
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpeditious access to cis-β-aryl, γ-alkyl disubstituted (±)-γ-butyrolactones via nickel-hydride catalysis†
O. Stephen
Ojo
 *,
Hannah J.
Steel
and
Haralampos N.
Miras
*,
Hannah J.
Steel
and
Haralampos N.
Miras
WestCHEM, School of Chemistry, University of Glasgow, The Joseph Black Building, Glasgow, G12 8QQ, UK. E-mail: Oluwarotimi.ojo@glasgow.ac.uk
First published on 1st August 2023
Abstract
The 1,4-reduction of β- and γ-substituted butenolides using 5 mol% of NiCl2·6H2O and NaBH4 in MeOH for rapid access to cis-β,γ-disubstituted γ-butyrolactones is described. The reaction was selective for cis-products, which were obtained in good to excellent yields. This study showcased the influence of steric hindrance and angle strain on the diastereoselectivity outcome of conjugate reductions facilitated by in situ generated nickel-hydride.
The γ-butyrolactone moiety is ubiquitous in natural products such as phaseolinic acid1a and in FDA-approved drugs such as pilocarpine, which is used for the treatment of glaucoma, and spironolactone, used for the treatment of high blood pressure and heart failure (Fig. 1). γ-Butyrolactone is a key motif in the structure of an experimental drug1b and can serve as a useful synthetic intermediate.1c,d
 | ||
| Fig. 1 γ-Butyrolactone moiety in natural products and medicinal drugs. | ||
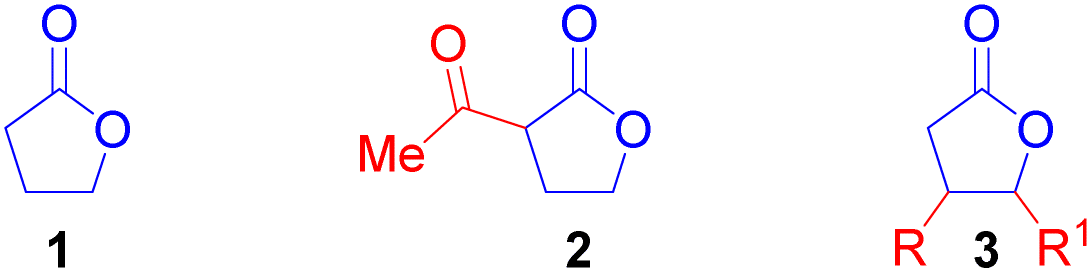
Ring-opening polymerisation (ROP) of cyclic esters has been used to synthesise degradable and chemically recyclable polyesters.1e Prior research has demonstrated the importance of γ-butyrolactone (1) as a highly desirable building block for the construction of poly(γ-butyrolactone).1f Despite some progress, the polymerisation of 1 is challenging due to the low ring strain in the 5-membered ring which has a negative change in enthalpy value that is too small to overcome the large negative entropy change associated with ROP.1g To solve this problem, the ring strain of 1 can be increased through the introduction of a substituent at any position on the lactone ring (Fig. 2).
 | ||
| Fig. 2 γ-Butyrolactones for ring opening polymerisation. | ||
This should enable easier incorporation of γ-butyrolactone units into polymer chains. For example, α-acetyl-γ-butyrolactone (2) displayed improved reactivity and displayed huge potential as a monomer in ROP.1h However, this potential and the applicability of γ-butyrolactones 2 and 3 as monomers in ROP could be restricted due to the multistep synthetic routes needed for their construction. Hence, this work provides a convenient, economical, and facile access to cis-disubstituted γ-butyrolactone of type 3, that could be explored as a monomer in ROP to produce functional and biodegradable polyesters. In the past decade, metal-hydrides2 such as Fe–H3a and Cu–H3b have been exploited for the hydrocarbonation of alkenes. In recent years, Ni–H has demonstrated the potential to functionalise alkenes with a directing group such as boronic esters4a and aryl groups4b,c next to the alkenyl moiety or remote olefins.4d,e
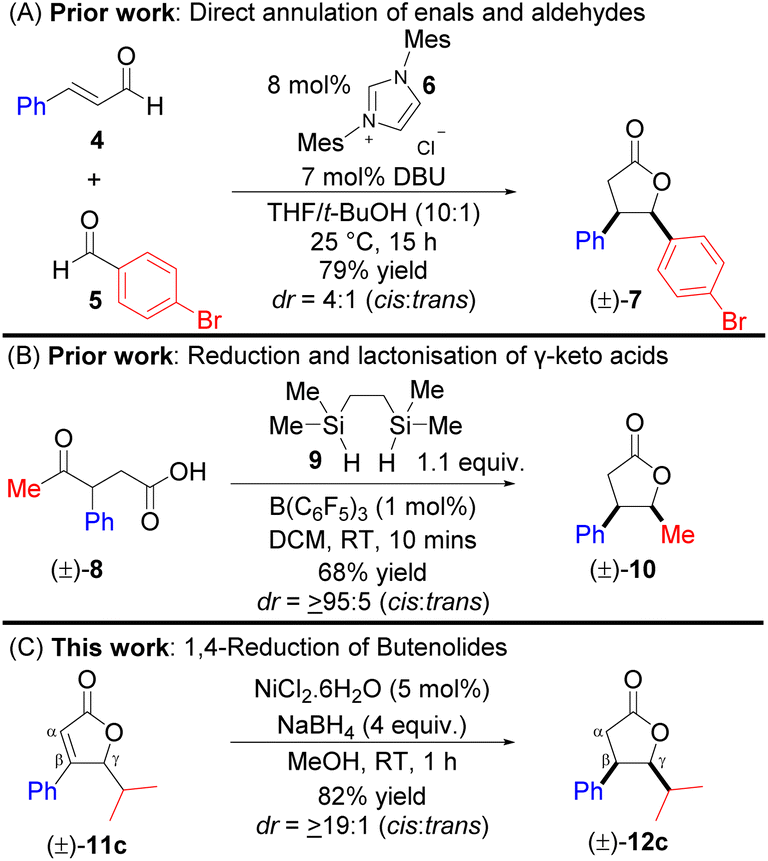
