
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvenient synthesis of N-alkyl-3,1-benzoxazin-2-ones from carbamate protected anthranil aldehydes and ketones via one-step alkylation/alkoxy rearrangement†
Guang
Tian‡
 ab,
Wei-Li
Jin‡
cb,
Chuanguang
Qin
*a and
Jie
Wang
*bcd
ab,
Wei-Li
Jin‡
cb,
Chuanguang
Qin
*a and
Jie
Wang
*bcd
aDepartment of Chemistry, Shanxi Key Laboratory of Polymer Science & Technology, MOE Key Laboratory of Supernormal Material Physics & Chemistry, School of Chemical & Chemical Engineering, Northwestern Polytechnical University, Xi'an 710129, China. E-mail: qinchg@nwpu.edu.cn
bState Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: jiewang@simm.ac.cn
cSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 28th June 2023
Abstract
A practical and step-economical protocol was developed to prepare N-alkyl-3,1-benzoxazin-2-one derivatives from anthranil aldehydes and ketones via one-step alkylation/alkoxy rearrangement, where three new chemical bonds and one ring were constructed in a single step. Control studies revealed a stepwise mechanism and that the alkoxy rearrangement was an intermolecular process.
3,1-Benzoxazin-2-one is a valuable structural motif found in both natural products and pharmaceuticals and exhibits various important biological activities.1 For example, the well-known efavirenz is a non-nucleoside reverse transcriptase inhibitor used to treat and prevent HIV/AIDS.2 L-371257 and its analogs are a class of compounds acting as oxytocin receptor antagonists.1c,3 Evodiamide A is a natural product isolated from the Chinese medicinal plant Evodia rutaecarpa (Juss.) and displays weak antibacterial activities (Scheme 1A).4
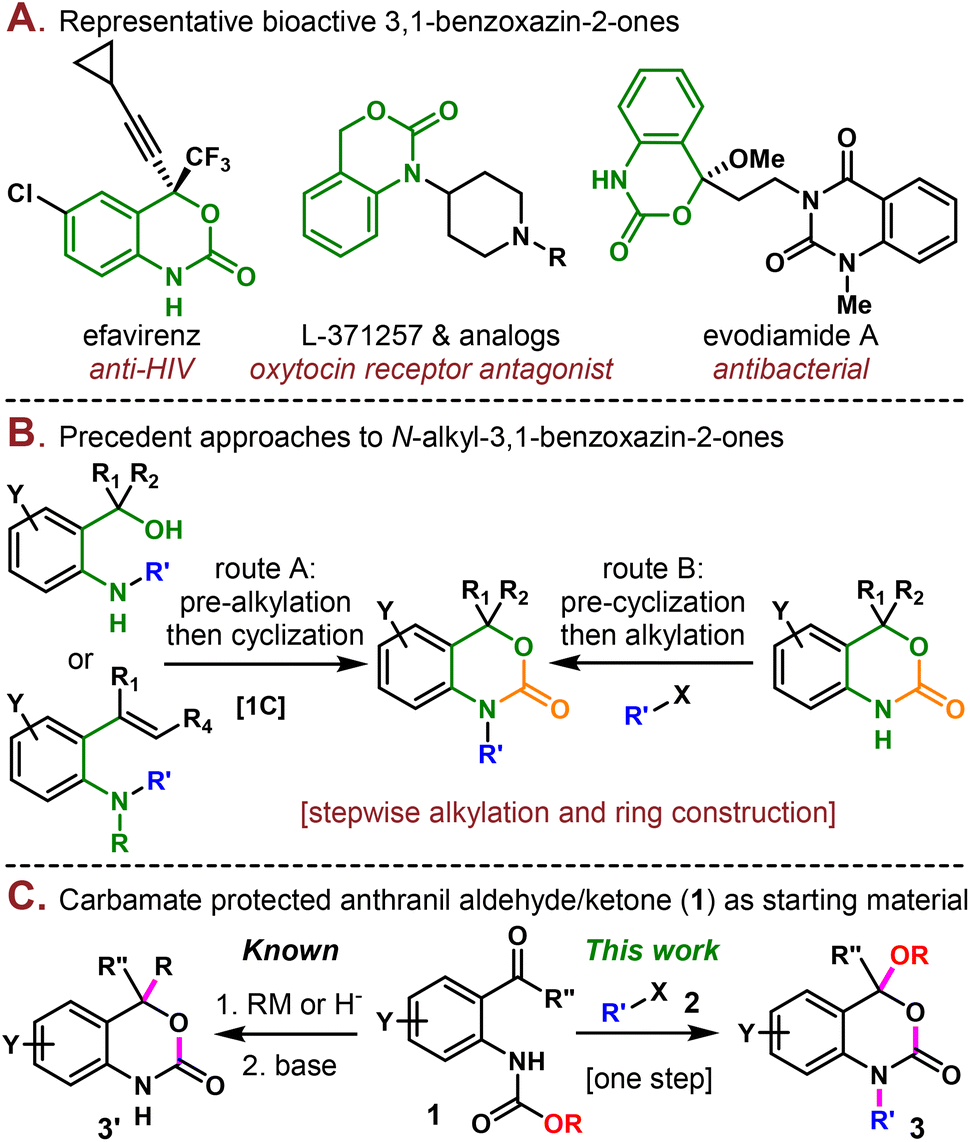 | ||
| Scheme 1 A) Biologically active compounds containing 3,1-benzoxazin-2-one motifs. (B) Previous approaches toward N-alkyl-3,1-benzoxazin-2-ones. (C) Synthesis of 3,1-benzoxazin-2-ones using the carbamate protected anthranil aldehyde/ketone as the starting material. | ||
Due to the significance of 3,1-benzoxazin-2-one in medicinal chemistry, a plethora of synthetic methodologies have been developed to construct this moiety.5 The N-alkylated 3,1-benzoxazin-2-one, in particular, is usually assembled using two different strategies (Scheme 1B). One of them relies on using o-aminoarylmethanol6 or o-aminostyrene7 as the starting material, which is first alkylated and then cyclized in the second step using a 1C carbonate equivalent, such as CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6b,c and CBr4.6a In the case of o-aminostyrene, additional steps are usually necessary to transform the olefin through electrophilic activation,7a epoxide addition,7b or boryl Michael addition.7c Alternatively, the 3,1-benzoxazin-2-one moiety could be first constructed by cyclization, followed by a second step of alkylation under basic conditions.8 Despite their synthetic validity, these routes typically involved multistep synthesis and required hazardous reagents such as Bu3SnH, phosgene, Me3Al, etc. Previous