
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-oxyamide analogues of protein kinase B (Akt) targeting anionic glycoglycerolipids and their antiproliferative activity on human ovarian carcinoma cells†
Marco
Zuccolo
a,
Giulia
Orsini
b,
Martina
Quaglia
c,
Luca
Mirra
d,
Cristina
Corno
d,
Nives
Carenini
d,
Paola
Perego
 d and
Diego
Colombo
*c
d and
Diego
Colombo
*c
aDipartimento di Scienze Agrarie e Ambientali, Università degli Studi di Milano, Via Celoria 2, 20133 Milano, Italy
bNOVA Institute of Chemical and Biological Technology António Xavier, New University of Lisbon, Av. da Repύblica, 2780-157 Oeiras, Portugal
cDipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano, Via Saldini 50, 20133 Milano, Italy. E-mail: diego.colombo@unimi.it
dMolecular Pharmacology Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, via Amadeo 42, 20133 Milan, Italy
First published on 26th July 2023
Abstract
N-Oxyamides of bioactive anionic glycoglycerolipids based on 2-O-β-D-glucosylglycerol were efficiently prepared. However, the oxidation step of the primary hydroxyl group of the glucose moiety in the presence of the N-oxyamide function appeared to be a difficult task that was nevertheless conveniently achieved for the first time by employing a chemoenzymatic laccase/TEMPO procedure. The obtained N-oxyamides exhibited a higher inhibition of proliferation of ovarian carcinoma IGROV-1 cells in serum-free medium than in complete medium, similarly to the corresponding bioactive esters. Stability and serum binding studies indicated that the observed reduced activity of the compounds in complete medium could be mainly due to a binding effect of serum proteins rather than the hydrolytic degradation of glycoglycerolipid acyl chains. Furthermore, the results of the cellular studies under serum-free conditions suggested that the N-oxyamide group could increase the antiproliferative activity of a glycoglycerolipid independently of the presence of the anionic carboxylic group. Cellular studies in other cell lines besides IGROV-1 also support a certain degree of selectivity of this series of compounds for tumor cells with Akt hyperactivation.
Introduction
Protein kinase B (Akt) is a component of the PI3K–Akt–mTOR signaling pathway which is part of the cellular network involved in cell survival. Because Akt activation has been linked to crucial events in cancer development and progression, such as resistance to anti-cancer drugs and metastatic spread, this kinase family represents an important target for cancer treatment.1,2 Based on the presence of a regulatory Pleckstrin Homology (PH) lipid binding domain in the Akt structure, characterized by positively charged amino acids,3 anionic glycoglycerolipids based on 2-O-β-D-glucosylglycerol (a natural compound named Lilioside B in which glucose is β-linked to the C-2 of glycerol)4 were recently synthesized and tested as Akt inhibitors targeting the PH domain.5,6 Their structures (1 and 2, Fig. 1) were inspired by the natural anionic lipids sulfoquinovosyldiacylglycerols (SQDG)7 and glucuronosyldiacylglycerols (GLCADG)8 mimicking phosphatidylinositol-3-phosphate (PI3P) (Fig. 1), a plasma membrane lipid derived from the action of phosphatidylinositol-3-kinase (PI3K) that is similar to phosphatidylinositol-3,4,5-triphosphate (PIP3), the natural ligand of the Akt lipid-binding PH domain.9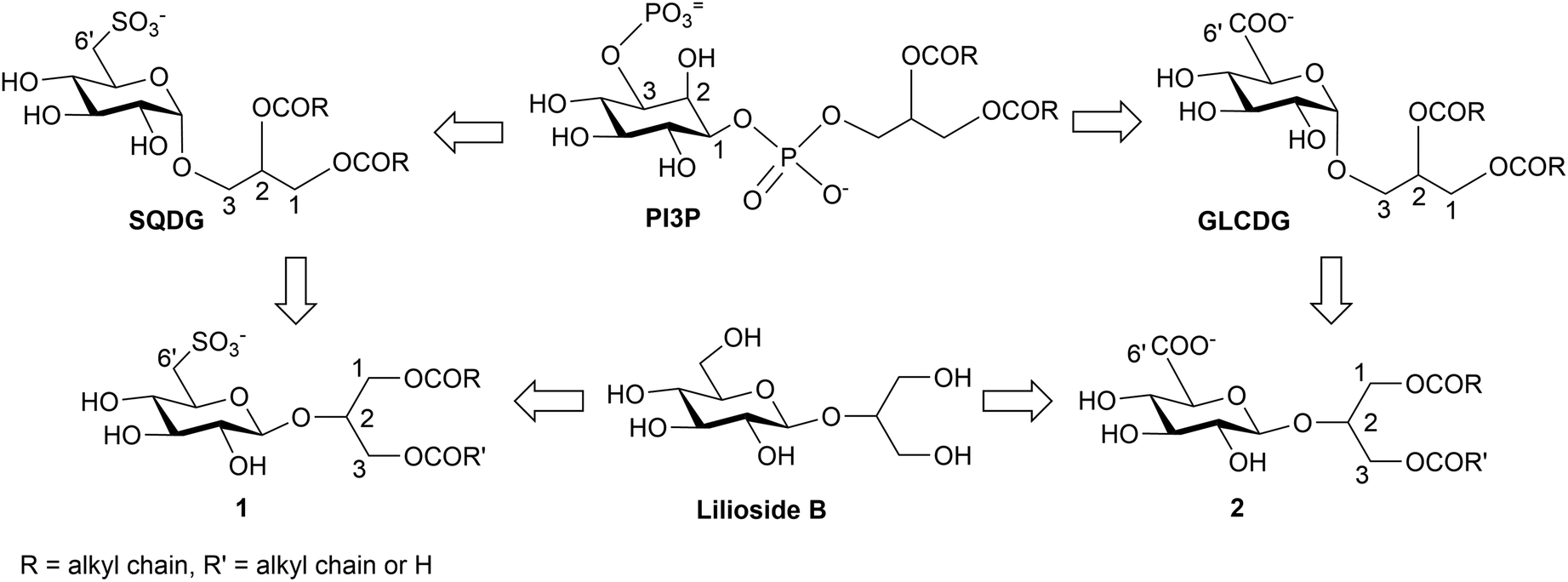 | ||
| Fig. 1 Structure of the synthetic anionic glycoglycerolipids 1 and 2 based on the merge of the natural compounds, Lilioside B, SQDG and GLCDG, mimicking PI3P. | ||
An antiproliferative activity in the human ovarian carcinoma IGROV-1 cell line was observed especially for compounds 1a,52a and 2b.6 Actually, a more marked growth inhibitory effect was produced when the drug incubation occurred in serum-free medium as compared to complete medium, thereby suggesting that serum affects the bioavailability of the compounds. Indeed, in serum-free medium, the IC50 value of compound 2a changed from 156.7 to 9.40 μM and that of compound 2b decreased from 49.0 to 3.35 μM (Fig. 2).
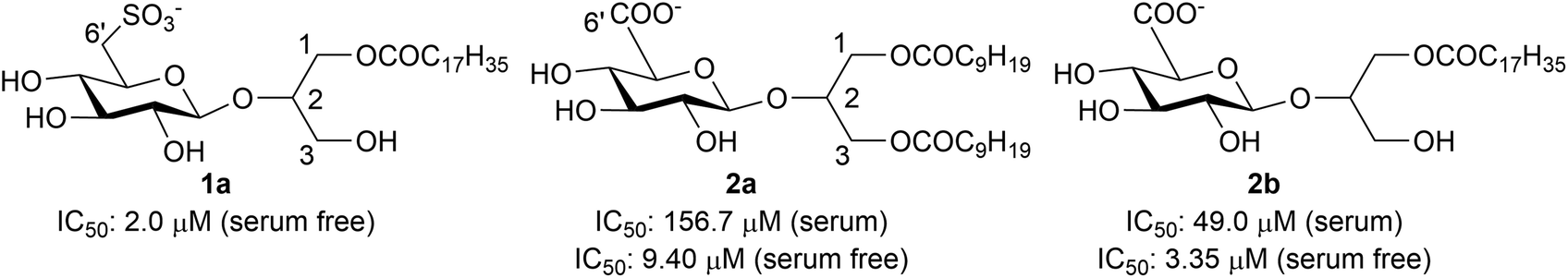 | ||
| Fig. 2 Structures of anionic glycoglycerolipids and their antiproliferative activity in the human ovarian carcinoma IGROV-1 cell line. Whilst 2a and 2b were assayed both in complete and serum-free medium, compound 1a was tested in serum-free medium only. | ||
In this work, we prepared N-oxyamide analogues of the anionic glucuronosylglycerolipid 2a and 2b. Namely, N-oxyamides 3a and 4a, together with the non-anionic N-oxyamides 3b and 4b and the known ester 5 (see the Experimental section) used as reference compounds, were synthesized in order to obtain anionic glycoglycerolipids with improved antiproliferative activity (Fig. 3). Actually, the conversion into N-oxyamide is an eligible modification to improve the metabolic stability of carboxylic derivatives as in the case of oligonucleotides,10 sugars,11 glycolipids12,13 and peptides14 which are reported to be very stable against enzymatic hydrolysis. Thus, stability and serum binding studies were also performed on selected compounds. Antiproliferative activity was assayed in the human ovarian carcinoma IGROV-1 cell line, in which Akt is hyperactivated,15 in complete or serum-free medium and the obtained results are discussed. In addition, the IGROV-1/Pt1 cisplatin-resistant cell lines, with increased Akt activation as compared to IGROV-1 cells and the TOV112D cell line in which Akt and other cell survival pathways are not activated, were used to prove the selectivity of our approach.
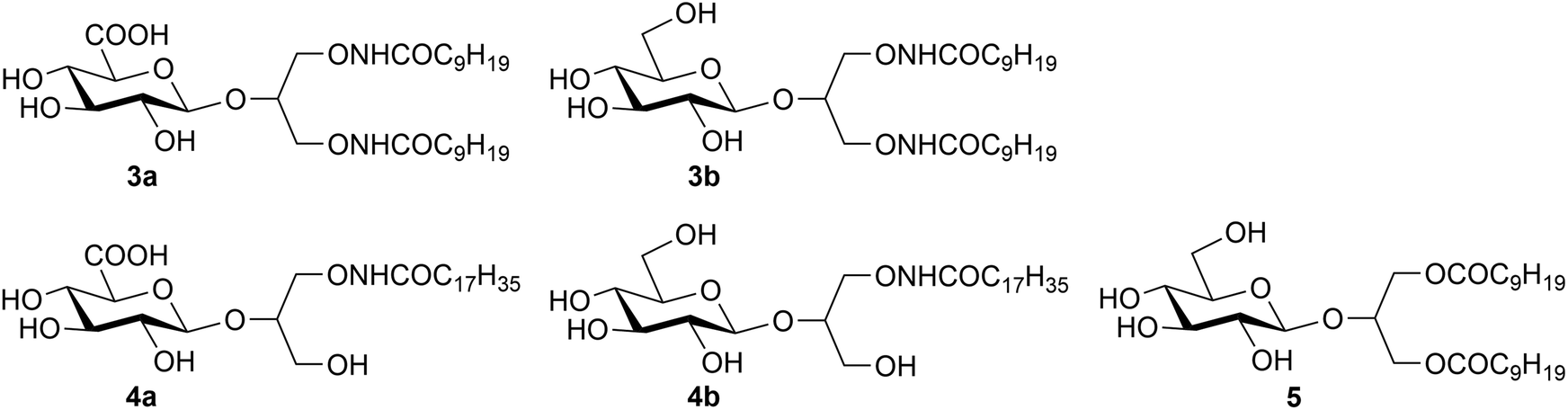 | ||
| Fig. 3 Structures of the tested compounds. | ||
Results and discussion
Chemistry
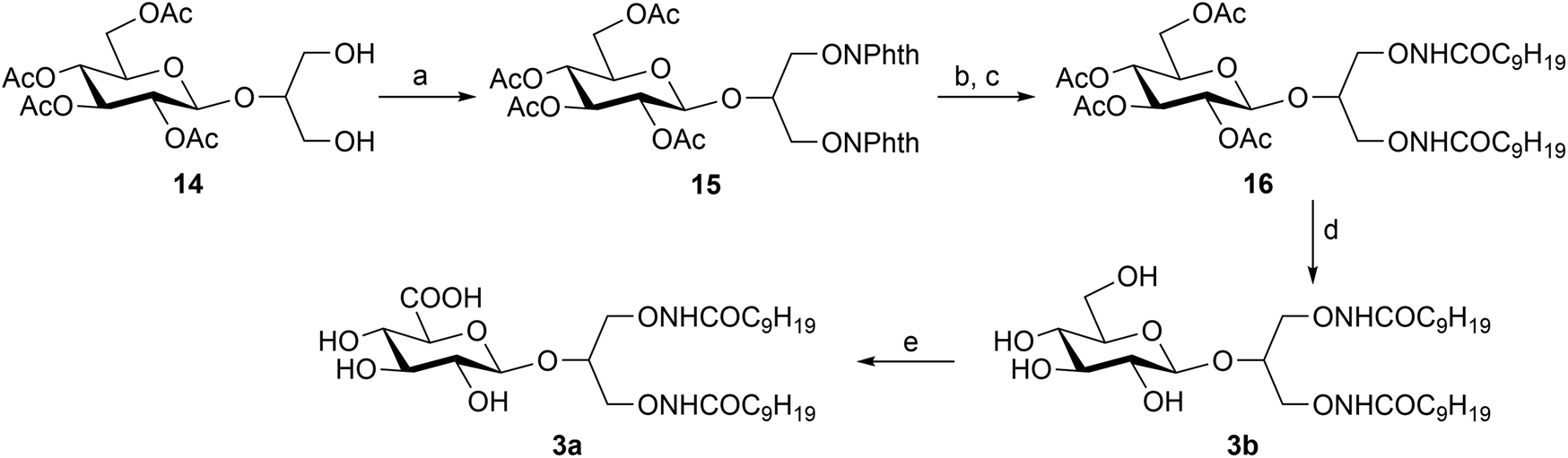
Following this synthetic pathway, compound 3a was prepared (Scheme 1) starting from the known 1,3-di-O-benzyl-2-O-β-D-glucopyranosyl-sn-glycerol (6)18 which was selectively oxidized with NaClO/NaClO2 at the C-6′ of the sugar moiety by a TEMPO-mediated procedure6 yielding glucuronide (7). After several attempts with different common reagents (e.g. tert-butanol with DCC), carboxyl esterification to tert-butyl ester 8 was achieved by using tert-butyl 2,2,2-trichloroacetimidate19 albeit in a quite low yield, due to the formation of various by-products in some cases identified by NMR as mixed tert-butyl ethers (data not shown). The obtained 8 was fully acetylated to 9 and then debenzylated by palladium-catalyzed hydrogenolysis yielding 10. The Mitsunobu reaction between this compound and N-hydroxyphthalimide yielded the di-O-phthalimido derivative 11 which was converted into a di-O-amino compound by hydrazine treatment. This compound was directly acylated, without previous purification, by treatment with decanoyl chloride and pyridine affording the fully protected N-oxyamide 12. The Zemplén deacetylation reaction yielded 13 which was finally converted into 3a by trifluoroacetic acid treatment at room temperature.