Synthesis of cis-(±)-disubstituted γ-butyrolactone has been achieved previously via linear precursors. For example, the direct annulation of enals (of type 4) and aldehydes (of type 5) was utilised for the stereoselective synthesis of disubstituted (±)-7, catalysed by N-heterocyclic carbene 6 (Scheme 1A).5a However, this synthetic method furnished all γ-butyrolactones with moderate diastereoselectivities (≤8/1; cis/trans). Recently, a report described a B(C6F5)3-catalysed reduction and lactonisation of γ-keto acids (±)-8 for the construction of γ-butyrolactone (±)-10 (Scheme 1B).5b The scope of the study was limited to substitutions such as Me, Ph (±)-8, allyl, or the propargyl group at the α-position of the ketone, although their corresponding γ-butyrolactones were obtained with excellent cis diastereoselectivity. Notably, four synthetic steps will be required to construct keto-acid (±)-8,5c and most importantly, different starting precursors will be needed for the synthesis of keto acid (±)-8 derivatives. As a subsequent study to our previous work,5d we herein described a convenient, economical, and straightforward access to cis-β(aryl),γ(alkyl)-disubstituted (±)-γ-butyrolactones of type 12cvia nickel-hydride 1,4-reduction of β,γ-disubstituted α,β-unsaturated lactones of type 11c (Scheme 1C).6a–c
 | ||
| Scheme 1 Construction of monocyclic cis-β,γ-disubstituted (±)-γ-butyrolactones. | ||
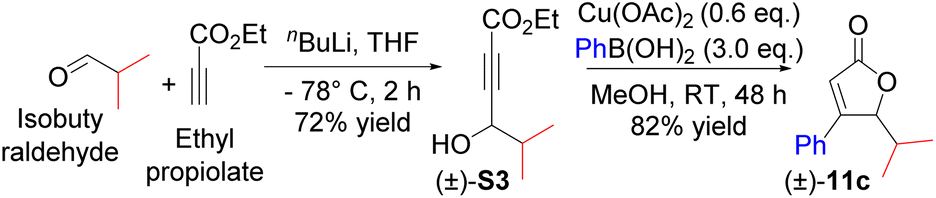
All the β,γ-disubstituted α,β-unsaturated lactones (butenolides) investigated (22 examples) in this study were obtained in two easy steps, starting from the cheap and commercially available ethyl propiolate and the corresponding aldehydes (Scheme 2). For example, the treatment of ethyl propiolate with nBuLi and subsequent addition of isobutyraldehyde provided (±)-S3. The two-step one-pot protocol of copper-catalysed conjugate addition of phenyl boronic acid and subsequent in situ cyclisation generated butenolide (±)-11c from (±)-S3.6d–e Initial attempts focused on the development of a strategy for the direct synthesis of γ-butyrolactone (±)-12c from alkynoate (±)-S3via nickel-catalysed hydroarylation, cyclisation and then 1,4-reduction using Ni(II) salts such as Ni(OAc)2·4H2O and NiCl2·6H2O instead of Cu(OAc)2. After 48 h, NaBH4 was added to the reaction; however, (±)-12c was obtained in a relatively low yield (<10%), with (±)-S3 mostly recovered. Based on this observation and in conjunction with previous reports,7a it seems that nickel-catalysed hydroarylation of alkynes proceed well when the alkyne bears a phenyl group rather than an ester group at the terminus end.7b Subsequent studies (Table 1) were designed and carried out with the aim of understanding the role of each reagent and to propose a plausible reaction mechanism. The starting material was recovered when the reaction was carried out in the absence of NiCl2·6H2O, and when NaBH4 was replaced with silanes as the hydride source (Table 1, entries 1–4).7c The order of addition of the reagents was critical to the feasibility of this reaction (Table 1, entries 5 vs. 6).