methods to construct the 3,1-benzoxazin-2-one (3′) framework starting from carbamate protected anthranil aldehydes/ketones (1) normally involved hydride reduction9 or organometallic addition10 to the aldehyde/ketone followed by base mediated cyclization, where the alkoxy part of the carbamate was wasted as a leaving group (Scheme 1C). Here, in this work, we present a step-economical methodology to prepare N-alkyl 3,1-benzoxazin-2-one derivatives (3) from 1via alkylation/alkoxy rearrangement in one step. In this reaction, the alkoxy group served not only as the leaving group but was also reincorporated into the final product. It is also noteworthy that three new carbon heteroatom bonds (one C–N and two C–O bonds) and one ring were constructed in a single step, which has rarely been reported in the synthesis of benzoxazinones.5h
6b,c and CBr4.6a In the case of o-aminostyrene, additional steps are usually necessary to transform the olefin through electrophilic activation,7a epoxide addition,7b or boryl Michael addition.7c Alternatively, the 3,1-benzoxazin-2-one moiety could be first constructed by cyclization, followed by a second step of alkylation under basic conditions.8 Despite their synthetic validity, these routes typically involved multistep synthesis and required hazardous reagents such as Bu3SnH, phosgene, Me3Al, etc. Previous methods to construct the 3,1-benzoxazin-2-one (3′) framework starting from carbamate protected anthranil aldehydes/ketones (1) normally involved hydride reduction9 or organometallic addition10 to the aldehyde/ketone followed by base mediated cyclization, where the alkoxy part of the carbamate was wasted as a leaving group (Scheme 1C). Here, in this work, we present a step-economical methodology to prepare N-alkyl 3,1-benzoxazin-2-one derivatives (3) from 1via alkylation/alkoxy rearrangement in one step. In this reaction, the alkoxy group served not only as the leaving group but was also reincorporated into the final product. It is also noteworthy that three new carbon heteroatom bonds (one C–N and two C–O bonds) and one ring were constructed in a single step, which has rarely been reported in the synthesis of benzoxazinones.5h
An attempt to install a prenyl group on benzyl (2-formylphenyl) carbamate (1a) using prenyl bromide (2a) under NaH/DMF conditions failed to obtain the desired product 4a even in the slightest amount (Table 1, entry 1). Instead, a significant amount (57%) of the deprotected alkylation product 5a was obtained together with a new compound bearing the same mass as 4a but missing the aldehyde functional group, whose structure was determined to be benzoxazinone 3a by NMR spectrometry and confirmed unambiguously through X-ray crystallographic analysis. We then optimized the reaction conditions using 1a and 2a as the model substrates. Firstly, regular strong bases such as tBuOK, LDA and LiHMDS were evaluated and no 3a could be detected, as indicated by crude NMR analysis (Table 1, entries 2–4). Solvent screening revealed that