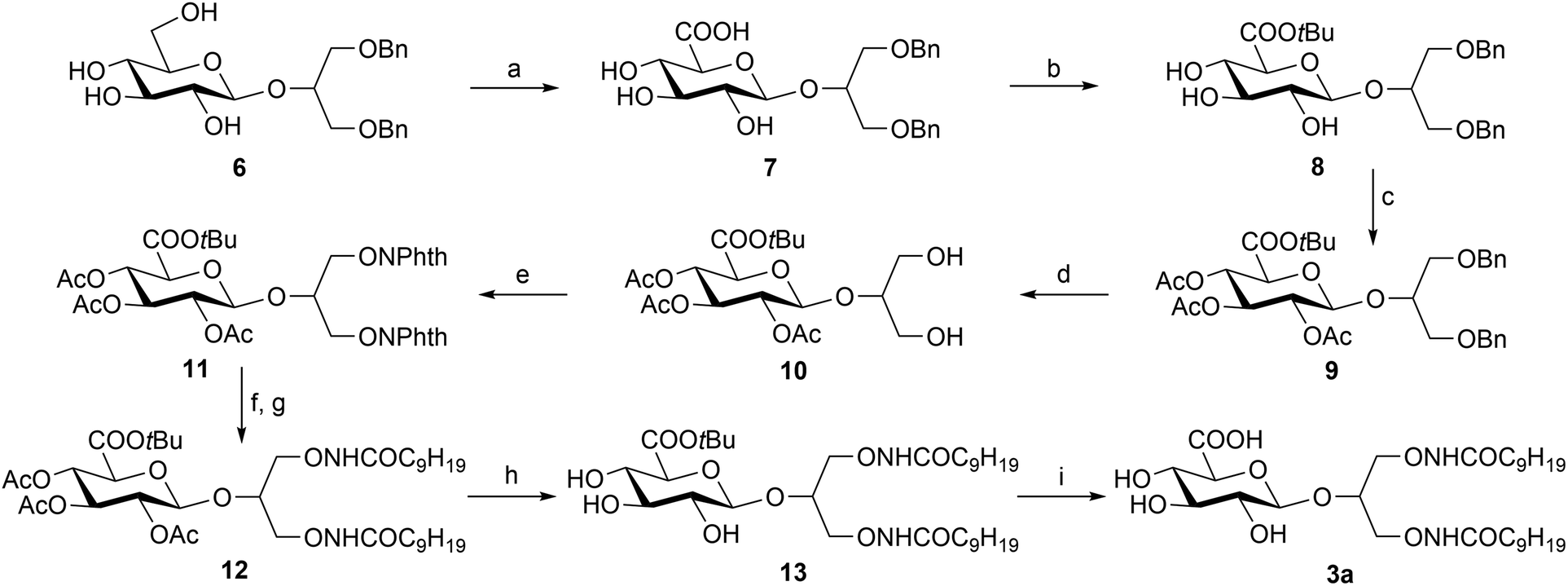 | ||
| Scheme 1 a. TEMPO, NaClO/NaClO2, CH3CN, 0.67 M phosphate buffer (pH 6.7), RT, (89%); b. tert-butyl 2,2,2-trichloroacetimidate, CH2Cl2/cyclohexane, RT, (33%); c. Ac2O, Py, RT, (80%); d. H2, 10% Pd/C, CH3OH, RT, (98%); e. PPh3, DIAD, N-hydroxyphthalimide, toluene/CH2Cl2, (86%); f. hydrazine hydrate, CH3OH; g. C9H19COCl, Py, CH2Cl2, RT, (45%, two step); h. CH3ONa, CH3OH, RT, (50%); i. TFA, CH2Cl2, RT, (67%). | ||
Nevertheless, a more concise synthetic route was also planned to increase the overall yield of the desired N-oxyamide 3a. This alternative synthetic strategy relies on the introduction of the carboxylic function into the last synthetic step after the generation of N-oxyamides. So, in this way, it should be possible to reduce the number of protection and deprotection steps required (see Scheme 2). The Mitsunobu reaction between the known 2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (14)20,21 and N-hydroxyphthalimide yielded the di-O-phthalimido derivative 15 which was converted into the unstable related di-O-amino derivative by treatment with hydrazine hydrate. The reaction time of this synthetic step became a very critical factor. We found that a short stirring time and rapid quenching of the reaction are mandatory for the success of the reaction. Actually the cleavage of the phthalimide occurred very rapidly within a few minutes after the addition of hydrazine hydrate, but migration of the acetyl groups from sugar to the free O-amino groups on the glycerol moiety also occurred. Because of this, the di-O-amino compound was directly acylated without isolation by treatment with decanoyl chloride and pyridine providing the fully protected N-oxyamide 16. Finally, this compound was successfully converted into compound 3b by the Zemplén transesterification. The first attempt of 3b oxidation (see Table 1) was performed using a catalytic amount (0.3 equivalent) of TEMPO and NaClO/NaClO2 as terminal oxidants according to Zhao's modification of Anelli's procedure.22 A similar oxidation procedure was already used in ref. 16 for the synthesis of castanospermine, indicating the possibility of performing TEMPO-mediated oxidation in the presence of the N-oxyamide. However, all attempts of oxidation under these conditions were unsuccessful leading to a mixture of degradation products. N-Halogenation might be involved in the degradation of the substrate. Indeed, the formation of N-alkoxy-N-chloroamides from the parent N-oxyamide in the presence of tert-butyl hypochlorite as a positive chlorine source has been described.23 In this context, we decided to perform the oxidation in the absence of the terminal oxidants. Indeed, in the TEMPO-mediated oxidation reactions, the active oxidant species is the oxoammonium generated in situ by oxidation of TEMPO with a suitable terminal oxidant.22 Thus, a second attempt of oxidation was performed using the stable TEMPO oxoammonium tetrafluoroborate salt (TEMPO+BF4−) as a non-catalytic oxidant.24 The oxidation of 3b was performed by adopting the reaction conditions reported by Davis and Flitsch for the oxidation of monosaccharide derivatives to uronic acids25 in a biphasic dichloromethane water system at 0 °C in the presence of tetrabutylammonium bromide and sodium bicarbonate as bases. Four equivalents of TEMPO+BF4− were used because of the competitive comproportionation between the unreacted oxoammonium salt and the reduced hydroxylamine formed as the by-product of the reaction.26 Under these conditions, it was not possible to obtain the complete conversion of the starting material, and product 3a was obtained in 30% yield. The incomplete conversion was attributed to the decomposition of TEMPO+BF4−. Indeed, it is known that in diluted basic solution, the oxoammonium salt can be converted in TEMPO with the formation of hydrogen peroxide as the by-product.27 Thus, in order to improve the conversion of the starting material, the reaction was performed using an excess of the oxidant reagent (6 eq.), but complete decomposition of the product was observed. Therefore, a screening was performed to find the reaction conditions favoring the desired oxidation of 3b (Table 1). The reaction was performed using different solvent mixtures, at different temperatures and pH values using various buffer systems, or pyridine as an organic base,28,29 but no significant improvements were obtained. In particular, we observed that the pH has a great influence on the reaction outcome. The best performances were observed using sodium bicarbonate solution and pH ranging from 8 to 9. The use of carbonate buffer with pH around 10 led to complete decomposition, while no reaction was observed using phosphate buffer at pH 7. This last result was not unexpected as primary alcohols carrying an oxygen atom at the β-position are known to exhibit very poor reactivity towards oxoammonium-mediated oxidations conducted in neutral and slightly acidic media.30 These substrates are generally oxidized when the reaction is performed in the presence of a base, leading to the formation of the corresponding dimeric esters.31,32 Moreover, we observed that the substrate and the product of the reaction were also sensitive to the amount of the oxidant, and an excess of TEMPO+BF4− invariably induced extensive decomposition.
 | ||
Scheme 2 a. PPh3, DIAD, N-hydroxyphthalimide, toluene/CH2Cl2, RT, (79%); b. NH2NH2·H2O, CH3OH/THF 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 0 °C; c. C9H19COCl, Py, CH2Cl2, −10 °C (73%, over two step); d. CH3ONa, CH3OH, RT, (81%); e. Trametes versicolor laccase, TEMPO, air, acetate buffer pH 4.5, 30 °C, (49%). :2, 0 °C; c. C9H19COCl, Py, CH2Cl2, −10 °C (73%, over two step); d. CH3ONa, CH3OH, RT, (81%); e. Trametes versicolor laccase, TEMPO, air, acetate buffer pH 4.5, 30 °C, (49%). | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Oxidants (eq.) | Additives/bases (eq.) | Solvent/buffer | Temp. (°C) | Time (h) | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reactions were performed using 3b (0.17 mmol) in 2.25 mL of solvents in the presence of oxidants, bases, and additives (as indicated in the table). b Isolated yields. c 3b recovered by flash chromatography (recovery >90%). d Further 11 U of T. versicolor laccase and 0.8 eq. of TEMPO were added after 3 and 8 days of the reaction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | TEMPO (0.3)/NaClO (0.2)/NaClO2 (3) | — | CH3CN/phosphate buff. (1.8 M, pH 6.8) 55:45 |
25 | 6 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | TEMPO + BF 4 − (4) | TBAB (0.06)/NaHCO 3 (3) |
DCM/H
2
O 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :3 :3
|
0 | 3 | 38 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | TEMPO+BF4− (6) | TBAB (0.06)/NaHCO3 (3) | DCM/H2O 5:3 |
0 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | TEMPO+BF4− (4) | TBAB (0.06)/NaHCO3 (3) | DCM/H2O 5:3 |
25 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | TEMPO+BF4− (4) | TBAB (0.06)/NaHCO3 (3) | DCM/H2O 5:3 |
−10 | 3 | 30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | TEMPO+BF4− (4) | TBAB (0.06) | DCM/H2O 5:3 |
0 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | TEMPO+BF4− (4) | TBAB (0.06) | CH3CN/H2O 9:1 |
0 | 24 | n.d.c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | TEMPO+BF4− (4) | TBAB (0.06) | DCM/phosphate buffer (1.8 M, pH 6.8) 5:3 |
0 | 24 | n.d.c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | TEMPO+BF4− (4) | TBAB (0.06) | DCM/phosphate buffer (1.8 M, pH 6.8) 5:3 |
25 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | TEMPO+BF4− (4) | — | CH3CN/phosphate buffer (1.8 M, pH 6.8) 55:45 |
0 | 24 | n.d.c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | TEMPO+BF4− (4) | TBAB (0.06) | DCM/carbonate buffer (1.8 M, pH 10) 5:3 |
0 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | TEMPO+BF4− (4) | TBAB (0.06)/pyridine (3) | DCM/H2O 5:3 |
0 | 3 | Dec. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | TEMPO (0.8)/T./versicolorlaccase (11 U)/air | — | Acetate buffer (20 mM, pH 4.5) | 30 | 10 days | 49 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Hence, we reconsidered the use of the catalytic TEMPO-mediated oxidation procedure. In particular, we selected a laccase–TEMPO system that was reported as a strategy for the mild regioselective oxidation of sugar primary hydroxy groups.33,34 Compound 3b was suspended in acetate buffer and stirred for ten days in the presence of a catalytic amount of TEMPO and commercially available Trametes versicolor laccase. Under these conditions (see the Experimental section), complete conversion of the starting material and reduced formation of by-products were observed allowing a significant increase of the product yield. However, the amphiphilic nature of 3a made its isolation and purification difficult, limiting its recovery to about 50%. Moreover, to reduce the reaction time, we considered to improve the low solubility of 3b in acetate buffer by using acetonitrile as a co-solvent as it has been reported that a concentration up to 10% in volume is well tolerated by this laccase without a detrimental effect on its activity,35 but, unfortunately, compound 3b exhibited a very low solubility even in pure acetonitrile.
20,21 was protected as decanoate at the 3-O position employing the known selectivity of the lipase from Candida antarctica catalyzed transesterification in the presence of trifluoroethyl decanoate as an acyl donor.36 This reaction proceeded with good regioselectivity affording the desired compound 17a and its 1-O isomer in a 90:10 ratio (by NMR). The configuration assignment of 17a was obtained by chemical correlation of 17b (Scheme 3) as reported in the Experimental section. The following Mitsunobu reaction yielded the 1-O-phthalimido derivative 18 which was converted into the unstable 1-O-amino derivative by treatment with hydrazine hydrate. This was directly acylated without isolation by treatment with octadecanoyl chloride and pyridine, providing the fully protected N-oxyamide 19. The mild removal of the protective acetyl groups of 19 was performed with triethylamine adopting the reaction conditions reported by Meier et al.37 in a methanol/triethylamine/water mixture, affording the deacetylated compound 20 in 38% yield. Unfortunately, it was not possible to obtain the complete conversion of the starting material as the prolonged stirring time led to the cleavage of the decanoyl protecting group and the formation of the fully deprotected compound 4b. Nevertheless, this compound was isolated in 18% yield and was employed as a non-anionic derivative of 4a for biological assays. The glucuronic derivative 21 was obtained in 66% yield through the same laccase-mediated oxidation previously used for the oxidation of the diacyl derivative. Finally, the desired derivative 4a was successfully obtained by the Zemplén transesterification.
