energies of Biological Importance, J. Chem. Theory Comput., 2007, 3, 289–300, DOI:10.1021/ct6002719.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox. GAUSSIAN 09 (Revision E.01), Gaussian Inc., Wallingford CT, 2013 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Definition of the hydrogen bond (IUPAC Recommendations 2011), Pure Appl. Chem., 2011, 83, 1637–1641, DOI:10.1351/PAC-REC-10-01-02.
Footnotes |
| † This paper is dedicated to Prof. Joan Bosch on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for compounds 3, 4, 5, 6, 7, 8 and 11. FT-IR, CD, NMR structural analysis and DFT calculations for compounds 14, 15, 16. Single-crystal X-ray data of 3. CCDC 2192947. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00906h |
| This journal is © The Royal Society of Chemistry 2023 |