the reaction was messy in DMSO, while 1,4-dioxane, CH2Cl2, DCE and toluene were viable choices giving 3a in 27–47% yields (entries 5–9). Although a slightly lower yield was obtained in toluene compared to CH2Cl2 (41% vs. 47%), the reaction in toluene was cleaner, with essentially only two spots on TLC (1a and 3a, 67% conversion) and neither of the unwanted 4a or 5a was detected. Thus, further efforts were made to improve the reaction conversion while fixing toluene as the solvent. Although raising the temperature did improve the conversion, the reaction was messy and the yield dropped to 33% (entry 10). Noticing that the reaction was heterogeneous due to the low solubility of NaH in toluene, 1.0 equiv. of 15-crown-5 was used as the additive and the yield could be improved to 75% (entry 11). Lastly, the addition of 0.6 equiv. of (nBn)4NI to enhance the electrophilicity of the alkylation reagent could further increase the yield to 81%. Under the optimized conditions, an isolated yield of 80% was obtained on a 0.3 mmol scale (entry 12).
|
|
||||
|---|---|---|---|---|
| Entrya | Base | Solvent | Conversion | Yield (3a/4a/5a)b |
| a Reaction conditions: carbamate 1a (0.1 mmol), bromide 2a (1.5 equiv.), and base (3.0 equiv.) were stirred in 1.0 mL of solvent at 0 °C for 1 h and then room temperature for another 2 h. b Yields determined by crude 1H NMR using trimethoxybenzene as the internal standard. c The reaction was run at 50 °C. d Addition of 15-crown-5 (1.0 equiv.). e Addition of 15-crown-5 (1.0 equiv.) and (nBu)4NI (0.6 equiv.). f Isolated yield on a 0.3 mmol scale. nd = not detected. | ||||
| 1 | NaH | DMF | >95% | 42%/nd/57% |
| 2 | tBuOK | DMF | 80% | nd/5%/<5% |
| 3 | LDA | DMF | 45% | nd/11%/nd |
| 4 | LiHMDS | DMF | 45% | nd/11%/nd |
| 5 | NaH | DMSO | >95% | nd/nd/nd |
| 6 | NaH | 1,4-Dioxane | 67% | 27%/nd/10% |
| 7 | NaH | CH2Cl2 | >95% | 47%/5%/<5% |
| 8 | NaH | DCE | >95% | 33%/45%/5% |
| 9 | NaH | Toluene | 67% | 41%/nd/nd |
| 10c | NaH | Toluene | >95% | 33%/nd/nd |
| 11d | NaH | Toluene | >95% | 75%/nd/12% |
| 12e | NaH | Toluene | >95% | 81% (80%)f/nd/11% |
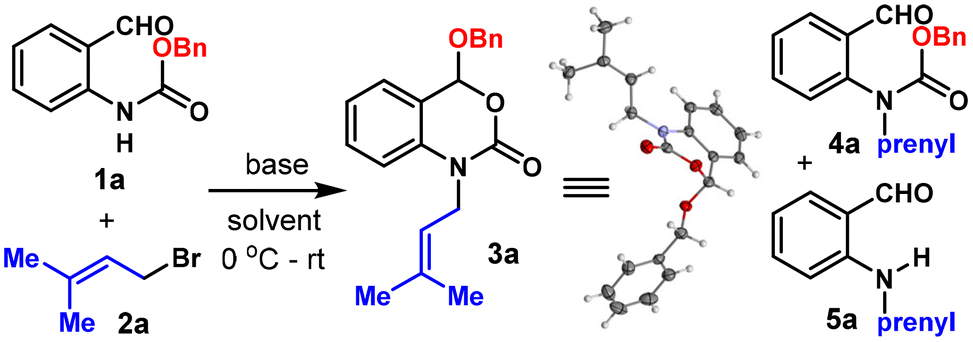
With the optimized conditions in hand, we set out to explore the scope of this reaction. Firstly, different carbamate protecting groups were evaluated (Scheme 2A). Other than Cbz, common carbamate protecting groups bearing the primary alkoxy group such as CO2Et, Troc, Alloc and p-nitrobenzyloxycarbonyl were well tolerated and afforded the corresponding benzoxazinone products 3b–3e in moderate to good yields (57%–68%). The Boc protecting group bearing a bulky tertiary tert-butoxy group didn't work well under the standard conditions, probably due to the relatively high stability of Boc under basic conditions.11 Nonetheless, under slightly modified conditions (50 °C, omit the addition of 15-crown-5 and TBAI), a useful yield of 48% could also be obtained (3f). Moreover, aryloxyl containing carbamate could be well tolerated, furnishing the corresponding product (3g) in 61% yield.