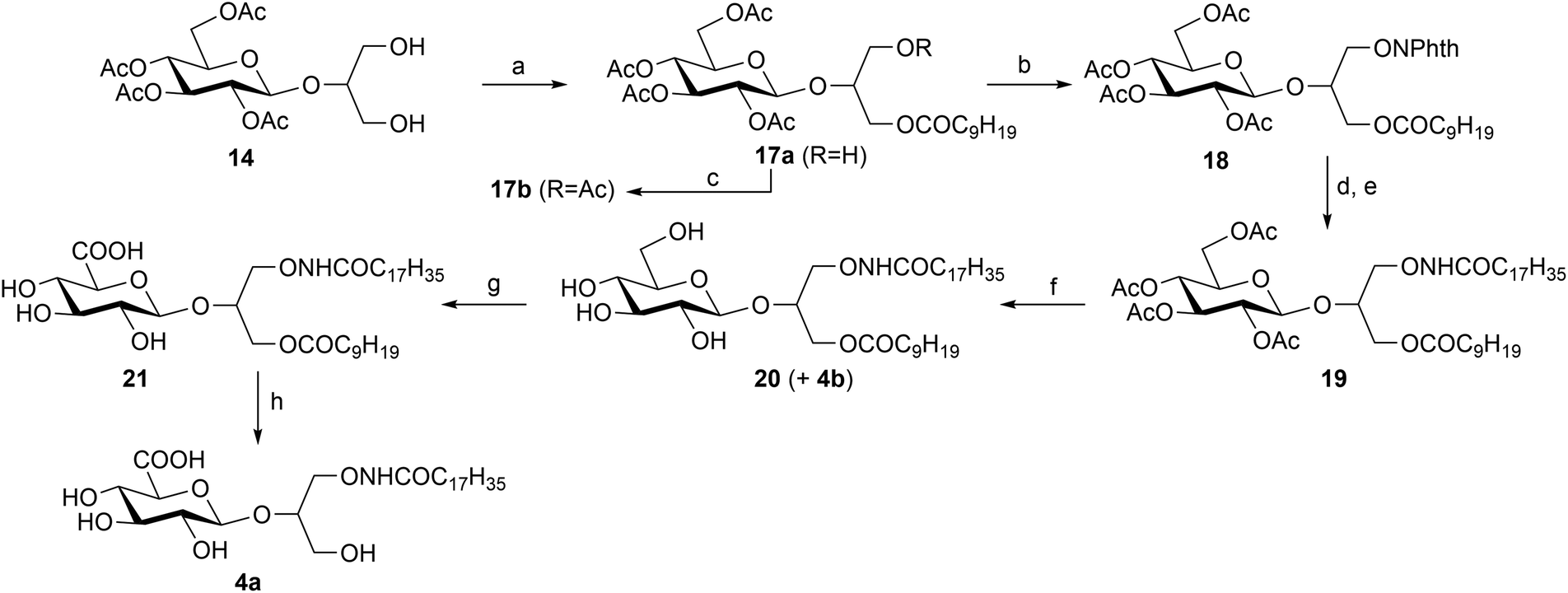 | ||
| Scheme 3 a. C9H19COOCH2CF3, Candida antarctica lipase, pyridine, 30 °C (76%); b. PPh3, DIAD, N-hydroxyphthalimide, toluene/CH2Cl2, RT, (92%); c. Ac2O, Py, RT. d. NH2NH2·H2O, CH3OH/THF 5:2, 0 °C; e. C17H35COCl, Py, CH2Cl2, −10 °C, (86%, over two step); f. Triethylamine/H2O/CH3OH, THF, RT, (38%); g. T. versicolor laccase, TEMPO, air, acetate buffer pH 4.5, 30 °C, (66%); h. CH3ONa, CH3OH, RT, (94%). | ||
Stability study
As the hydrolytic activity of serum components, including albumin, vs. ester compounds such as drugs or lipids is known,38–40 in order to compare the hydrolytic resistance of N-oxyamide vs. ester glycoglycerolipids, a stability study was conducted. Compounds 3b and 5 were selected as representatives of oxyamides and esters, respectively, and were subjected to conditions similar to those used for cellular studies in complete serum and checked by TLC and 1H-NMR analysis (see the Experimental section). The results indicated that, different from 5, 3b was detectable again after 24 h, confirming the greater stability of N-oxyamides vs. esters for this class of glycoglycerolipids.Serum binding study
Plasma protein binding is known to affect the concentration of a number of drugs;41 thus the binding of the oxyamide compounds to serum proteins was evaluated. In particular, compound 3a, as a representative of the oxyamides, was used for a test mimicking the cellular experimental conditions (see the Experimental section). Among the most commonly reported methods (equilibrium dialysis – ED, rapid equilibrium dialysis – RED, ultrafiltration – UF, and ultracentrifugation – UC),42,43 UF was used to recover the unbound 3a. As non-specific binding could interfere with the analysis,43 we prepared samples with the same concentration of 3a but with an increasing amount of serum in order to check the trend of binding. Actually, we observed the diminishing of unbound 3a with the increase of the serum amount by 1H-NMR analysis (see the Experimental section). This result could support the reduced activity of the tested compounds in the cellular experiments performed in the presence of serum.Cellular studies
The antiproliferative activity of the synthesized N-oxyamides was examined using the IGROV-1 ovarian carcinoma cell line known to harbor hyperactivated Akt15 either in complete medium or serum-free medium. The percentages of the inhibition of IGROV-1 cell growth in drug-treated samples versus solvent-treated samples, after 24 h of exposure to compounds 3a, 3b, 4a, 4b, and 5 either in complete medium or serum-free medium (see the Experimental section), are reported in the representative dose–response curves (Fig. 4).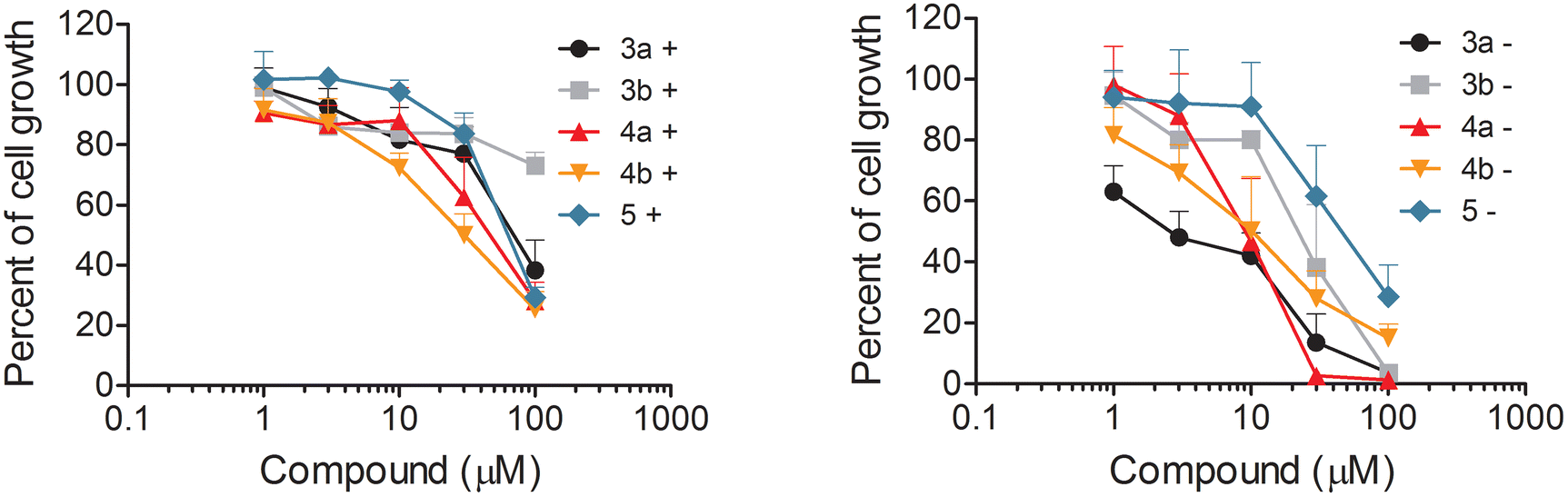 | ||
| Fig. 4 Cell sensitivity to the studied compounds. IGROV-1 cells were seeded and 24 h later, they were exposed to the compounds in the presence (+) or the absence (−) of fetal bovine serum. Forty-eight hours later, the cells were counted with an automatic cell counter. The graphs report the mean (±SEM) of at least 3 independent experiments. | ||
IC50 values of compounds 3–5 and the previously tested compounds 2a and 2b are shown in Table 2 in order to compare their effects on cell growth inhibition under both experimental conditions.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | Functional group (R)c | Linkage (X)b | IC50 μM ± SDa | IC50 μM ± SDa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Serum (+) | Serum (−) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a IC50 values represent the compound concentration producing 50% inhibition of cell growth (±SD) in the presence (+) or in the absence (−) of fetal bovine serum. b Type of linkage between fatty acid and glycerol. c Functional group at the C-6 of glucose. d Data from ref. 6. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Octadecanoylmonoacyl derivatives | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2b | COOH | O | 49.0 ± 7.60 | 3.35 ± 0.35 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4a | COOH | ONH | 41.2 ± 20.1 | 5.85 ± 1.43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4b | CH2OH | ONH | 31.5 ± 15.9 | 10.4 ± 8.26 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Decanoyldiacyl derivatives | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2a | COOH | O | 156.7 ± 23.0 | 9.40 ± 5.00 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3a | COOH | ONH | 71.5 ± 34.3 | 6.98 ± 4.94 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3b | CH2OH | ONH | n.d. | 13.7 ± 7.13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CH2OH | O | 63.4 ± 4.65 | 33.2 ± 21.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

All the new oxyamides 3a, 3b, 4a, and 4b were less active in the complete medium experiments (Table 2), as already reported for esters 2a and 2b,6 indicating that the observed negative serum influence could mainly be due to a serum protein binding effect rather than the hydrolytic degradation of glycoglycerolipid acyl chains. In the serum-free medium, oxyamides 3a and 4a were as active as the corresponding esters 2a and 2b showing similar IC50 values (6.98 and 5.85 vs. 9.40 and 3.35, respectively, Table 2). Of note, the new monooxyamide 4a and dioxyamide 3a showed similar IC50 values (6.98 and 5.85, respectively, Table 2), whereas monoester 2b seemed more active than diester 2a (3.35 and 9.40, respectively, Table 2).
Interestingly, even oxyamides 3b and 4b lacking the carboxylic function were active even though they showed twice the IC50 values of the corresponding anionic glycoglycerolipids 3a and 4a (13.7 and 10.4 vs. 6.98 and 5.85, respectively, Table 2). In contrast, the non-anionic ester 5, lacking both the carboxylic and oxyamide functions, was the less active compound showing the worst IC50 value (33.2, Table 2), which was about 4–5 fold higher than that of 3a (6.98, Table 2).
Based on these results, it seems that the N-oxyamide group exerts a generally positive effect on the IGROV-1 anti-proliferative activity of glycoglycerolipids that is similar to that caused by the carboxylic (anionic) function, even if the effect of the two groups seems to be not additive (see 4avs. 2b and 3avs. 2a, Table 2).
The antiproliferative effect of compounds 4a and 4b was examined in two additional ovarian carcinoma cell lines. Specifically, we used the cisplatin-resistant variant IGROV-1/Pt1 showing increased activation of the Akt pathway as compared to the IGROV-1 parental cell line44 and the TOV112D cell line known to be characterized by the lack of alterations of the PI3K/Akt pathway.45 When the cells were exposed to the compounds in the absence of serum, we observed a higher activity of both in the IGROV-1/Pt1 cells (IC50 was 2.00 ± 0.6 μM and 3.78 ± 1.4 μM for 4a and 4b, respectively; P < 0.05 by Student's t test for comparison vs. IGROV-1 values), consistent with a higher activation of the Akt pathway (Fig. 5). Conversely (Fig. 6), the activity of both compounds was reduced in TOV112D cells, and the IC50 values were 20.51 ± 4.6 μM and 55.85 ± 10.3 μM for 4a and 4b, respectively (P < 0.05 by Student's t test for comparison vs. IGROV-1/Pt1 values). Such findings are in agreement with the lack of Akt-dependence of TOV112D cells and highlight the interest in our compounds as inhibitors of tumor cells with deregulated survival pathways.
 | ||
| Fig. 5 Cell sensitivity to 4a and 4b of IGROV-1/Pt1 cells with increased activation of the Akt pathway in the absence of fetal bovine serum. The graphs report the mean (±SEM) of triplicate experiments. | ||
 | ||
| Fig. 6 Cell sensitivity to 4a and 4b of TOV112D cells with lack of alterations of the PI3K/Akt pathway in the absence of fetal bovine serum. The graphs report the mean (±SEM) of triplicate experiments. | ||
As the aminoxy amide NH function is a very good hydrogen bond donor,12,46 the insertion of an N-oxyamide function into the glycoglycerolipid structure could modify the mode of interaction of these compounds with the cell membrane or the possible molecular target. Thus, we subjected oxyamides 3a, 3b, 4a, and 4b to inhibition cell-free assays (ELISA test, Fig. 5) to check their inhibitory effect on Akt.