 | ||
| Scheme 2 Two-step synthesis of (±)-11c. | ||
|
|
|||
|---|---|---|---|
| Entry | Conditions |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13b :13b |
Yieldc |
| a Reactions were carried out at the 0.75 mmol scale. b Determined by 1H NMR analysis of the crude sample. c Isolated yield based on three reaction runs. d Formation of nickel boride by stirring NiCl2·6H2O and NaBH4 in MeOH for 5 min. After effervescence subsided, a black precipitate was formed, after which (±)-11c was added. Ni(OAc)2·4H2O also performed well in this reaction. | |||
| 1 | NaBH4, MeOH | — | — |
| 2 | NiCl2·6H2O (5 mol%) PMHS (4 eq.), MeOH | — | — |
| 3 | NiCl2·6H2O (5 mol%) PhSiH3(4 eq.), MeOH | — | — |
| 4 | NiCl2·6H2O (5 mol%) PhMe2SiH (4 eq.), MeOH | — | — |
| 5 | NiCl 2 ·6H2O (5 mol%) NaBH4(4 eq.), MeOH |
≥19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
82 |
| 6d | NiCl2·6H2O (5 mol%) NaBH4(4 eq.), MeOH | — | — |
| 7 | NiCl2·6H2O (5 mol%) NaBH4(4 eq.), THF | — | — |
| 8 | NiCl2·6H2O (5 mol%) NaBH4 (4 eq.), PhMe | — | — |
| 9 | NiCl2·6H2O (10 mol%) NaBH4(4 eq.), MeOH | ≥19:1 |
88 |
| 10 | NiCl2·6H2O (15 mol%) NaBH4(4 eq.), MeOH | ≥19:1 |
91 |

Compound (±)-12c was generated diastereoselectively in excellent yield when NaBH4 was added to a stirring light-green mixture of (±)-11c and NiCl2·6H2O. In contrast, the addition of (±)-11c to the stirring black suspension of pre-mixed NiCl2·6H2O and NaBH4 gave no product.8a This nullifies the notion that nickel boride facilitates the 1,4-reduction of the butenolide. The importance of a protic solvent towards the feasibility of the reaction was also noted (Table 1, entry 5 vs. entries 7 and 8). A higher mol% of nickel in the reaction resulted in improved yields (Table 1, entries 9 and 10).
The summary of the results in Table 1 postulates a butenolide ligated nickel hydride complex B (Scheme 3), since NiCl2·6H2O, NaBH4 and MeOH worked in tandem.8b,4c This ligation enabled the inner-sphere delivery of the hydride, followed by protonation of the Nickel enolate species D or D′ (Fig. 3), thereby generating the cis product in a highly diastereoselective manner.9
 | ||
| Scheme 3 Plausible mechanism. See Fig. S4† for more details. | ||
 | ||
| Fig. 3 CH3OH vs. CD3OD as solvent. | ||
The proposed mechanism in Scheme 3 was experimentally validated by investigating linear alkyl (Me, Et, iPr, and C5H11) substituents at the γ-position (Table 2). While 12a, b, and d were furnished in 3:1 dr, 12c was generated as the sole product. This observation can be attributed to the steric hindrance caused by the alteration of the tetrahedral angle.8d In addition, we ascertained the correlation between steric hindrance and high diastereoselectivity by exploring cyclic rings at the γ-position (Table 2, 12e–h). As the flexibility of the ring increases with increasing size (decrease in transannular strain), this allows for conformations that can disrupt the nickel-hydride species, forcing the delivery of the hydride from either the re-face or the si-face but not both.