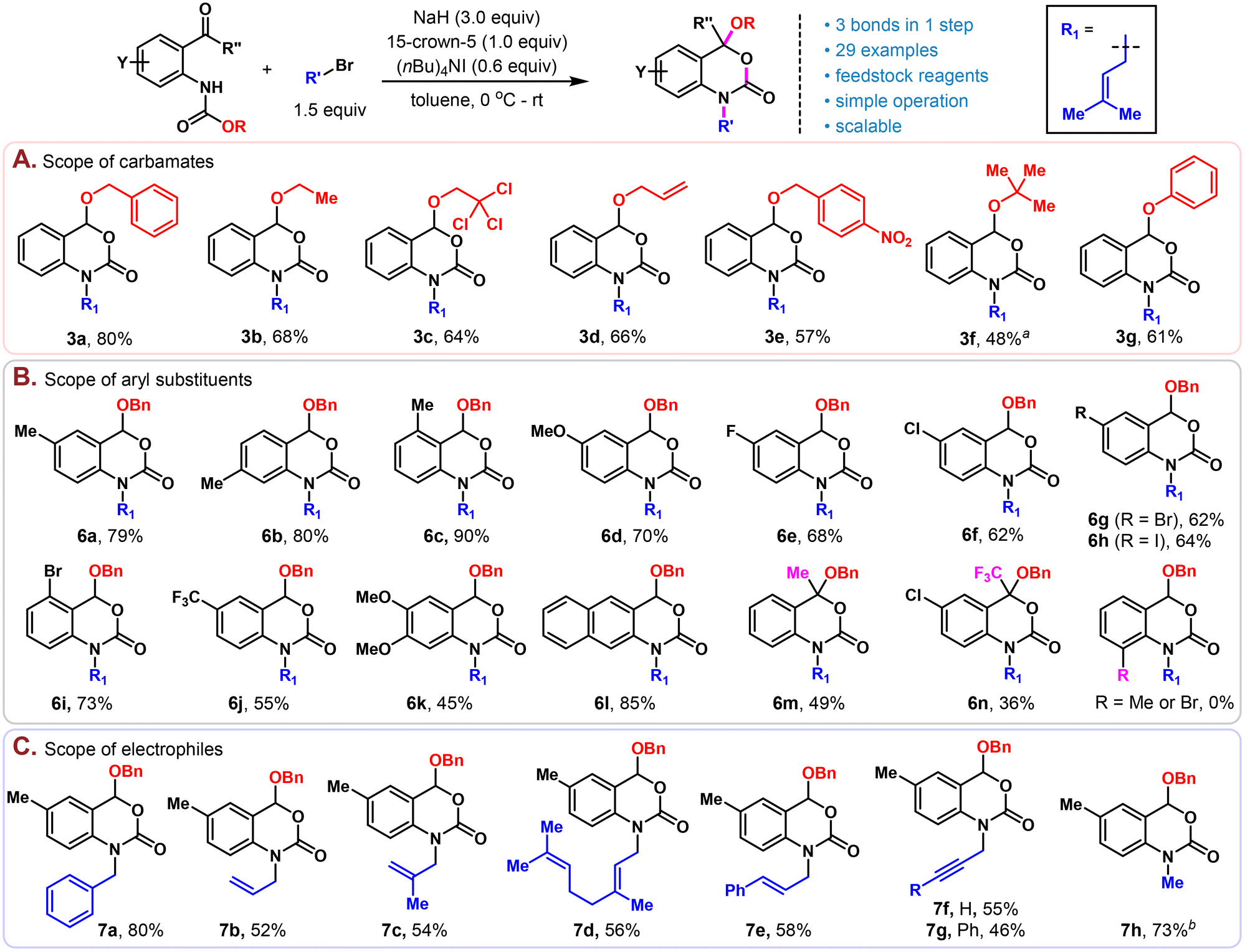 | ||
| Scheme 2 Substrate scope. Reaction conditions: carbamate 1a (1.0 equiv.), bromide 2a (1.5 equiv.), NaH (3.0 equiv.), 15-crown-5 (1.0 equiv.) and (nBu)4NI (0.6 equiv.) were stirred in toluene (1.0 mL) at 0 °C for 1 h and then at room temperature for another 2 h. aThe reaction was run at 50 °C in the absence of 15-crown-5 and (nBu)4NI. bMeI was used in the absence of (nBu)4NI. | ||
Next, substitutions on the benzene ring of anthranil aldehyde were investigated (Scheme 2B). Moderate electron-rich substrates (6a–6d) were converted to the benzoxazinone products in good to excellent yields (70%–90%). All types of halides (6e–6i) including aryl iodide were well tolerated, setting up an orthogonal reactive site for further derivatization. Strong electron withdrawing (6j) and donating groups (6k) led to products in moderate yields (55% and 45%), possibly due to the substantial influence of these functional groups on the nucleophilicity of nitrogen and the electrophilic nature of the aldehyde. By replacing the benzene group with naphthylene, the corresponding naphtho[2,3-d][1,3]oxazin-2-one 6l could also be prepared in a high yield (85%). Compared to aldehydes, anthranil ketone substrates would provide products possessing a quaternary carbon at C4, which is structurally more convincing and difficult to realize due to steric hindrance and retarded electrophilicity at the carbonyl group. Gratifyingly, our reaction also proceeded uneventfully with both methyl ketone and trifluoromethyl ketone substrates and provided the corresponding products (6m and 6n) in moderate yields without further