Cell-free evaluation of Akt inhibition
Oxyamides 3a, 3b, 4a and 4b were tested for inhibitory activity against Akt (Akt1) using an in vitro ELISA to measure kinase activity (see the Experimental section). The obtained results at 10 μM sample concentration as reported in Fig. 7 show that diacyloxyamides 3a and 3b were more active than the monoacyl derivatives 4a and 4b. Comparing the results of oxyamides 3avs. 3b and 4avs. 4b, it seems that, in both cases, the presence or the absence of the anionic carboxylic group does not modify significantly the inhibition of Akt (75% vs. 81% and 21% vs. 34%, respectively, Fig. 7), suggesting that the lack of anionic carboxylate was compensated by the presence of the oxyamidic function. | ||
| Fig. 7 Akt inhibition in cell-free assays Akt inhibition was assayed by ELISA by measuring the O.D. at 450 nm. Oxyamides 3a, 3b, 4a, and 4b, were examined besides perifosine, used as a reference compound, at 10 μM concentration. The data represent the mean (±SEM) of three values. Statistical analysis was performed by one-way ANOVA followed by Bonferroni's correction for multiple comparisons. **, P < 0.01; ***, P < 0.001. | ||
Conclusions
In this work, we aimed to prepare anionic glycoglycerolipids having acyl chains linked to the glycerol moiety with an N-oxyamide linkage, and the oxidation of a primary hydroxyl group in the presence of the N-oxyamide function, rarely reported in the literature to the best of our knowledge, turned out to be quite a difficult task. However, a careful optimization of the experimental conditions allowed us to obtain the desired new N-oxyamide glucuronides 3a and 4a in satisfactory yields using a laccase/TEMPO-mediated chemoenzymatic approach. These compounds together with the corresponding non-anionic N-oxyamides, namely the newly obtained glycoglycerolipids 3b and 4b, were tested for their antiproliferative activity in the human ovarian carcinoma IGROV-1 cell line in complete and serum-free medium. The cellular experiments also revealed that N-oxyamides (3a and 4a) were less active when tested in the complete serum medium as already observed in the case of the corresponding esters (2a and 2b), suggesting that binding with serum components could be mainly responsible for their reduced overall activity as suggested also by the stability and serum binding studies. The results in the absence of serum showed that the new anionic oxyamides (3a and 4a) were almost as active as the ester counterparts (2a and 2b), and also that the compounds having only the oxyamide acyl chain(s) but not the carboxylic group (3b and 4b) were just a little bit less active than the N-oxyamide glucuronides 3a and 4a, suggesting a role of the oxyamide function in maintaining the anti-proliferative activity that conversely was significantly reduced for compound 5 lacking both the groups. An ELISA assay showed an inhibitory effect of all the new compounds against Akt, especially for diacyl derivatives, suggesting that this crucial kinase could be their target as already demonstrated for the ester-based anionic glycoglycerolipid.6 In conclusion, in this paper, an efficient way for the oxidation of primary hydroxyl in the presence of N-oxyamide was demonstrated in the case of a glycoglycerolipid scaffold, affording in this way new anionic N-oxyamide glycoglycerolipids. The biological data showed that also the non-anionic N-oxyamide glycoglycerolipids were able to inhibit the growth of the human ovarian carcinoma cells IGROV-1 under the experimental conditions used, confirming their already explored potentiality and applications.12,13,47 In this regard, the analysis of the antiproliferative activity of compound 4a suggests a certain degree of selectivity of this series, because the best activity was observed in the cells with increased activation of the Akt pathway (i.e., IGROV-1/Pt1) as compared to the corresponding cell line with a lower extent of activation (IGROV-1) and to a cell line lacking Akt activation (i.e. TOV112D).Experimental
Chemistry
1,3-Di-O-benzyl-2-O-β-D-glucuronopyranosyl-sn-glycerol (7). To a suspension of 1,3-di-O-benzyl-2-O-β-D-glucopyranosyl-sn-glycerol (6)18 (2.5 g, 5.7 mmol) in a 55
:45 mixture of CH3CN/0.67 M sodium phosphate buffer (56 mL, pH 6–7), TEMPO (0.266 g, 1.71 mmol) and 20% aqueous NaClO2 (7.6 mL, 16.8 mmol) were added. Then, 15% aqueous NaClO (0.7 mL, 1.14 mmol) was added dropwise at room temperature. The reaction mixture was stirred for 5 h, then 2.5 mL of NaClO2 solution and 1.8 mL of NaClO solution were further added and the reaction mixture was stirred overnight. Furthermore, 2 mL of NaClO2 solution and 0.4 mL of NaClO solution were added after 15 h. 0.5 M Na2S2O3 (25 mL) was added and the reaction mixture was concentrated under vacuum to reduce the volume. The reaction mixture was basified to pH 10 with 1 M NaOH (25 mL) and extracted with Et2O (3 × 20 mL) to remove TEMPO by-products. The aqueous phase was then acidified by dropping conc. HCl and extracted with CH2Cl2 (4 × 40 mL). The combined organic layers were dried (Na2SO4), filtered and the solvent was removed under vacuum. The crude product (2.3 g, 89%) obtained as a white foam was used for the next step without further purification. 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 3.46–3.67 (m, 5H, H-2′, H-1a, H-1b, H-3a and H-3b), 3.62 (dd, 1H, J3′,2′ = 8.7 Hz, J3′,4′ = 8.7 Hz, H-3′), 3.76 (dd, 1H, J4′,5′ = 8.7 Hz, H-4′), 3.88 (d, 1H, H-5′), 4.00 (m, 1H, H-2), 4.36–4.45 (m, 4H, 2CH2Ph), 4.52 (d, 1H, J1′,2′ = 7.0 Hz, H-1′), 7.14–7.27 (m, 10H, 2Ph). 13C-NMR (CDCl3): δ = 69.7 (C1 or C3), 69.9 (C3 or C1), 71.4 (C4′), 72.9 (C2′), 73.2 and 72.3 (2 CH2Ph), 74.3 (C5′), 75.5 (C3′), 77.5 (C2), 127.7, 127.8, 127.9, 128.4, 137.6 and 137.7 (12C, 2Ph), 171.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). HRMS (negative-ion mode): m/z 447.1654 [M − H]−, calcd for C23H28O9, m/z 448.1733 [M].
O). HRMS (negative-ion mode): m/z 447.1654 [M − H]−, calcd for C23H28O9, m/z 448.1733 [M].
1,3-Di-O-benzyl-2-O-β-D-glucuronopyranosyl-sn-glycerol tert-butylester (8). A solution of tert-butyl 2,2,2-trichloroacetamidate (0.320 mL, 1.72 mmol) in dry cyclohexane (5.5 mL) was added dropwise at 0 °C to a solution of 7 (0.7 g, 1.56 mmol) in dry CH2Cl2 (50 mL). The reaction mixture was stirred at room temperature. Further amounts of tert-butyl 2,2,2-trichloroacetamidate (0.087 mL, 0.46 mmol) in dry cyclohexane (1.5 mL) were added dropwise at 0 °C after 5 and 23 h. Pure tert-butyl 2,2,2-trichloroacetamidate (0.100 mL, 0.53 mmol) was finally added after 27 h. After 48 h, the solvent was removed under vacuum and the crude product was purified by flash chromatography (CH2Cl2/CH3OH from 98
:2 to 95:5) affording 8 (0.257 g, 33%) as a colorless oil. [α]20D: −14° (CHCl3, c 1.0); 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 1.49 (s, 9H, 3CH3–tBu), 3.45 (dd, 1H J2′,1′ = 7.9 Hz, J2′,3′ = 9.0 Hz, H-2′), 3.58 (dd, 1H, J3′,4′ = 8.4 Hz, H-3′), 3.61–3.69 (m, 4H, H-1a, H-1b, H-3a and H-3b), 3.69 (d, 1H, J5′,4′ = 9.5 Hz, H-5′), 3.73 (dd, 1H, H-4′), 4.09 (m, 1H, H-2), 4.50 (d, 1H, H-1′), 4.50–4.57 (m, 4H, 2CH2Ph), 7.24–7.37 (m, 10H, 2Ph). 13C-NMR (CDCl3): δ = 28.0 (3CH3–tBu), 70.3 (C1 or C3), 70.6 (C3 or C1), 71.4 (C4′), 72.9 (C2′), 73.6 (2 CH2Ph), 74.7 (C5′), 75.4 (C3′), 77.7 (C2), 83.2 (C–tBu), 103.2 (C1′), 127.8, 127.9, 128.4, 128.5, 137.7 and 137.8(12C, 2Ph), 168.5 (CO). HRMS (positive-ion mode): m/z 527.2264 [M + Na]+, calcd for C27H36O9, m/z 504.2359 [M], m/z 527.2252 [M + Na]+.
1,3-Di-O-benzyl-2-O-(2′,3′,4′-tri-O-acetyl-β-D-glucuronopyranosyl)-sn-glycerol tert-butylester (9). To a solution of 8 (0.250 g, 0.49 mmol) in dry pyridine (1.87 mL), Ac2O (1.87 mL) was added dropwise at room temperature. The reaction mixture was stirred for 3 h until complete consumption of the starting material (TLC, CH2Cl2/MeOH 95
:5). The solvent was removed under vacuum and the crude 9 (0.252 g, 81%), obtained as a white waxy solid, was used for the next step without further purification. 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 1.43 (s, 9H, 3CH3, tBu), 1.93, 2.00 and 2.01 (3s, 9H, 3CH3CO), 3.55 (dd, 2H, J1(3)a,1(3)b = 10.5 Hz, J1(3)a,2 = 6.5 Hz, H-1a and H-3a), 3.68 (dd, 2H, J1(3)b,2 = 4.0 Hz, H-1b and H-3b), 3.88 (d, 1H, J5′,4′ = 9.5 Hz, H-5′), 4.06 (m, 1H, H-2), 4.46–4.54 (m, 4H, 2CH2Ph), 4.83 (d, 1H, J1′,2′ = 7.9 Hz, H-1′), 5.01 (dd, 1H, J2′,3′ = 9.5 Hz, H-2′), 5.19 (dd, 1H, J3′,4′ = 9.5 Hz, H-3′), 5.25 (dd, 1H, H-4′), 7.23–7.38 (m, 10H, 2Ph). 13C-NMR (CDCl3): δ = 20.6 (3CH3CO), 27.8 (3CH3–tBu), 69.5 (C4′), 70.1 (C1 or C3), 71.1 (C3 or C1), 71.4 (C2′), 72.6 (C3′), 73.3 (C5′), 73.5 (2 CH2Ph), 78.3 (C2), 82.9 (C–tBu), 100.7 (C1′), 127.5, 127.6, 127.7, 128.4, 138.1 and 138.2 (12C, 2Ph), 165.6 (CO), 169.0, 169.3, 170.3 (3CH3CO). HRMS (positive-ion mode): m/z 653.2570 [M + Na]+, calcd for C33H42O12, m/z 630.2676 [M], m/z 653.2568 [M + Na]+.
2-O-(2′,3′,4′-Tri-O-acetyl-β-D-glucuronopyranosyl)-sn-glycerol tert-butylester (10). To a solution of 9 (0.239 g, 0.38 mmol) in dry MeOH, 10% Pd/C (0.119 g) was added under an inert atmosphere. The flask was evacuated and backfilled with H2 three times and then stirred at room temperature until complete consumption of the starting material (TLC, hexane/acetate 50
:50, CH2Cl2/MeOH 95:5). The mixture was filtered on a pad of Celite and the solvent was removed under vacuum. The crude 10, obtained as a white waxy solid (0.175 g, quantitative yield), was used for the next step without further purification. 1H-NMR (CD3OD): δ = 1.45 (s, 9H, 3CH3–tBu), 1.97, 2.00 and 2.02 (3s, 9H, 3CH3CO), 3.53–3.66 (m, 4H, H-1a, H-1b, H-3a and H-3b), 3.73 (m, 1H, H-2), 4.16 (d, 1H, J5′,4′ = 10.0 Hz, H-5′), 4.88–4.94 (m, 2H, H-1′ and H-2′), 5.12 (dd, 1H, J4′,3′ = 10.0 Hz, H-4′), 5.27 (dd, 1H, J3′,2′ = 10.0 Hz, H-3′). 13C-NMR (CD3OD): δ = 20.5 and 20.6 (3CH3CO), 28.1 (3CH3–tBu), 62.6 (C1 or C3), 63.1 (C3 or C1), 70.8 (C4′), 72.8 (C2′), 73.8 (C5′), 73.9 (C3′), 83.8 (C2), 84.4 (C–tBu), 101.8 (C1′), 167.9 (CO), 170.8, 171.3, 171.5 (3CH3CO). HRMS (positive-ion mode): m/z 473.1631 [M + Na]+, calcd for C19H30O12, m/z 450.1737 [M], m/z 473.1629 [M + Na]+.