| a Reaction conditions: (±)-11a–h (0.75 mmol), NiCl2·6H2O (0.0375 mmol), NaBH4 (3.00 mmol, 4.0 eq.), MeOH (10 mL), RT, 1 h. dr values were determined by 1H NMR analysis of the crude sample. Yields are those of the isolated cis-products 12a–h. |
|---|

|
The impact of angle strain of the γ-substituents on diastereoselectivity is noticeable when the dr of 12c is compared to that of 12e and/or 12f.8eTable 3 displays γ-butyrolactones with different aryl substitutions at the β-position. The results showed that their diastereoselectivities were solely dependent on the type of alkyl substituents at the γ-position (e.g., 12ivs.12o or 12pvs.12t) and they all followed similar trends as shown in Table 2. Next, we explored a butenolide with a ketone functional group that is susceptible to NaBH4 to test our Ni–H hypothesis. Using 4.0 equivalents of NaBH4, we obtained 12v. Noteworthily, using 1.0 equivalent of NaBH4, 12j was produced predominantly in a moderate yield and with excellent dr, accompanied by ketone reduction products (1:1).

Previously, we established that NaBH4 alone cannot facilitate the 1,4-reduction of butenolides (Table 1, entry 1). Perhaps, the 1,4-reduction was facilitated by Ni–H catalysis. The lack of reactivity of compound 11w (Scheme 4A) demonstrated that an additional substituent at the γ-position possibly disrupted the Ni–H species (as shown in B and C, R = Et, Scheme 3), presumably hindering the delivery of the hydride. The relative stereochemistry of the γ-butyrolactones was assigned cis based on the observed NOESY spectra (for 12c, 12g and 12h) and the trend in the 3JH–H coupling constant between the key protons on the lactone at the β- and γ-positions (see Fig. S5†). The synthetic utility of this class of cis-disubstituted γ-butyrolactone has been shown previously.8c12d was converted into a biologically active natural product, phaseolinic acid, in two steps.
 | ||
| Scheme 4 (A) Effect of additional substituent at the γ-position and (B) synthetic utility of γ-butyrolactone 12d. | ||
Conclusions
Our previous study2d described a Cu–H catalysed 1,4-reduction of β-substituted butenolides. Herein, we demonstrated the 1,4-reduction of β- and γ-substituted butenolides via Ni–H catalysis, which generated cis-disubstituted γ-butyrolactones. This study revealed that γ-butyrolactones with iPr, or cyclopentane or cyclohexane at the γ-position can be obtained in excellent diastereoselectivities and yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the School of Chemistry, University of Glasgow is gratefully acknowledged. Thanks to Giulia Pellegrino for laboratory assistance.References
- (a) R. Bandichhor, B. Nosse and O. Reiser, Paraconic Acids - The Natural Products from Lichen Symbiont, Top. Curr. Chem., 2005, 243, 43–72 CrossRef CAS; (b) N. S. Zhangabylov, L. Y. Dederer, L. B. Gorbacheva, S. V. Vasil'eva, A. S. Terekhov and S. M. Adekenov, Sesquiterpene lactone arglabin influences DNA synthesis in P388 leukemia cells in vivo, Pharm. Chem. J., 2004, 38, 651–653 CrossRef CAS; (c) T. Ok, A. Jeon, J. Lee, H. L. Jung, S. H. Chang and H. S. Lee, Enantiomerically pure synthesis of β-substituted γ-butyrolactones: A key intermediate to concise synthesis of pregabalin, J. Org. Chem., 2007, 72, 7390–7393 CrossRef CAS PubMed; (d) M. Bielitza and J. Pietruszka, An enantioselective Mukaiyama aldol reaction as the key step towards the tetrahydropyran core of psymberin via a γ-butyrolactone intermediate, Synlett, 2012, 1625–1628 CAS; (e) M. Hong and E. Y.-X. Chen, completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerisation of γ-butyrolactone, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed; (f) M. Hong and E. Y.-X. Chen, towards truly sustainable polymers: a metal-free recyclable polyester from biorenewable non-strained γ-butyrolactone, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed; (g) Q. Song, J. Zhao, G. Zhang, F. Peruch and S. Carlotti, ring-opening (co)polymerisation of γ-butyrolactone, Polym. J., 2020, 52, 3–11 CrossRef CAS; (h) X. Deng, J. Yao and M. Yuan, Polymerization of lactides and lactones 11. Ring-opening polymerization of α-acetyl-γ-butyrolactone and copolymerization with β-butyrolactone, Eur. Polym. J., 2000, 36, 2739–2741 CrossRef CAS.