optimization, the latter of which (6n) constituted the alkylated version of an important class of efavirenz analogs.12 However, 6-substituted (Me or Br) substrates are not compatible in this chemistry. Under the same conditions, only the alkylation products were obtained and could not be converted to the corresponding 3,1-benzoxazin-2-ones by prolonging the reaction time or increasing the temperature. This is possibly because the steric repulsion of the 6-substituent and the alkyl group on the nitrogen would position the carbamate group in a conformation that is stereoelectronically unfavourable for the subsequent intramolecular alkoxide attack (see Scheme 3D for the proposed mechanism).
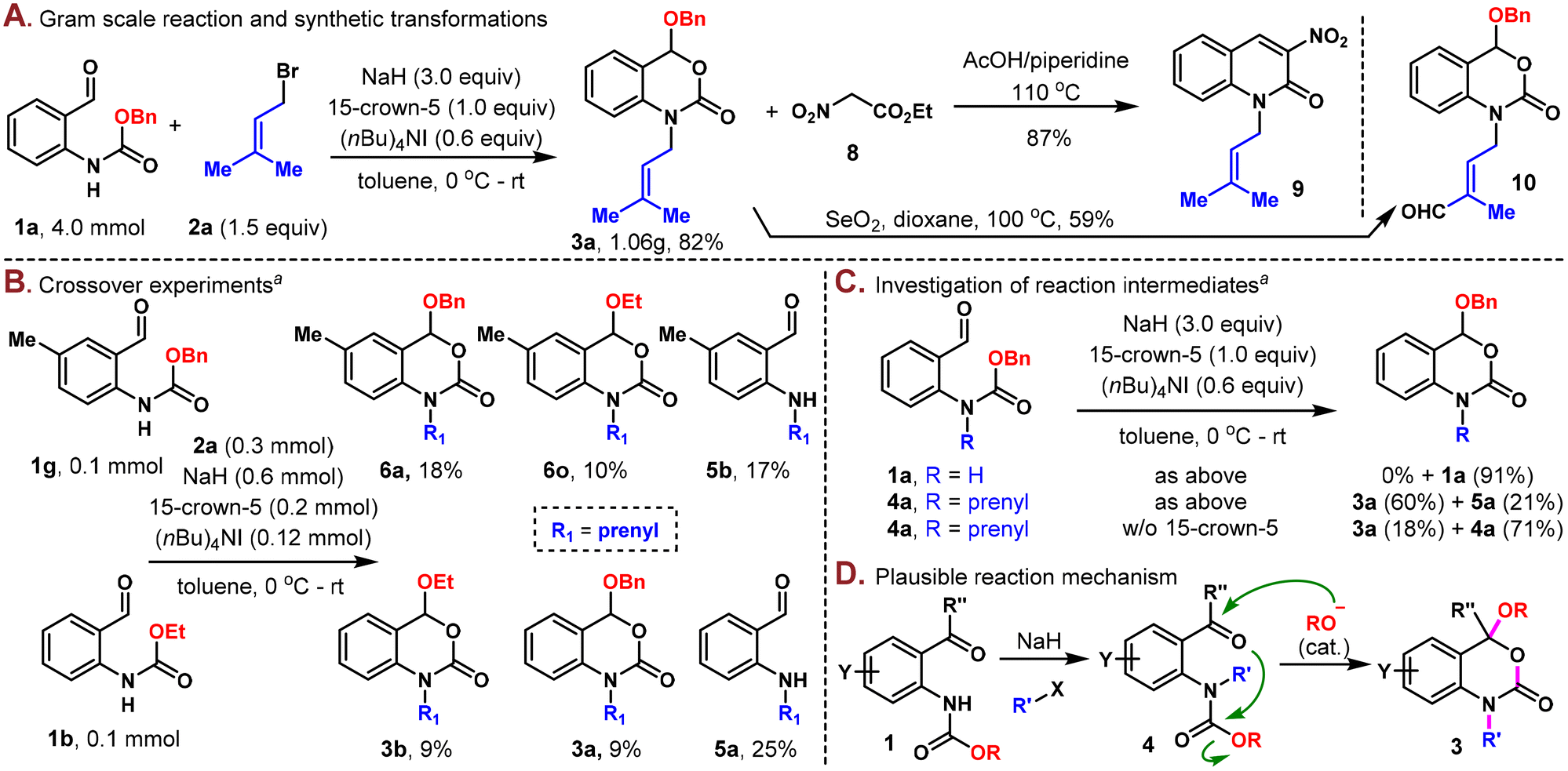 | ||
| Scheme 3 A) Gram-scale synthesis and synthetic transformations. (B) Crossover experiments. (C) Investigation of the reaction intermediates. (D) Plausible reaction mechanism. aYield determined by crude 1H NMR using trimethoxybenzene as the internal standard. | ||
Lastly, various alkylation reagents were tested with Cbz protected 5-methylanthranil aldehyde as the substrate under the standard conditions (Scheme 2C). Benzyl bromide (7a), allyl bromide (7b) and those with mono- (7c and 7e) or di- (7d) substitution were all good reaction partners for this chemistry. Propargyl bromides with or without terminal substitution (7f and7g) also worked smoothly, providing products with an additional handle for further structural modification and cross-linking with various azides for chemical probe development.13 Other than activated alkyl bromides, non-activated alkyl halides such as MeI could also form the alkylated product 7h in 73% yield.