1,3-Di-O-phthalimido-2-O-(2′,3′,4′-tri-O-acetyl-β-D-glucuronopyranosyl)-sn-glycerol tert-butyl ester (11). To a solution of 10 (0.171 g, 0.38 mmol) in a mixture of dry CH2Cl2 (1.5 mL) and dry toluene (3 mL), PPh3 (0.398 g, 1.52 mmol) was added at room temperature. The reaction mixture was cooled to 0 °C before DIAD (0.307 g, 1.52 mmol) and N-hydroxyphthalimide (0.247 g, 1.52 mmol) were added. The reaction mixture was slowly warmed to room temperature and then stirred at room temperature 2 h until complete consumption of the starting material (TLC, CH2Cl2/MeOH 95
:5). The solvent was removed under vacuum and the crude product was purified twice by flash chromatography (petroleum ether/AcOEt, from 40:60 to 30:70), affording 11 as a white/yellow waxy solid (0.240 g, 85%). [α]20D: +24.2 (CHCl3, c 1.0); 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 1.40 (s, 9H, 3CH3, tBu), 1.98, 2.00 and 2.09 (3s, 9H, 3CH3CO), 3.99 (d, 1H, J5′,4′ = 9.9 Hz, H-5′), 4.29 (dd, 1H, J1(3)a,1(3)b = 11.1 Hz, J1(3)a,2 = 7.0 Hz, H-1a or H-3a), 4.48 (dd, 1H, J1(3)b,2 = 4.0 Hz, H-1b or H-3b), 4.44 (m, 1H, H-2), 4.55 (dd, 1H, J3(1)a,3(1)b = 11.5 Hz, J3(1)a,2 = 7.4 Hz, H-3a or H-1a), 4.74 (dd, 1H, J3(1)b,2 = 2.5 Hz, H-3b or H-1b), 4.93 (dd, 1H, J2′,1′ = 7.9 Hz, J2′,3′ = 9.3 Hz, H-2′), 5.14 (d, 1H, H-1′), 5.17 (dd, 1H, J4′,3′ = 9.3 Hz, H-4′), 5.24 (dd, 1H, H-3′), 7.70–7.88 (m, 8H, 2Phth). 13C-NMR (CDCl3): δ = 20.6 (3CH3CO), 27.7 (3CH3, tBu), 69.4 (C4′), 70.9 (C2′), 72.5 (C3′), 73.1 (C5′), 75.1 (C2), 76.7 (C1 or C3), 79.0 (C3 or C1), 82.9 (C–tBu), 100.7 (C1′), 123.6, 128.8, 129.0, 134.5 (12C, 2Phth), 163.2 and 163.5 (4 CO Phth), 165.5 (CO), 169.1, 169.7, 170.1 (3CH3CO). HRMS (positive-ion mode): m/z 763.1965 [M + Na]+, calcd for C35H36N2O16, m/z 740.2065 [M], m/z 763.1957 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-(2′,3′,4′-tri-O-acetyl-β-D-glucuronopyranosyl)-sn-glycerol tert-butylester (12). To a suspension of 11 (0.235 g, 0.32 mmol) in dry MeOH (5.7 mL), hydrazine hydrate (0.038 g, 0.76 mmol) was added dropwise at 0 °C. The reaction mixture was stirred for 2 h at 0 °C and then 3 h at room temperature until complete consumption of the starting material (TLC CH2Cl2/MeOH 95
:5). The solvent was removed under vacuum and the crude 1,3-di-O-amino-2-O-(2′,3′,4′-tri-O-acetyl-β-D-glucuronopyranosyl)-sn-glycerol tert-butylester was used for the next step without further purification. To a solution of the crude diamino derivative in dry CH2Cl2 (2.8 mL), a 10% (v/v) solution of pyridine in CH2Cl2 (1.4 mL) and a 15% (v/v) solution of decanoyl chloride in CH2Cl2 (1.16 mL, 0.86 mmol) was added at −10 °C. The reaction mixture was stirred at room temperature for 30 minutes. The reaction mixture was diluted with 30 mL of CH2Cl2 and washed with 1 M HCl (1 × 20 mL), water (1 × 20 mL), NaHCO3 saturated solution (1 × 20 mL) and finally with water again (2 × 20 mL). Aqueous layers were re-extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were dried (Na2SO4), filtered and the solvent removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 98:2 to 95:5), affording 12 as a white solid (0.106 g, 42%). [α]20D: −22.4 (CHCl3, c 1.0); 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 0.87 (m, 6H, 2 CH3), 1.19–1.35 (m, 24H, 12 CH2), 1.45 (s, 9H, 3CH3, –tBu), 1.56–1.72 (m, 4H, 2 CH2), 2.02, 2.04 and 2.06 (3s, 9H, 3CH3CO), 2.05–2.17 (m, 4H, 2 CH2), 3.86–4.13 (m, 4H, H-1a, H-1b, H-3a and H-3b), 4.03 (d, 1H, J5′,4′ = 9.6 Hz, H-5′), 4.27 (m, 1H, H-2), 4.89 (br d, 1H J1′,2′ = 7.4 Hz, H-1′), 5.01 (dd, 1H, J2′,3′ = 9.2 Hz, H-2′), 5.23 (dd, 1H, J4′,3′ = 9.2 Hz, H-4′), 5.26 (dd, 1H, H-3′), 9.18 and 9.82 (2br s, 2H, 2NH). 13C-NMR (CDCl3): δ = 14.0 (2 CH3), 20.5, 20.6 and 20.7 (3CH3CO), 22.6 (2 CH2), 25.3–25.6 (br, 2 CH2), 27.8 (3CH3–tBu), 29.0–29.5 (8 CH2), 31.8 (2 CH2), 33.0 (br, 2 CH2), 68.9 (C4′), 71.2 (C2′), 71.9 (br, C5′), 72.4 (C3′), 75.1 (br, C2), 75.2 (br, C1 or C3), 75.8 (br, C3 or C1), 84.1 (C–tBu), 101.1 (C1′), 166.9 (CO), 169.0, 169.8 (br), 170.0 (3 CH3CO), 171.6 (br, 2 NHCO). HRMS (positive-ion mode): m/z 811.4568 [M + Na]+, calcd for C39H68N2O14, m/z 788.4671 [M], m/z 811.4563 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-β-D-glucuronopyranosyl-sn-glycerol tert-butylester (13). To a suspension of 12 (0.086 g, 0.11 mmol) in dry MeOH (4.4 mL), 1 M MeONa (1.2 mL) was added dropwise at room temperature. The reaction mixture was stirred at room temperature until complete consumption of the starting material (TLC, CH2Cl2/MeOH 95
:5). The reaction mixture was neutralized with DOWEX 50 × 8 H+, filtered and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 95:5 to 90:10), affording compound 13 as a white solid (0.036 g, 50%). [α]20D: −23.7 (MeOH, c 1.0); 1H-NMR (CD3OD): δ = 0.89 (m, 6H, 2 CH3), 1.22–1.37 (m, 24H, 12 CH2), 1.50 (s, 9H, 3CH3–tBu), 1.56–1.66 (m, 4H, 2 CH2), 2.04–2.14 (m, 4H, 2 CH2), 3.26 (dd, 1H, J2′,1′ = 7.8 Hz, J2′,3′ = 9.1 Hz, H-2′), 3.38 (dd, 1H, J3′,4′ = 9.1 Hz, H-3′), 3.47 (dd, 1H, J4′,5′ = 9.7 Hz, H-4′), 3.77 (d, 1H, H-5′), 3.91–4.12 (m, 4H, H-1a, H-1b, H-3a and H-3b), 4.18 (m, 1H, H-2), 4.58 (d, 1H, H-1′). 13C-NMR (CD3OD): δ = 14.4 (2 CH3), 23.7 (2 CH2), 26.6 (2 CH2), 28.3 (3CH3–tBu), 30.1–30.7 (8 CH2), 33.0 (2 CH2), 33.7 (CH2), 33.8 (CH2), 73.2 (C4′), 74.8 (C2′), 76.7 (br, C1 or C3), 76.9 (C2), 77.0 (C5′), 77.3 (br, C3 or C1), 77.5 (C3′), 83.6 (C–tBu), 104.6 (C1′), 170.4 (CO), 172.9 and 173.0 (2 NHCO). HRMS (positive-ion mode): m/z 685.4250 [M + Na]+, calcd for C33H62N2O11, m/z 662.4354 [M], m/z 685.4246 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-β-D-glucuronopyranosyl-sn-glycerol (3a). Compound 13 (0.020 g, 0.030 mmol) was stirred with 15% (v/v) trifluoroacetic acid in CH2Cl2 (0.5 mL) at room temperature until complete consumption of the starting material. The solvent was removed under vacuum and the crude product was purified by flash chromatography (CH2Cl2/MeOH from 85
:15 to 80:20). The obtained pure compound was dissolved in CH2Cl2, stirred with DOWEX 50 × 8 H+ (previously washed only with deionized water) and filtered. The resin was washed with a 1:1 mixture of EtOAc/i-PrOH for a complete recovery of the desired compound. The solvent was removed under vacuum affording 3a as a white solid (0.012 g, 66%). [α]20D: −5.15° (MeOH, c 0.66); 1H-NMR (CD3OD): δ = 0.89 (m, 6H, 2 CH3), 1.21–1.38 (m, 24H, 12 CH2), 1.54–1.64 (m, 4H, 2 CH2), 2.04–2.13 (m, 4H, 2 CH2), 3.29 (dd, 1H, J2′,1′ = 7.8 Hz, J2′,3′ = 9.2 Hz, H-2′), 3.42 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 3.51 (dd, 1H, J4′,5′ = 9.7 Hz, H-4′), 3.88 (d, 1H, H-5′), 3.90–4.11 (m, 4H, H-1a, H-1b, H-3a and H-3b), 4.21 (m, 1H, H-2), 4.61 (d, 1H, H-1′). 13C-NMR (CD3OD): δ = 14.4 (2 CH3), 23.7 (2 CH2), 26.6 (2 CH2), 30.1–30.6 (8 CH2), 33.0 (2 CH2), 33.7 (2 CH2), 73.1 (C4′), 74.7 (C2′), 76.2 (br, C5′), 76.6 (C1 or C3), 76.8 (C2), 77.3 (C3 or C1), 77.5 (C3′), 104.5 (C1′), 172.6 (br, CO), 172.9 and 173.0 (2 NHCO). HRMS (negative-ion mode): m/z 605.3653 [M − H]−, calcd for C29H54N2O11, m/z 606.3728 [M].
1,3-Di-O-phthalimido-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (15). To a solution of 14
20,21 (2 g, 4.73 mmol) in a mixture of dry CH2Cl2 (15 mL) and dry toluene (30 mL), PPh3 (5.16 g, 19.67 mmol) was added at room temperature. The reaction mixture was cooled to 0 °C before DIAD (3.87 mL, 19.67 mmol) and N-hydroxyphthalimide (3.21 g, 19.67 mmol) were added. The reaction mixture was slowly warmed and then stirred at room temperature for 2 h, until complete consumption of the starting material (TLC, CH2Cl2/MeOH 95:5, EtOAc/petroleum ether 70:30). The reaction mixture was concentrated under vacuum. The resulting dark red viscous oil was dissolved in about 15 mL of Et2O and allowed to crystallize at 5 °C overnight. The crystalline solid was collected by filtration using a Büchner filter and further purified by flash chromatography (petroleum ether/EtOAc 30:70) affording 15 as a white solid (2.67 g, 79%). [α]20D: −22° (CHCl3, c 1.0). 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 1.98, 2.00, 2.05 and 2.08 (4s, 12H, 4CH3CO), 3.80 (ddd, 1H, J5′,4′ = 9.8 Hz, J5′,6a′ = 2.3 Hz, J5′,6b′ = 5.0 Hz, H-5′), 4.09 (dd, 1H, J6′a,6′b = 12.3 Hz, H-6′a), 4.18 (dd, 1H, H-6′b), 4.31 (dd, 1H, J1(3)a,1(3)b = 11.0 Hz, J1(3)a,2 = 6.6 Hz, H-1a or H-3a), 4.43 (m, 1H, H-2), 4.46 (dd, 1H, J1(3)b,2 = 4.1 Hz, H-1b or H-3b), 4.53 (dd, 1H, J3(1)a,3(1)b = 11.6 Hz, J3(1)a,2 = 7.4 Hz, H-3a or H-1a), 4.69 (dd, 1H, J3(1)b,2 = 2.8 Hz, H-3b or H-1b), 4.91 (dd, 1H, J2′,1′ = 8.0 Hz, J2′,3′ = 9.5 Hz, H-2′), 5.00 (dd, 1H, J4′,3′ = 9.5 Hz, H-4′), 5.12 (d, 1H, H-1′), 5.24 (dd, 1H, H-3′), 7.72–7.87 (m, 8H, 2Phth). 13C-NMR (CDCl3): δ = 20.6 (4CH3CO), 61.9 (C6′), 68.5 (C4′), 71.2 (C2′), 71.7 (C5′), 72.9 (C3′), 75.2 (C2), 76.8 (C1 or C3), 79.0 (C3 or C1), 100.8 (C1′), 123.6, 128.9, 129.0, 134.5 (12C, 2Phth), 163.2 and 163.5 (4 CO Phth), 169.4, 169.6, 170.1, 170.7 (4 CH3CO). HRMS (positive-ion mode): m/z 735.1646 [M + Na]+, calcd for C33H32N2O16, m/z 712.1752 [M], m/z 735.1644 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (16). To a suspension of 15 (1.69 g, 2.37 mmol) in a mixture of dry THF (16.9 mL) and dry MeOH (42.3 mL), a freshly prepared solution of hydrazine hydrate (100 μL mL−1) in dry MeOH (2.3 mL, 4.74 mmol) was added dropwise at 0 °C and the reaction mixture was stirred at the same temperature, under an inert atmosphere, for 5 minutes (TLC CH2Cl2/MeOH 95
:5). The reaction was diluted with CH2Cl2 (250 mL) and washed with water (150 mL). The aqueous layer was re-extracted with CH2Cl2 (3 × 30 mL), and the combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude 1,3-di-O-amino-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol was used for the next step without further purification. To a solution of the crude diamino derivative in dry CH2Cl2 (34 mL), a 10% (v/v) solution of pyridine in dry CH2Cl2 (11.5 mL, 14.22 mmol) and a 15% (v/v) solution of decanoyl chloride in dry CH2Cl2 (9.8 mL, 7.11 mmol) were added at −10 °C. The reaction mixture was stirred at the same temperature for 15 minutes, until complete consumption of the starting material (TLC CH2Cl2/MeOH 95:5). The reaction mixture was diluted with 100 mL of CH2Cl2 and washed with 1 M HCl (1 × 150 mL), and the aqueous layers were re-extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude product was washed with hexane (3 × 3 mL) to remove most of the decanoic acid and then purified by flash chromatography (EtOAc/petroleum ether +2% v/v MeOH from 70:30 to 80:20) affording 16 (1.32 g, 73%) as a white waxy solid. [α]20D: −9.9° (CHCl3, c 1.0). 1H-NMR (CDCl3) (residual CHCl3 fixed at 7.26 ppm): δ = 0.86 (m, 6H, 2 CH3), 1.17–1.35 (m, 24H, 12 CH2), 1.56–1.66 (m, 4H, 2 CH2), 1.99, 2.02, 2.04 and 2.09 (4s, 12H, 4CH3CO), 2.07–2.14 (m, 4H, 2 CH2), 3.74 (br ddd, 1H, J5′,4′ = 9.7 Hz, J5′,6′a = 4.2 Hz, J5′,6′b < 1.0 Hz, H-5′), 3.93–4.13 (m, 5H, H-1a, H-1b, H-3a, H-3b and H-2), 4.17 (dd, 1H, J6′a,6′b = 12.4 Hz, H-6′a), 4.31 (brdd, 1H, H-6′b), 4.77 (br d, J1′,2′ = 7.9 Hz, H-1′), 4.94 (dd, 1H, J2′,3′ = 9.5 Hz, H-2′), 5.06 (dd, 1H, J4′,3′ = 9.5 Hz, H-4′), 5.21 (dd, 1H, H-3′), 9.58 and 10.10 (2br s, 2H, 2NH). 13C-NMR (CDCl3): δ = 14.1 (2 CH3), 20.6, 20.7 and 20.8 (4CH3CO), 22.7 (2 CH2), 25.4 (br, 2 CH2), 29.2–29.7 (8 CH2), 31.9 (2 CH2), 33.0 (br, 2 CH2), 61.5 (C6′), 68.3 (C4′), 71.4 (C2′), 72.0 (C5′), 72.5 (C3′), 75.6–75.8 (br, C1, C2 and C3), 100.6 (C1′), 169.5, 169.9, 170.1 and 171.1 (4 CH3CO), 171.3 (br, 2 NHCO). HRMS (positive-ion mode): m/z 783.4253 [M + Na]+, calcd for C37H64N2O14, m/z 760.4358 [M], m/z 783.4250 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-β-D-glucopyranosyl-sn-glycerol (3b). To a suspension of 16 (0.66 g, 0.87 mmol) in dry MeOH (30 mL), a freshly prepared solution of sodium methoxide 0.075 M (7.0 mL, 0.52 mmol) was added dropwise at room temperature. The reaction mixture was stirred for 4 hours, until complete consumption of the starting material (TLC CH2Cl2/MeOH 90
:10). The reaction mixture was acidified with DOWEX 50 × 8 H+, filtered and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 90:10 to 85:15) affording compound 3b (0.417 g, 81%) as a white waxy solid. [α]20D: −35.3° (CHCl3, c 1.0). 1H-NMR (CDCl3: CD3OD 70:30): δ = 0.83 (m, 6H, 2 CH3), 1.14–1.32 (m, 24H, 12 CH2), 1.50–1.60 (m, 4H, 2 CH2), 1.99–2.07 (m, 4H, 2 CH2), 3.23–3.34 (m, 3H, H-2′, H-4′, H-5′), 3.38 (dd, 1H, J3′,4′ = 8.5 Hz, J3′,2′ = 8.5 Hz, H-3′), 3.63 (dd, 1H, J6′a,6′b = 11.7 Hz, J6′a,5′ = 5.9 Hz, H-6′a), 3.84–4.06 (m, 5H, H-1a, H-1b, H-3a, H-3b and H-6′b), 4.11 (m, 1H, H-2), 4.43 (d, 1H, J1′,2′ = 7.6 Hz, H-1′). 13C-NMR (CDCl3: CD3OD 70:30): δ = 14.2 (2 CH3), 22.9 (2 CH2), 25.8 (2 CH2), 29.3–30.0 (8 CH2), 32.1 (2 CH2), 33.1 (CH2), 33.2 (CH2), 62.1 (C6′), 70.7 (C4′), 73.9 (C2′), 75.7 (C1 or C3), 76.2 (C2), 76.4 (C3 or C1), 76.8 (C5′), 76.9 (C3′), 103.6 (C1′), 171.9 and 172.3 (2 NHCO). HRMS (positive-ion mode): m/z 615.3830 [M + Na]+, calcd for C29H56N2O10, m/z 592.3935 [M], m/z 615.3827 [M + Na]+.