- For reports where NiCl2·6H2O and NaBH4 were used for the 1,4-reduction of the butenolide moiety in natural product synthesis, see: (a) A. V. Bekish, K. N. Prokhorevich and O. G. Kulinkovich, transformation of esters into 2-Substituted allyl halides via tertiary cyclopropanols: application in the stereoselective synthesis of (2S,3S,7S,)-3,7-dimethyl-2-pentadecyl acetate, the sex pheromone of the pine Sawfly Neodiprion sertifer, Eur. J. Org. Chem., 2006, 5069–5075 CrossRef CAS; (b) F. Kido, Y. Noda and A. Yoshikoshi, new access to dl-Paniculide A using α-Phenylthio-β-Vinylbutenolide as synthetic block, Tetrahedron, 1987, 43, 5467–5474 CrossRef CAS; (c) T. Honda, T. Matsukawa and K. Takahashi, efficient total synthesis of (-)-stemoamide, Org. Biomol. Chem., 2011, 9, 673–675 RSC; (d) Y. Yamamoto and N. Kirai, synthesis of 4-aryl-substituted butenolides and pentinolides by copper-catalysed hydroarylation, Heterocycles, 2010, 80, 269–279 CrossRef CAS; (e) Y. Yamamoto, N. Kirai and Y. Harada, Cu-catalysed stereoselective conjugate addition of arylboronic acids to alkynoates, Chem. Commun., 2008, 2010–2012 RSC.
- (a) J. C. Lo, J. Gui, Y. Yabe, C. M. Pan and P. S. Baran, Functionalised olefin cross-coupling to construct carbon–carbon bonds, Nature, 2014, 516, 343–348 CrossRef CAS PubMed; (b) S. D. Friis, M. T. Pirnot and S. L. Buchwald, asymmetric hydroarylation of vinylarenes using a synergistic combination of CuH and Pd catalysis, J. Am. Chem. Soc., 2016, 138, 8372–8375 CrossRef CAS PubMed.
- (a) S. Bera and X. Hu, Nickel-catalysed regioselective hydroalkylation and hydroarylation of Alkenyl Boronic Esters, Angew. Chem., Int. Ed., 2019, 58, 13854–13859 CrossRef CAS PubMed; (b) J. He, P. Song, X. Xu, S. Zhu and Y. Wang, migratory reductive acylation between alkyl halides or alkenes and alkyl carboxylic acids by Nickel catalysis, ACS Catal., 2019, 9, 3253–3259 CrossRef CAS; (c) Y.-G. Chen, B. Shuai, X.-T. Xu, Y.-Q. Li, Q.-L. Yang, H. Qiu, K. Zhang, P. Fang and T.-S. Mei, Nickel-catalysed enantioselective hydroarylation and hydroalkenylation of styrenes, J. Am. Chem. Soc., 2019, 141, 3395–3399 CrossRef CAS PubMed; (d) Y. He, Y. Cai and S. Zhu, mild and regioselective benzylic C−H functionalization: Ni-catalysed reductive arylation of remote and proximal olefins, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS PubMed; (e) W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, tandem isomerisation and C-H activation: regioselective hydroheteroarylation of allylarenes, Org. Lett., 2013, 15, 5358–5361 CrossRef CAS PubMed.
- (a) S. S. Sohn, E. L. Rosen and J. W. Bode, N-heterocyclic carbene-catalysed generation of homoenolates: γ-Butyrolactones by direct annulations of enals and aldehydes, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed; (b) H. Xie, J. Lu, Y. Gui, L. Gao and Z. Song, (HMe2SiCH2)2: a useful reagent for B(C6F5)3-catalysed reduction–lactonization of keto acids: concise syntheses of (–)-cis-whisky and (–)-cis-cognac lactones, Synlett, 2017, 2453–2459 CAS; (c) K. Nozaki, H. Komaki, Y. Kawashima, T. Hiyama and T. Matsubara, predominant 1,2-Insertion of styrene in the Pd-catalysed alternating copolymerization with carbon monoxide, J. Am. Chem. Soc., 2001, 123, 534–544 CrossRef CAS PubMed; (d) O. S. Ojo, D. L. Hughes and C. J. Richards, An expedient copper-catalysed asymmetric synthesis of γ-lactones and γ-lactams. Application to the synthesis of lucidulactone A, Org. Biomol. Chem., 2023, 21, 4144–4149 RSC.