The scalability and robustness of this protocol were further demonstrated through the gram-scale preparation of 3a in 82% yield (1.06 g) on a 4.0 mmol scale (Scheme 3A). Additional exploration of the utility of these N-alkyl-3,1-benzoxazin-2-one derivatives was also carried out. To this end, product 3a was converted to N-alkyl quinolin-2-one 9 when condensed with ethyl nitroacetate (8) at elevated temperature. The prenyl group could be selectively oxidized with SeO2 to generate the synthetically versatile aldehyde (10) with the 3,1-benzoxazin-2-one moiety unaltered.
Intuitively, this one-pot reaction involved stepwise N-alkylation and alkoxy rearrangement. However, the exact order of these steps and the mechanism of alkoxy transfer remained elusive. Thus, several control experiments were designed and implemented to shed light on the reaction mechanism. First of all, a crossover experiment with substrates 1g and 1b showed that scrambling took place, resulting in four cyclized products 6a, 6o, 3b, and 3a in 18%, 10%, 9% and 9% yields, respectively, which illustrated an intermolecular mechanism (Scheme 3B). Next, under the standard conditions in the absence of 2a, aldehyde 1a was recovered in 91% yield, while 4a was successfully converted to 3a in 60% yield along with 21% of the de-Cbz product 5a. The same reaction was sluggish in the absence of 15-crown-5, with only 18% of 3a obtained and 71% of 4a untouched, as indicated by crude NMR analysis (Scheme 3C). These experiments indicated that 4a was a possible intermediate in the reaction and that the N-alkylation took place prior to the alkoxy rearrangement step.