1,3-Di-O-decanoylamino-2-O-β-D-glucuronopyranosyl-sn-glycerol (3a). In an open reaction vial, to a suspension of 3b (0.05 g, 0.084 mmol) in 20 mM acetate buffer (4 mL, pH 4.5), TEMPO (0.010 g, 0.064 mmol) and Trametes versicolor laccase (0.022 g, 11 U) were added at room temperature. The reaction mixture was stirred for 10 days in open air at 28–30 °C until complete consumption of the starting material (TLC, CH2Cl2/MeOH 85
:15). A further amount of TEMPO (0.010 g) and laccase (0.022 g) were added after 3 and 8 days of the reaction. The reaction mixture was diluted with water (10 mL), acidified with 0.1 M HCl up to pH 2, and extracted with diethyl ether (4 × 10 mL) and CH2Cl2 (1 × 10 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 90:10 to 80:20). The collected fractions were treated with DOWEX 50 × 8 H+, filtered and the solvent was removed under vacuum affording compound 3a (0.025 g, 49%).
3-O-Decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (17a). To a solution of 14 (1.04 g, 2.46 mmol) in dry THF (27 mL), trifluoroethyl decanoate36 (1.88 g, 0.71 mmol) and Candida antarctica lipase (3.0 g, 0.0005 PLU) were added at room temperature. The reaction mixture was stirred at 45 °C for 24 hours, until complete consumption of the starting material (TLC EtOAc/petroleum ether 70
:30). The enzyme was removed by filtration using a Büchner filter, and the solvent was removed under vacuum. The crude product, containing 10% of the 1-O isomer (by NMR), was purified by flash chromatography (petroleum ether/EtOAc 50:50) affording pure 17a (1.08 g, 76%) as a white waxy solid. [α]20D: −4.4° (CHCl3, c 1.0). 1H-NMR (CDCl3): δ = 0.88 (t, J = 6.8 Hz, 3H, CH3), 1.24–1.32 (m, 12H, 6 CH2), 1.57–1.64 (m, 2H, CH2), 2.00, 2.02, 2.05 and 2.08 (4s, 12H, 4CH3CO), 2.31 (t, J = 7.7 Hz, 2H, CH2), 3.58–3.69 (m, 2H, H-1a, H-1b), 3.72 (ddd, J5′,4′ = 10.0, J5′,6′b = 5.3 Hz, J5′,6′a = 2.4 Hz, 1H, H-5′), 3.87–3.94 (m, 1H, H-2), 4.06–4.15 (m, 2H, H-3a, H-6′a), 4.24 (dd, J6′a,6′b = 12.3 Hz, 1H, H-6′b), 4.30 (dd, J = J3b,3a = 11.8 Hz, J3b,2 = 4.9 Hz, 1H, H-3b), 4.69 (d, J1′,2′ = 8.0 Hz, 1H, H-1′), 5.00 (dd, J2′,3′ = 9.6 Hz, 1H, H-2′), 5.06 (dd, J4′,3′ = 9.4 Hz, 1H, H-4′), 5.21 (dd, 1H, H-3′). 13C-NMR (CDCl3): δ = 14.1 (CH3), 20.5–20.6 (4 CH3CO), 22.6 (CH2), 24.8 (CH2), 29.1–29.4 (4 CH2), 31.8 (CH2), 34.1 (CH2), 62.0 (C6′), 62.2 (C1), 62.8 (C3), 68.3 (C4′), 71.6 (C2′), 72.0 (C5′), 72.6 (C3′), 79.4 (C2), 100.9 (C1′), 169.4, 169.6, 170.2 and 170.6 (4 CH3CO), 173.6 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 599.2689 [M + Na]+, calcd for C27H44O13, m/z 576.2782 [M], m/z 599.2674 [M + Na]+.
1-O-Acetyl-3-O-decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (17b). An enriched chromatographic fraction of 17a (0.011 g, 0.02 mmol) containing about 23% (by NMR) of the corresponding diastereoisomer was acetylated by acetic anhydride (0.004 mL, 0.04 mmol) and pyridine (0.3 mL) treatment. After 12 hours, the solvent was removed under vacuum and the crude product was subjected to 1H-NMR analysis in deuterated chloroform. Here, we report the resonances of the anomeric protons of the reaction mixture enriched in 17b. 1H-NMR (CDCl3): δ = 4.64 ppm 17b (about 77%), 4.63 ppm (about 23%). In the same way, pure 3-O-decanoyl-2-O-β-D-glucopyranosyl-sn-glycerol (possessing 2R configuration) prepared according to ref. 18 (0.0085 g, 0.02 mmol), was fully acetylated by acetic anhydride (0.004 mL, 0.04 mmol) and pyridine (0.3 mL) treatment, affording, after usual work-up, 17b. [α]20D: −10.0° (CHCl3, c 1.0). 1H-NMR (CDCl3): δ = 0.87 (t, J = 6.8 Hz, 3H, CH3), 1.24–1.32 (m, 12H, 6 CH2), 1.56–1.64 (m, 2H, CH2), 2.00, 2.02, 2.03, 2.07 and 2.08 (5s, 15H, 5CH3CO), 2.30 (t, J = 7.6 Hz, 2H, CH2), 3.70 (ddd, J5′,4′ = 10.1, J5′,6′b = 5.2 Hz, J5′,6′a = 2.4 Hz, 1H, H-5′), 4.06 (m, 1H, H-2), 4.08–4.20 (m, 4H, H-1a, H-1b, H-3a, H-6′a), 4.20–4.29 (m, 2H, H-3b, H-6′b), 4.64 (d, J1′,2′ = 8.0 Hz, 1H, H-1′), 4.97 (dd, J2′,3′ = 9.6 Hz, 1H, H-2′), 5.06 (dd, J4′,3′ = 9.7 Hz, 1H, H-4′), 5.19 (dd, 1H, H-3′). 13C-NMR (CDCl3): δ = 14.1 (CH3), 20.5–20.7 (5 CH3C
O), 22.6 (CH2), 24.8 (CH2), 29.1–29.4 (4 CH2), 31.8 (CH2), 34.0 (CH2), 61.9 (C6′), 63.0 (C3), 63.4 (C1), 68.3 (C4′), 71.3 (C2′), 71.9 (C5′), 72.7 (C3′), 75.7 (C2), 100.8 (C1′), 169.1, 169.4, 170.2, 170.5 and 170.6 (5 CH3CO), 173.3 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 641.2795 [M + Na]+, calcd for C29H46O14, m/z 618.2888 [M], m/z 641.2780 [M + Na]+. The comparison of the two spectra showed that the resonances of the main isomer from 17a acetylation were superimposable to that of pure 17b, confirming in this way the 2R configuration of 17a (Fig. S1†).
1-O-Phthalimido-3-O-decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (18). To a solution of 17a (0.925 g, 1.60 mmol) in a mixture of dry CH2Cl2 (7 mL) and dry toluene (14 mL), PPh3 (0.881 g, 3.36 mmol) was added at room temperature. The reaction mixture was cooled to 0 °C before DIAD (0.662 mL, 3.36 mmol) and N-hydroxyphthalimide (0.548 g, 3.36 mmol) were added. The reaction mixture was slowly warmed and then stirred at room temperature for 1 hour, until complete consumption of the starting material (TLC, petroleum ether/EtOAc 50
:50). The solvent was removed under vacuum, and the crude product was purified by flash chromatography (petroleum ether/EtOAc/toluene 50:50:10) affording 18 as a white waxy solid (1.06 g, 92%). [α]20D: +4.0° (CHCl3, c 1.0). 1H-NMR (CDCl3): δ = 0.87 (t, J = 6.9 Hz, 3H, CH3), 1.18–1.32 (m, 12H, 6 CH2), 1.53–1.60 (m, 2H, CH2), 2.00, 2.02, 2.08 and 2.09 (4s, 12H, 4CH3CO), 2.29 (brt, 2H, CH2), 3.79 (ddd, J5′,4′ = 10.0, J5′,6′b = 4.9 Hz, J5′,6′a = 2.4 Hz, 1H, H-5′), 4.11–4.21 (m, 2H, H-3a, H-6′a), 4.21–4.36 (m, 5H, H-1a,b, H-3b, H-2, H-6′b), 5.00 (d, J1′,2′ = 8.0 Hz, 1H, H-1′), 5.04 (dd, J2′,3′ = 9.2 Hz, 1H, H-2′), 5.08 (dd, J4′,3′ = 9.3 Hz, 1H, H-4′), 5.25 (dd, 1H, H-3′), 7.74–7.81 (m, 2H, Phth), 7.81–7.88 (m, 2H, Phth). 13C-NMR (CDCl3): δ = 14.1 (CH3), 20.6–20.7 (4 CH3CO), 22.6 (CH2), 24.8 (CH2), 29.1 (CH2), 29.2 (2 CH2), 29.4 (CH2), 31.8 (CH2), 34.0 (CH2), 62.0 (C6′), 63.1 (C3), 68.5 (C4′), 71.2 (C2′), 71.8 (C5′), 72.9 (C3′), 75.2 (C2), 78.9 (C1), 100.8 (C1′), 123.7, 128.8 and 134.7 (6C, Phth), 163.3 (2 CO Phth), 169.4, 169.7, 170.2 and 170.7 (4 CH3CO), 173.3 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 744.2850 [M + Na]+, calcd for C35H47NO15, m/z 721.2946 [M], m/z 744.2838 [M + Na]+.
1-O-Octadecanoylamino-3-O-decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (19). To a suspension of 18 (1.04 g, 1.44 mmol) in a mixture of dry THF (25 mL) and dry MeOH (10 mL), hydrazine hydrate (0.070 mL, 1.44 mmol) was added dropwise at 0 °C and the reaction mixture was stirred at the same temperature, under an inert atmosphere, for 5 minutes (TLC petroleum ether/EtOAc 50
:50). The reaction was diluted with CH2Cl2 (100 mL) and washed with water (50 mL). The aqueous layer was re-extracted with CH2Cl2 (3 × 30 mL), the combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude 1-O-amino-3-O-decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol was used for the next step without further purification. To a solution of the crude amine derivative in dry CH2Cl2 (20 mL), a 10% (v/v) solution of pyridine in dry CH2Cl2 (3.5 mL, 4.32 mmol) and a 15% (v/v) solution of octadecanoyl chloride in dry CH2Cl2 (4.9 mL, 2.16 mmol) were added at −10 °C. The reaction mixture was stirred at the same temperature for 5 minutes, until complete consumption of the starting material (TLC petroleum ether/EtOAc 50:50). The reaction mixture was diluted with CH2Cl2 (20 mL), washed once with a mixture of 1 M HCl (50 mL) and brine (30 mL), and the aqueous layers were re-extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude product was purified by flash chromatography (petroleum ether/EtOAc/toluene 50:50:10), affording 19 (1.07 g, 87%) as a white waxy solid. [α]20D: −16.0° (CHCl3, c 0.6). 1H-NMR (CDCl3): δ = 0.86 (m, 6H, 2CH3), 1.07–1.42 (m, 40H, 20 CH2), 1.51–1.68 (m, 4H, CH2), 1.99, 2.01, 2.04 and 2.07 (4s, 12H, 4CH3CO), 2.02–2.11 (m, 2H, CH2), 2.28 (t, J = 7.6 Hz, 2H, CH2), 3.73 (ddd, J5′,4′ = 10.1, J5′,6′b = 5.3 Hz, J5′,6′a = 2.4 Hz, 1H, H-5′), 3.91 (dd, J1a,1b = 12.1, J1,2 = 7.3 Hz, 1H, H-1a), 4.02–4.15 (m, 4H, H-1b, H-2, H-3a, H-6′a), 4.18–4.30 (m, 2H, H-3b, H-6′b), 4.73 (d, J1′,2′ = 7.9 Hz, 1H, H-1′), 4.99 (dd, J2′,3′ = 9.7 Hz, 1H, H-2′), 5.04 (dd, J4′,3′ = 9.7 Hz, 1H, H-4′), 5.22 (dd, 1H, H-3′), 8.66 (brs, 1H, NH). 13C-NMR (CDCl3): δ = 14.1 (2 CH3), 20.5, 20.6 and 20.7 (4 CH3CO), 22.6 (2 CH2), 24.8 (CH2), 25.3 (br, CH2) 28.9–29.8 (16 CH2), 31.8 (CH2), 31.9 (CH2), 33.1 (br, CH2) 34.0 (CH2), 61.9 (C6′), 62.8 (C3), 68.3 (C4′), 71.7 (C2′), 71.9 (C5′), 72.4 (C3′), 76.8 (C2), 77.2 (C1), 100.9 (C1′), 169.4, 170.1 and 170.6 (4 CH3CO), 171.2 (br, NHCO), 173.3 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 880.5416 [M + Na]+, calcd for C45H79NO14, m/z 857.5501 [M], m/z 880.5393 [M + Na]+.
1-O-Octadecanoylamino-3-O-decanoyl-2-O-β-D-glucopyranosyl-sn-glycerol (20). To a solution of 19 (0.2 g, 0.23 mmol) in a mixture of THF (2 mL) and methanol (0.8 mL), water (1 mL) and freshly distilled Et3N (0.4 mL) were added at room temperature. The reaction mixture was stirred at 30 °C for 15 hours (TLC CH2Cl2/MeOH 95
:5). Then it was diluted with CH2Cl2 (20 mL), washed once with a mixture of 0.1 M HCl (10 mL) and brine (10 mL), and the aqueous layers were re-extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 90:10 to 80:20) affording 20 (0.061 g, 38%) as a white waxy solid. The unreacted starting material 19 (0.026 g, 13%) and deacylated product 4b (0.022 g, 18%) were also collected as white waxy solids after chromatographic separation. [α]20D: −16.0° (CHCl3, c 1.0). 1H-NMR (CDCl3:CD3OD 70:30): δ = 0.84 (m, 6H, 2CH3), 1.15–1.32 (m, 40H, 20 CH2), 1.51–1.62 (m, 4H, CH2), 2.03 (t, J = 7.5 Hz, 2H, CH2), 2.29 (t, J = 7.6 Hz, 2H, CH2), 3.23–3.34 (m, 3H, H-2′, H-4′, H-5′), 3.38 (dd, J3′,2′ = 8.7, J3′,4′ = 8.7 Hz, 1H, H-3′), 3.67 (dd, J6′a,6′b = 12.0 Hz, J6′a,5′ = 5.4 Hz, 1H, H-6′a), 3.83 (dd, J6′b,5′ = 2.5 Hz, 1H, H-6′b), 3.88 (dd, J1a,1b = 11.6 Hz, J1a,2 = 7.8 Hz, 1H, H-1a), 4.06 (dd, J1b,2 = 2.6 Hz, 1H, H-1b), 4.08–4.12 (m, 1H, H-2), 4.16 (dd, J3a,3b = 11.5 Hz, J3a,2 = 5.7 Hz, 1H, H-3a), 4.23 (dd, J3b,2 = 4.3 Hz, 1H, H-3b), 4.46 (d, J1′,2′ = 7.7 Hz, 1H, H-1′). 13C-NMR (CDCl3:CD3OD 70:30): δ = 14.2 (2 CH3), 23.0 (2 CH2), 25.2 (CH2), 25.9 (CH2), 29.5 (CH2), 29.6 (CH2), 29.6 (2 CH2), 29.7 (2 CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (2 CH2), 30.1 (6 CH2), 32.2 (CH2), 32.3 (CH2), 33.3 (br, CH2) 34.4 (CH2), 62.3 (C6′), 64.1 (C3), 70.7 (C4′), 74.1 (C2′), 76.8 (C5′), 77.0 (C1), 77.1 (C2), 77.1 (C3′), 104.0 (C1′), 171.9 (NHCO), 174.6 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 712.4977 [M + Na]+, calcd for C37H71NO10, m/z 689.5078 [M], m/z 712.4970 [M + Na]+.
1-O-Octadecanoylamino-3-O-decanoyl-2-O-β-D-glucuronopyranosyl-sn-glycerol (21). In an open reaction vial, to a suspension of 20 (0.03 g, 0.043 mmol) in 20 mM acetate buffer (1 mL, pH 4.5), TEMPO (0.005 g, 0.032 mmol) and Trametes versicolor laccase (0.011 g, 11 U) were added at room temperature. The reaction mixture was stirred for 5 days in open air at 28–30 °C, until complete consumption of the starting material (TLC CH2Cl2/MeOH 85
:15). A further amount of TEMPO (0.005 g) and laccase (0.011 g) was added after 2, 3 and 4 days of the reaction. The reaction mixture was diluted with water (10 mL), acidified with 0.1 M HCl up to pH 2, and extracted with diethyl ether (4 × 10 mL) and CH2Cl2 (1 × 10 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH from 90:10 to 80:20). The collected fractions were treated with DOWEX 50 × 8 H+, filtered and the solvent was removed under vacuum affording compound 21 (0.02 g, 66%) as a white waxy solid. [α]20D: −16°: (CHCl3, c 0.9). 1H-NMR (CD3OD): δ = 0.89 (m, 6H, 2 CH3), 1.23–1.38 (m, 40H, 20 CH2), 1.59 (m, 4H, CH2), 2.08 (t, J = 7.4 Hz, 2H, CH2), 2.32 (t, J = 7.5 Hz, 2H, CH2), 3.27 (dd, J2′,3′ = 9.4 Hz, J2′,1′ = 7.9 Hz, 1H, H-2′), 3.40 (dd, J3′,4′ = 9.1 Hz, 1H, H-3′), 3.52 (dd, J4′,5′ = 9.7 Hz, 1H, H-4′), 3.81 (d, 1H, H-5′), 3.92 (dd, J1a,1b = 11.4 Hz, J1a,2 = 6.5 Hz, 1H, H-1a), 4.03 (dd, J1b,2 = 3.3 Hz, 1H, H-1b), 4.13–4.20 (m, 2H, H-3a, H-2), 4.25 (dd, J3a,3b = 13.1 Hz, J3a,2 = 6.9 Hz, 1H, H-3b), 4.56 (d, 1H, H-1′). 13C-NMR (CD3OD): δ = 14.4 (2 CH3), 23.7 (2 CH2), 25.9 (CH2), 26.6 (CH2), 30.1 (CH2), 30.2 (CH2), 30.4 (4 CH2), 30.6 (2 CH2), 30.7–30.8 (8 CH2), 33.0 (2 CH2), 33.7 (CH2), 34.9 (CH2), 64.9 (C3), 73.1 (C4′), 74.8 (C2′), 76.7 (C5′), 77.3 (C1), 77.5 (C3′), 77.6 (C2), 104.8 (C1′), 172.5 (COOH), 172.9 (br, NHCO), 175.2 (CH2CO decanoyl). HRMS (negative-ion mode): m/z 702.4792 [M − H]−, calcd for C37H69NO11, m/z 703.4871 [M].
1-O-Octadecanoylamino-2-O-β-D-glucuronopyranosyl-sn-glycerol (4a). To solid 21 (0.015 g, 0.021 mmol), a freshly prepared solution of sodium methoxide 0.037 M (0.91 mL, 0.034 mmol) was directly added dropwise at room temperature. The reaction mixture was stirred for 3 h, until complete consumption of the starting material (TLC CH2Cl2/MeOH 80
:20). The reaction mixture was acidified with DOWEX 50 × 8 H+, filtered and the solvent was removed under vacuum. The crude product was purified by precipitation from diethyl ether affording compound 4a (0.011 g, 95%) as a white waxy solid. [α]20D: −32.0° (CH3OH, c 1.0). 1H-NMR (CD3OD): δ = 0.89 (t, J = 6.9 Hz, 3H, CH3), 1.25–1.35 (m, 28H, 14 CH2), 1.59 (m, 2H, CH2), 2.08 (t, J = 7.4 Hz, 2H, CH2), 3.28 (dd, J2′,3′ = 9.2 Hz, J2′,1′ = 7.8 Hz, 1H, H-2′), 3.41 (dd, J3′,4′ = 9.1 Hz, 1H, H-3′), 3.51 (dd, J4′,5′ = 9.8 Hz, 1H, H-4′), 3.65 (m, 2H, H-3a, H-3b), 3.83 (d, 1H, H-5′), 3.92 (dd, J1a,1b = 10.6 Hz, J1a,2 = 7.0 Hz, 1H, H-1a), 3.94–4.00 (m, 1H, H-2), 4.06 (dd, J1b,2 = 3.2 Hz, 1H, H-1b), 4.57 (d, 1H, H-1′). 13C-NMR (CD3OD): δ = 14.4 (CH3), 23.7 (CH2), 26.6 (CH2), 30.2 (CH2), 30.4 (CH2), 30.5 (CH2), 30.6 (2 CH2), 30.7–30.8 (7 CH2), 33.1 (CH2), 33.7 (CH2), 63.1 (C3), 73.1 (C4′), 74.7 (C2′), 76.4 (C5′), 77.4 (C3′), 77.5 (C1), 80.5 (C2), 104.5 (C1′), 172.7 (COOH), 172.9 (br, NHCO). HRMS (negative-ion mode): m/z 548.3432 [M − H]−, calcd for C27H51NO10, m/z 549.3513 [M].
1-O-Octadecanoylamino-2-O-β-D-glucopyranosyl-sn-glycerol (4b). Compound 4b (0.022 g, 18%) was obtained as a white waxy solid by-product from the synthesis of 20. [α]20D: −27.7° (CHCl3
:CH3OH = 9:1, c 0.45). 1H-NMR (CDCl3): δ = 0.83 (t, J = 6.9 Hz, 3H, CH3), 1.11–1.37 (m, 28H, 14 CH2), 1.56 (m, 2H, CH2), 2.02 (t, J = 7.6 Hz, 2H, CH2), 3.24–3.35 (m, 3H, H-2′, H-5′, H-4′), 3.39 (dd, J3′,2′ = 8.7 Hz, J3′,4′ = 8.7 Hz, 1H, H-3′), 3.58 (dd, J3a,2 = 5.6 Hz, J3a,3b = 12.0 Hz, 1H, H-3a), 3.63 (dd, J3b,2 = 4.1 Hz, 1H, H-3b), 3.66 (dd, J6′a,5′ = 5.4 Hz, J6′a,6′b = 12.0 Hz, 1H, H-6′a), 3.83 (dd, J6′b,5′ = 2.3 Hz, 1H, H-6′b), 3.87 (dd, 1H, J1a,1b = 11.0 Hz, J1a,2 = 7.3 Hz, H-1a), 3.93 (m, 1H, H-2), 4.01 (dd, J1b,2 = 2.9 Hz, 1H, H-1b), 4.43 (d, J1′,2′ = 7.8 Hz, 1H, H-1′). 13C-NMR (CDCl3): δ = 14.2 (CH3), 23.0 (CH2), 25.9 (CH2), 29.5 (CH2), 29.7 (2 CH2), 29.9 (2 CH2), 30.0 (7 CH2), 32.3 (CH2), 33.3 (CH2), 61.9 (C6′), 62.4 (C3), 70.5 (C4′), 74.1 (C2′), 76.7 (C1), 76.9 (C5′), 77.0 (C3′), 80.1 (C2), 103.3 (C1′), 172.0 (br, NHCO). HRMS (positive-ion mode): m/z 558.3621 [M + Na]+, calcd for C27H53NO9, m/z 535.3720 [M], m/z 558.3613 [M + Na]+.
6 was more efficiently prepared by acylation of 14 to the corresponding didecanoyl derivative and its subsequent mild deacetylation with hydrazine hydrate in aqueous ethanol at room temperature48 according to the following procedure.
To a solution of 14 (0.5 g, 1.18 mmol) in dry CH2Cl2 (10 mL), pyridine (0.57 mL, 7.08 mmol) and decanoyl chloride in dry CH2Cl2 (0.74 mL, 3.55 mmol) were added dropwise at −10 °C. The reaction mixture was stirred at the same temperature for 1 hour, until complete consumption of the starting material (TLC CH2Cl2/MeOH 95:5; petroleum ether/EtOAc 2:1). The reaction mixture was diluted with CH2Cl2 (30 mL), washed once with a mixture of 1 M HCl (30 mL), water (30 mL), a saturated solution of sodium bicarbonate (30 mL), and brine (30 mL). The organic layer was dried over Na2SO4, and the solvent was removed under vacuum. The crude product was subjected to flash chromatography (petroleum ether/EtOAc 20:10) affording pure 1,3-di-O-decanoyl-2-O-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-sn-glycerol (0.78 g, 90%) as a white waxy solid. [α]20D: −9.6° (CHCl3, c 1.0). 1H-NMR (CDCl3): δ = 0.88 (m, 6H, 2 CH3), 1.20–1.35 (m, 24H, 12 CH2), 1.60 (m, 4H, 2 CH2), 2.00, 2.02, 2.03 and 2.09 (4s, 12H, 4CH3CO), 2.31 (m, 4H, 2 CH2), 3.69 (ddd, J5′,4′ = 10.1, J5′,6′b = 5.1 Hz, J5′,6′a = 2.4 Hz, 1H, H-5′), 4.03–4.08 (m, 1H, H-2), 4.08–4.16 (m, 3H, H-1a,b or H-3a,b and H-6′a), 4.18 (dd, J1(3)a,2 = 11.6 Hz, J1(3)a,1(3)b = 4.0 1H, H-1a or H-3a), 4.21–4.28 (m, 2H, H-1b or H-3b and H-6′b), 4.64 (d, J1′,2′ = 8.0 Hz, 1H, H-1′), 4.98 (dd, J2′,3′ = 9.6 Hz, 1H, H-2′), 5.07 (dd, J4′′,3′ = 9.7 Hz, 1H, H-4′) 5.19 (dd, 1H, H-3′). 13C-NMR (CDCl3): δ = 14.1 (2 CH3), 20.6 and 20.7 (4 CH3CO), 22.7 (2 CH2), 24.8 (2 CH2), 29.2–29.4 (8 CH2), 31.8 (2 CH2), 34.0 (CH2), 34.1 (CH2), 61.9 (C6′), 63.0 (C1 or C3), 63.1 (C1 or C3), 68.3 (C4′), 71.2 (C2′), 71.9 (C5′), 72.7 (C3′), 75.7 (C2), 100.8 (C1′), 169.2, 169.4, 170.3, 170.7 (4 CH3CO), 173.4 (CH2CO decanoyl). HRMS (positive-ion mode): m/z 753.4041 [M + Na]+, calcd for C37H62O14, m/z 730.4140 [M], m/z 753.4032 [M + Na]+.
To a solution of the previously prepared didecanoyl derivative (0.1 g, 0.14 mmol) in 85% (v/v) aqueous ethanol (5 mL), hydrazine hydrate (0.027 mL, 0.55 mmol) was added at room temperature. The reaction mixture was stirred for 3 hours, until complete consumption of the starting material TLC (CH2Cl2/MeOH 95:5, petroleum ether/EtOAc 20:10). The reaction mixture was concentrated under vacuum, diluted with cold water (10 mL), and extracted with CH2Cl2 (6 × 10 mL). The combined organic layers were dried over Na2SO4 and the solvent was removed under vacuum. The crude product was purified by flash chromatography (CH2Cl2/MeOH 90:10) affording 1,3-di-O-decanoyl-2-O-β-D-glucopyranosyl-sn-glycerol (5) (0.058 g, 74%) as a white waxy solid having the same characteristics as reported in ref. 6.
:15). Afterwards, the complete disappearing of 5 but not of 3b was observed (see the ESI, Fig. S2†). Then the mixtures were freeze-dried and 600 μL of CDCl3 and MeOD (1:1) were added to the lyophilized residue of each sample. The suspension was filtered and the solution obtained was analysed by 1H-NMR (see the ESI, Fig. S3†). Whilst the spectrum of treated ester 5 showed no more signals related to the starting compound in the 3b spectrum, the characteristic resonances of this oxyamide were detectable again.
500 rpm for 30 min. The filtrates, collected in the filter collection tubes, were then acidified with a 0.1 M HCl solution to a slightly acidic pH, so that our compound, bearing a carboxylic group, could be found in its undissociated form for a clearer NMR analysis. The samples were then freeze-dried and prepared for NMR analysis. 500 μL of a 1:1 solution of CDCl3 and MeOD were added to each one of the collection tubes along with 5 μL of 0.14 M DMSO in CDCl3, used as an internal standard. The solutions obtained were then analysed by 1H-NMR (see the ESI, Fig. S4 and S5†).
Biology
Stock preparation. All compounds were dissolved in DMSO at 50 mM and directly added to the cell culture medium for cell sensitivity assays. For ELISA, the compounds were prepared as 10× solutions and then diluted in kinase buffer.
Cell culture and cell growth inhibition assay. The human ovarian carcinoma IGROV-1, IGROV/Pt1,15 and TOV112D (ATCC, CRL 11731) cell lines were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37 °C under a 5% CO2 atmosphere. For cell sensitivity assays, all cell lines (10
000 cells per cm2) were plated in complete medium using 12-well plates. Twenty-four hours later, the cells were exposed to the solvent (DMSO) or different concentrations of the compounds for 24 h either in complete medium or serum-free medium. After treatment, the cells were washed with saline and complete medium was added and for 48 h. The cells were then harvested using trypsin and counted using a Coulter Counter (Z1, Beckman Coulter). The percentages of inhibition in drug-treated versus solvent-treated samples are reported in dose–response curves. IC50 represents the drug concentration inhibiting growth by 50%. Each experiment was performed 3 times or with triplicate samples.
Author contributions
MZ: conceptualization, methodology, investigation, writing – original draft, and writing – review and editing. GO: conceptualization and investigation. MQ: investigation, writing – original draft, and writing – review and editing. LM: investigation. CC: investigation and visualization. NC: investigation. PP: conceptualization, resources, writing – original draft, writing – review and editing, and funding acquisition. DC: conceptualization, methodology, investigation, resources, writing – original draft, writing – review and editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Industriale Chimica srl for funding the fellowship to M. Z. and M. Q. in memoriam of the founder Dr Fulvio Benigni as well as Associazione Italiana per la Ricerca sul Cancro (grant number 24725). The authors also thank Prof. Fiamma Ronchetti and Dr Paola Rota for the helpful discussion.References
- G. M. Nitulescu, D. Margina, P. Juzenas, Q. Peng, O. T. Olaru, E. Saloustros, C. Fenga, D. A. Spandidos, M. Libra and A. M. Tsatsakis, Int. J. Oncol., 2016, 48, 869 CrossRef CAS PubMed.
- G. Cassinelli, V. Zuco, L. Gatti, C. Lanzi, N. Zaffaroni, D. Colombo and P. Perego, Curr. Med. Chem., 2013, 20, 1923 CrossRef CAS PubMed.
- I. Hers, E. E. Vincent and J. M. Tavaré, Cell. Signalling, 2011, 23, 1515 CrossRef CAS PubMed.
- M. Kaneda, K. Mizutani, Y. Takahashi, G. Kurono and Y. Nishikawa, Tetrahedron Lett., 1974, 15, 3937 CrossRef.
- B. Costa, M. Dangate, M. Vetro, G. Donvito, L. Gabrielli, L. Amigoni, G. Cassinelli, C. Lanzi, M. Ceriani, L. De Gioia, G. Filippi, L. Cipolla, N. Zaffaroni, P. Perego and D. Colombo, Bioorg. Med. Chem., 2016, 24, 3396 CrossRef CAS PubMed.
- M. Vetro, B. Costa, G. Donvito, N. Arrighetti, L. Cipolla, P. Perego, F. Compostella, F. Ronchetti and D. Colombo, Org. Biomol. Chem., 2015, 13, 1091 RSC.
- C. Benning, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1998, 49, 53 CrossRef CAS PubMed.
- Y. Okazaki, H. Otsuki, T. Narisawa, M. Kobayashi, S. Sawai, Y. Kamide, M. Kusano, T. Aoki, M. Y. Hirai and K. Saito, Nat. Commun., 2013, 4, 1510 CrossRef PubMed.
- S. Jean and A. A. Kiger, J. Cell Sci., 2014, 127, 923 CrossRef CAS PubMed.
- Y. Gong, S. Peyrat, H. Sun and J. Xie, Tetrahedron, 2011, 67, 7114 CrossRef CAS.
- Z. Song, X. P. He, G. R. Chen and J. Xie, Synthesis, 2011, 17, 2761 Search PubMed.
- N. Chen and J. Xie, J. Org. Chem., 2014, 79, 10716 CrossRef CAS PubMed.
- N. Chen and J. Xie, Org. Biomol. Chem., 2016, 14, 1102 RSC.
- F. Chen, B. Ma, Z. C. Yang, G. Lin and D. Yang, Amino Acids, 2012, 43, 499 CrossRef CAS PubMed.
- P. Perego, S. Romanelli, N. Carenini, I. Magnani, R. Leone, A. Bonetti, A. Paolicchi and F. Zunino, Ann. Oncol., 1998, 9, 423 CrossRef CAS PubMed.
- E. G. Bowen and D. J. Wardrop, Org. Lett., 2010, 12, 5330 CrossRef CAS PubMed.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739 RSC.
- D. Colombo, F. Compostella, F. Ronchetti, A. Scala, L. Toma, H. Tokuda and H. Nishino, Bioorg. Med. Chem., 1999, 7, 1867 CrossRef CAS PubMed.
- J. Thierry, C. Yue and P. Potier, Tetrahedron Lett., 1998, 39, 1557 CrossRef CAS.
- D. Colombo, F. Ronchetti, A. Scala, I. M. Taino, F. Marinone Albini and L. Toma, Tetrahedron: Asymmetry, 1994, 5, 1377 CrossRef CAS.
- F. Marinone Albini, C. Murelli, G. Patritti and M. Rovati, Synth. Commun., 1994, 24, 1651 CrossRef.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564 CrossRef CAS.
- M. Kawase, T. Kitamura and Y. Kikugawa, J. Org. Chem., 1989, 54, 3394 CrossRef CAS.
- X. H. Wei, Z. H. Li, L. B. Zhao, P. Zhang, H. C. Zhou and Y. B. Wang, RSC Adv., 2019, 9, 32081 RSC.
- N. J. Davis and S. L. Flitsch, Tetrahedron Lett., 1993, 34, 1181 CrossRef CAS.
- A. Israeli, M. Patt, M. Oron, A. Samuni, R. Kohen and S. Goldstein, Free Radicals Biol. Med., 2005, 38, 317 CrossRef CAS PubMed.
- T. Miyazawa, T. Endo and M. Okawara, J. Org. Chem., 1985, 50, 5389 CrossRef CAS.
- J. M. Bobbitt, A. L. Bartelson, W. F. Bailey, T. A. Hamlin and C. B. Kelly, J. Org. Chem., 2014, 79, 1055 CrossRef CAS PubMed.
- N. Merbouh, J. M. Bobbitt and C. Bruckner, Tetrahedron Lett., 2001, 42, 8793 CrossRef CAS.
- J. M. Bobbitt, J. Org. Chem., 1998, 63, 9367 CrossRef CAS.
- N. Merbouh, J. M. Bobbitt and C. Brückner, J. Org. Chem., 2004, 69, 5116 CrossRef CAS PubMed.
- M. Yamaguchi, T. Takata and T. Endo, Tetrahedron Lett., 1988, 29, 5671 CrossRef CAS.
- M. Marzorati, B. Danieli, D. Haltrich and S. Riva, Green Chem., 2005, 7, 310 RSC.
- L. Baratto, A. Candido, M. Marzorati, F. Sagui, S. Riva and B. Danieli, J. Mol. Catal. B: Enzym., 2006, 39, 3 CrossRef CAS.
- K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448 RSC.
- D. Colombo, F. Ronchetti, A. Scala, I. M. Taino and L. Toma, Tetrahedron: Asymmetry, 1996, 7, 771 CrossRef CAS.
- L. Meier, G. C. Monteiro, R. A. M. Baldissera and M. M. Sá, J. Braz. Chem. Soc., 2010, 21, 859 CrossRef CAS.
- G. B. Reis, J. C. Rees, A. A. Ivanova, Z. Kuklenyik, N. M. Drew, J. L. Pirkle and J. R. Bar, J. Mass Spectrom. Adv. Clin. Lab, 2021, 22, 34 CrossRef CAS PubMed.
- J. Córdova, J. D. Ryan, B. B. Boonyaratanakornkit and D. S. Clark, Enzyme Microb. Technol., 2008, 42, 278 CrossRef.
- M. Koitka, J. Höchel, H. Gieschen and H. H. Borchert, J. Pharm. Biomed. Anal., 2010, 51, 664 CrossRef CAS PubMed.
- T. Bohnert and L. S. Gan, J. Pharm. Sci., 2013, 102, 2953 CrossRef CAS PubMed.
- D. Dimitrijevic, E. Fabian, D. Funk-Weyer and R. Landsiedel, Biochem. Biophys. Res. Commun., 2023, 651, 114 CrossRef CAS PubMed.
- C. Wang and N. S. Williams, J. Pharm. Biomed. Anal., 2013, 75, 112 CrossRef CAS PubMed.
- V. Benedetti, P. Perego, G. L. Beretta, E. Corna, S. Tinelli, S. C. Righetti, R. Leone, P. Apostoli, C. Lanzi and F. Zunino, Mol. Cancer Ther., 2008, 7, 679 CrossRef CAS PubMed.
- A. J. Hanrahan, N. Schultz, M. L. Westfal, R. A. Sakr, D. D. Giri, S. Scarperi, M. Janakiraman, N. Olvera, E. V. Stevens, Q. B. She, C. Aghajanian, T. A. King, E. De Stanchina, D. R. Spriggs, A. Heguy, B. S. Taylor, C. Sander, N. Rosen, D. A. Levine and D. B. Solit, Cancer Discovery, 2012, 2, 56 CrossRef CAS PubMed.
- L. Xiang, W. Yun-Dong and Y. Dan, Acc. Chem. Res., 2008, 41, 1428 CrossRef PubMed.
- N. Chen, Z. H. Yu, D. Zhou, X. L. Hu, Y. Zang, X. P. He, J. Li and J. Xie, Chem. Commun., 2016, 52, 2284 RSC.
- D. Pagano, A. Cutignano, E. Manzo, F. Tinto and A. Fontana, Carbohydr. Res., 2016, 424, 21 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all the compounds, Fig. S1–S5. See DOI: https://doi.org/10.1039/d3ob00891f |
| This journal is © The Royal Society of Chemistry 2023 |