- For reports where NiCl2·6H2O and NaBH4 were used for the 1,4-reduction of the butenolide moiety in natural product synthesis, see: (a) A. V. Bekish, K. N. Prokhorevich and O. G. Kulinkovich, transformation of esters into 2-Substituted allyl halides via tertiary cyclopropanols: application in the stereoselective synthesis of (2S,3S,7S,)-3,7-dimethyl-2-pentadecyl acetate, the sex pheromone of the pine Sawfly Neodiprion sertifer, Eur. J. Org. Chem., 2006, 5069–5075 CrossRef CAS; (b) F. Kido, Y. Noda and A. Yoshikoshi, new access to dl-Paniculide A using α-Phenylthio-β-Vinylbutenolide as synthetic block, Tetrahedron, 1987, 43, 5467–5474 CrossRef CAS; (c) T. Honda, T. Matsukawa and K. Takahashi, efficient total synthesis of (-)-stemoamide, Org. Biomol. Chem., 2011, 9, 673–675 RSC; (d) Y. Yamamoto and N. Kirai, synthesis of 4-aryl-substituted butenolides and pentinolides by copper-catalysed hydroarylation, Heterocycles, 2010, 80, 269–279 CrossRef CAS; (e) Y. Yamamoto, N. Kirai and Y. Harada, Cu-catalysed stereoselective conjugate addition of arylboronic acids to alkynoates, Chem. Commun., 2008, 2010–2012 RSC.
- For a review on nickel-catalysed arylation of alkynes using aryl boronic acids, see: (a) S. M. Gillbard and H. W. Lam, Nickel-catalysed arylative cyclisations of alkyne- and allene-tethered electrophiles using arylboron reagents, Chem. – Eur. J., 2022, 28, e202104230 CAS; (b) For an example of nickel-catalysed arylation of alkynoates using aryl iodide and a reductant, see: D. K. Rayabarapu and C.-H. Cheng, Synthesis of seven-membered lactones via Nickel- and Zinc-catalysed highly regio- and stereoselective cyclisation of 2-Iodobenzyl alcohols with propiolates, J. Am. Chem. Soc., 2002, 124, 5630–5631 CrossRef CAS PubMed; (c) Z. M. Heiden and A. P. Lathem, Establishing the hydride donor abilities of main group hydride, Organometallic, 2015, 34, 1818–1827 CrossRef CAS.
- (a) G. Proietti, K. J. Prathap, X. Ye, R. T. Olsson and P. Dinér, Nickel boride catalysed reductions of nitro compounds and azides: Nanocellulose-supported catalysts in tandem reactions, Synthesis, 2022, 133–146 CAS; (b) J. C. Jenkins and M. Brookhart, A mechanistic investigation of the polymerisation of ethylene catalysed neutral Ni(II) complexes derived from bulky anilinotropone ligands, J. Am. Chem. Soc., 2004, 126, 5827–5842 CrossRef CAS PubMed; (c) J. L. Nallasivam and R. A. Fernandes, A protecting-group-free synthesis of (+)-nephrosteranic, (+)-protolichesterinic, (+)-nephrosterinic, (+)-phaseolinic, (+)-rocellaric acids and (+)-methylenolactocin, Org. Biomol. Chem., 2017, 15, 708–716 RSC; (d) R. M. Beesley, C. K. Ingold and J. F. Thorpe, The formation and stability of spiro -compounds. Part I. spiro-compounds from cyclohexane, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC; (e) T. Dudev and C. Lim, Ring strain energies from ab Initio calculations, J. Am. Chem. Soc., 1998, 120, 4450–4458 CrossRef CAS.
- W. N. O. Wylie, A. J. Lough and R. H. Morris, Mechanistic Investigation of the Hydrogenation of Ketones Catalyzed bya Ruthenium(II)Complex Featuring an N-Heterocyclic Carbene with aTethered Primary Amine Donor: Evidence for an Inner Sphere Mechanism, Organometallics, 2011, 30, 1236–1252 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00895a |
| This journal is © The Royal Society of Chemistry 2023 |