Based on these experiments, a plausible reaction mechanism is outlined, as shown in Scheme 3D. Carbamate 1 first underwent N-alkylation to produce the alkylated product 4. The basic reaction conditions would then cause partial decomposition of the carbamate group and release a small amount of alkoxide,14 which would proceed to attack the aldehyde or ketone and induce downstream intramolecular cyclization9,10 to form the thermodynamically stable N-alkyl-3,1-benzoxazin-2-one product. The same alkoxide was continuously regenerated in this process and actually involved in the reaction as a catalyst. The role of 15-crown-5 was believed to be multifaceted, namely, facilitating the N-alkylation step and accelerating both carbamate decomposition and subsequent alkoxide attack on the aldehyde/ketone.
Conclusions
In summary, we have developed a simple method to synthesize N-alkyl-3,1-benzoxazin-2-one derivatives from carbamate protected anthranil aldehydes and ketones via a one-step alkylation/alkoxy rearrangement reaction. This protocol uses feedstock reagents for constructing three carbon-hetero bonds and one ring in a single step and has a broad substrate scope. Control studies revealed that N-alkylation took place prior to the ring closing step and that the alkoxy transfer was an intermolecular process. The modular and highly step-economical nature would make this chemistry attractive for benzoxazinone analog synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22101290), the Chinese Academy of Sciences and the Shanghai Institute of Materia Medica.References
- (a) S. J. Robinson, B. I. Morinaka, T. Amagata, K. Tenney, W. M. Bray, N. C. Gassner and P. Crews, J. Med. Chem., 2010, 53, 1651–1661 CrossRef CAS PubMed; (b) V. Namasivayam, M. Vanangamudi, V. G. Kramer, S. Kurup, P. Zhan, X. Liu and S. N. Byrareddy, J. Med. Chem., 2018, 62, 4851–4883 CrossRef PubMed; (c) P. D. Williams, B. V. Clineschmidt, J. M. Erb, R. M. Freidinger, M. T. Guidotti, E. V. Lis and D. R. Reiss, J. Med. Chem., 1995, 38, 4634–4636 CrossRef CAS PubMed; (d) P. Zhang, E. A. Terefenko, A. Fensome, J. Wrobel, R. Winneker, S. Lundeen, K. B. Marschke and Z. Zhang, J. Med. Chem., 2002, 45, 4379–4382 CrossRef CAS PubMed; (e) A. Fensome, R. Bender, R. Chopra, J. Cohen, M. A. Collins, V. Hudak, K. Malakian, S. Lockhead, A. Olland, K. Svenson, E. A. Terefenko, R. J. Unwalla, J. M. Wilhelm, S. Wolfrom, Y. Zhu, Z. Zhang, P. Zhang, R. C. Winneker and J. Wrobel, J. Med. Chem., 2005, 48, 5092–5095 CrossRef CAS PubMed; (f) T. Mizutani, S. Ishikawa, T. Nagase, H. Takahashi, T. Fujimura, T. Sasaki, A. Nagumo, K. Shimamura, Y. Miyamoto, H. Kitazawa, M. Kanesaka, R. Yoshimoto, K. Aragane, S. Tokita and N. Sato, J. Med. Chem., 2009, 52, 7289–7300 CrossRef CAS PubMed.
- M. M. Bastos, C. C. P. Costa, T. C. Bezerra, F. C. da Silva and N. Boechat, Eur. J. Med. Chem., 2016, 108, 455–465 CrossRef CAS PubMed.
- P. D. Williams, M. G. Bock, B. E. Evans, R. M. Freidinger, S. N. Gallicchio, M. T. Guidotti and C. J. Woyden, Bioorg. Med. Chem. Lett., 1999, 9, 1311–1316 CrossRef CAS PubMed.
- X. L. Su, S. Xu, Y. Shan, M. Yin, Y. Chen, X. Feng and Q. Z. Wang, Fitoterapia, 2018, 127, 186–192 CrossRef CAS.
- (a) P. D. Champlain, J.-L. Luche, R. A. Marty and P. D. Mayo, Can. J. Chem., 1976, 54, 3749–3756 CrossRef; (b) S. S. Nikam, P.-W. Yuen, B. E. Kornberg, B. Tobias and M. F. Rafferty, J. Org. Chem., 1997, 62, 9331–9334 CrossRef CAS; (c) Y. Nishiyama, Y. Naitoh and N. Sonoda, Synlett, 2004, 886–888 CrossRef CAS; (d) E. Hernandez, J. M. Velez and C. P. Vlaar, Tetrahedron Lett., 2007, 48, 8972–8975 CrossRef CAS PubMed; (e) K. Kobayashi, S. Fukamachi and H. Konishi, Heterocycles, 2008, 75, 2301–2307 CrossRef CAS; (f) O. R. Suárez-Castillo, C. I. Bautista-Hernández, M. Sánchez-Zavala, M. Meléndez-Rodríguez, A. Sierra-Zenteno, M. S. Morales-Ríos and P. Joseph-Nathan, Heterocycles, 2012, 85, 2147–2171 CrossRef; (g) Y. M. Yu, Y. N. Huang and J. Deng, Org. Lett., 2017, 19, 1224–1227 CrossRef CAS PubMed; (h) S. Sun, C. Zhou, J. T. Yu and J. Cheng, Org. Lett., 2019, 21, 6579–6583 CrossRef CAS PubMed; (i) H. Xiong, X. Wu, H. Wang, S. Sun, J. T. Yu and J. Cheng, Adv. Synth. Catal., 2019, 361, 3538–3542 CrossRef CAS; (j) M. Vayer, M. Pastor, C. Kofink and N. Maulide, Org. Lett., 2022, 24, 27–32 CrossRef CAS PubMed; (k) X. Li, J. Benet-Buchholz, E. C. Escudero-Adan and A. W. Kleij, Angew. Chem., Int. Ed., 2023, 62, e202217803 CrossRef CAS PubMed.
- (a) Y. Zhao, B. Huang, C. Yang, Q. Chen and W. Xia, Org. Lett., 2016, 18, 5572–5575 CrossRef CAS PubMed; (b) T. Niemi, I. Fernandez, B. Steadman, J. K. Mannisto and T. Repo, Chem. Commun., 2018, 54, 3166–3169 RSC; (c) T. V. Tran, Y. Shen, H. D. Nguyen, S. Deng, H. Roshandel, M. M. Cooper, J. R. Watson, J. A. Byers, P. L. Diaconescu and L. H. Do, Green Chem., 2022, 24, 9245–9252 RSC.
- (a) K. Kobayashi, S. Fukamachi, D. Nakamura, O. Morikawa and H. Konishi, Heterocycles, 2008, 75, 95–105 CrossRef CAS; (b) H. Fan, Y. Wan, P. Pan, W. Cai, S. Liu, C. Liu and Y. Zhang, Chem. Commun., 2019, 56, 86–89 RSC; (c) E. M. Larin, A. Torelli, J. Loup and M. Lautens, Org. Lett., 2021, 23, 2720–2725 CrossRef CAS PubMed.
- (a) J. C. Kern, E. A. Terefenko, A. Fensome, R. Unwallla, J. Wrobel, Y. Zhu, J. Cohen, R. Winneker, Z. Zhang and P. Zhang, Bioorg. Med. Chem. Lett., 2007, 17, 189–192 CrossRef CAS PubMed; (b) K. C. Nicolaou, A. Krasovskiy, U. Majumder, V. E. Trepanier and D. Y. Chen, J. Am. Chem. Soc., 2009, 131, 3690–3699 CrossRef CAS PubMed; (c) V. Palomo, D. I. Perez, C. Roca, C. Anderson, N. Rodriguez-Muela, C. Perez, J. A. Morales-Garcia, J. A. Reyes, N. E. Campillo, A. M. Perez-Castillo, L. L. Rubin, L. Timchenko, C. Gil and A. Martinez, J. Med. Chem., 2017, 60, 4983–5001 CrossRef CAS PubMed.
- J. L. Garcia Ruano, C. Pedregal and J. H. Rodriguez, Tetrahedron, 1989, 45, 203–214 CrossRef.
- (a) L. A. Radesca, Y. S. Lo, J. R. Moore and M. E. Pierce, Synth. Commun., 1997, 27, 4373–4384 CrossRef CAS; (b) G.-J. Mei, C.-Y. Bian, G.-H. Li, S.-L. Xu, W.-Q. Zheng and F. Shi, Org. Lett., 2017, 19, 3219–3222 CrossRef CAS PubMed.
- N. J. Tom, W. M. Simon, H. N. Frost and M. Ewing, Tetrahedron Lett., 2004, 45, 905–906 CrossRef CAS.
- A. J. Cocuzza, D. R. Chidester, B. C. Cordova, S. Jeffrey, R. L. Parsons, L. T. Bacheler, S. Erickson-Viitanen, G. L. Trainor and S. S. Ko, Bioorg. Med. Chem. Lett., 2001, 11, 1177–1179 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- L. W. Dittert and T. Higuchi, J. Pharm. Sci., 1963, 52, 852–857 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2258994. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00812f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |