
C–H functionalization of pyridines
Susmita
Maity†
a,
Asish
Bera†
ab,
Ayantika
Bhattacharjya†
a and
Pradip
Maity
 *ab
*ab
aOrganic Chemistry Division, CSIR-National Chemical Laboratory, Pune 411008, India. E-mail: p.maity@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 15th June 2023
Abstract
Pyridine and its reduced form (piperidine) are the most common nitrogen heterocycles in FDA-approved drugs. Additionally, their presence in alkaloids, ligands for transition metals, catalysts, and organic materials with various properties makes them among the most important structural cores. Despite its importance, direct and selective functionalization of pyridine remains scarce due to its electron-poor nature and nitrogen coordination power. Instead, functionalized pyridine rings were primarily constructed from suitably substituted acyclic precursors. The focus on sustainable chemistry with minimum waste generation encourages chemists to develop direct C–H functionalization. This review summarizes different approaches to tackle the reactivity and regio- and stereoselectivity aspects for direct pyridine C–H functionalization.
 Susmita Maity | Susmita Maity is currently working as Project Associate-I in the Organic Chemistry Division at the CSIR-National Chemical Laboratory under the supervision of Dr Pradip Maity. She pursued her B.Sc. and M.Sc. from Vidyasagar University in 2020 and 2022, respectively. She is working on catalytic asymmetric aerobic oxidations. |
 Asish Bera | Asish Bera completed his B.Sc. (Chemistry) in 2012 from Vidyasagar University and his M.Sc. (Chemical Science) in 2014 from IIT Madras, India. In 2016, he joined the CSIR-National Chemical Laboratory (NCL) under the supervision of Dr Pradip Maity. His Ph.D. thesis is based on novel phosphite catalyst synthesis and its application in enantioselective radical reactions. |
 Ayantika Bhattacharjya | Ayantika Bhattacharjya obtained her B.Sc. from Vidyasagar University in 2020 and M.Sc. from Midnapore College Autonomous (Vidyasagar University) in 2022. She is currently working as Project Associate-I in the Organic Chemistry Division at the CSIR-NCL under the supervision of Dr Pradip Maity. Her current research interest focuses on the asymmetric functionalization of imines. |
 Pradip Maity | Dr Pradip Maity received his B.Sc. degree in chemistry from Calcutta University in 2001 and his M.Sc. from IIT Kanpur, India. Then he moved to the US for Ph.D. from Florida Atlantic University in 2006 with Professor Salvatore D. Lepore. After completion, he joined UT Southwestern Medical Center in Dallas as a postdoctoral fellow with Professor Uttam K. Tambar in 2011. He began his independent research career at the CSIR-National Chemical Laboratory, Pune, as a Senior Scientist in 2015. His research group is interested in developing a sustainable catalysis platform for asymmetric pyridine functionalization and medicinal chemistry. |
Six-membered N-heterocyclic (pyridine and piperidines) cores are the first and second most frequent heterocyclic structures that appear in FDA-approved drugs.1 Besides, pyridines serve as crucial ligands for transition metals,2 are present in a large number of alkaloids with biological importance,3 act as organocatalysts,4 and feature in many functional materials.5 Hence, synthesis of functionalized pyridines attracts considerable attention from synthetic communities.6 Traditionally, highly functionalized pyridine rings are built from acyclic precursors. Although many elegant methods were developed over the years, they fundamentally suffer from multi-step synthesis of suitable acyclic precursors and the final elimination reaction sequence to generate waste. With green and sustainable methods in focus to reduce environmental footprints, regio- and stereoselective direct C–H functionalization becomes one of the most critical approaches.
Aromatic C–H can be directly functionalized by electrophilic aromatic substitutions (SEAr; Friedel–Crafts reaction). However, pyridine, an electron-deficient aromatic, needs forced conditions. The Lewis acid activation of electrophiles is also problematic since the nitrogen of pyridine competes via coordination with the Lewis acid. As a result, pyridine deactivates the Lewis acid and becomes further electron deficient for the Friedel–Crafts reaction.
In the last few decades, transition metal catalysed regioselective functionalization of aryl C–H bonds has achieved extraordinary progress.7 Electron-deficient pyridines are less reactive for C–H functionalization and are instead used mainly as directing groups owing to their unreactive C–H bonds. The strong coordinating nature of the pyridine nitrogen makes it a good ligand but further deactivates it for C–H activation.
The lack of reports resulted in only one comprehensive review in 2017 summarizing the C–H functionalization of six-membered N-heteroaromatic compounds, including pyridines.6c The significance of direct pyridine functionalization has led to the development of numerous elegant strategies since then. This review will briefly discuss different approaches based on reactivity patterns and present recent developments for pyridine C–H functionalization.
1. Direct functionalization
This section will discuss the reports for direct C–H functionalization of pyridine without pre-activation of the pyridine nitrogen.1.1. Friedel–Crafts reaction with electrophiles
SEAr with electron-deficient pyridine works only at elevated temperatures with C3 selectivity, as C3 is the least deactivated center. Brominations and nitrations are known to work at 300 °C, but acylation with acyl chloride and a Lewis acid does not work.8The Lewis acid preferentially coordinates with the pyridine nitrogen over acyl chloride, deactivating the pyridine further (Scheme 1). The functional group tolerance at this high temperature is an issue, and therefore the direct SEAr approach did not enjoy widespread applications.
 | ||
| Scheme 1 Electronic nature of pyridine and its deactivation via Lewis acid. | ||
1.2. Electrophiles via C–H deprotonation
The electron-deficient nitrogen of pyridine makes the C2–H acidic and prone to deprotonation with a strong base. The deprotonated pyridine can react with electrophiles to generate various C2-functionalized pyridines. The Knochel group extensively studied the formation and reactivities of deprotonated pyridines at different positions.9 Hevia, McLellan, and co-workers used a strong magnesium amide base for deprotonation (Scheme 2). The monomeric magnesium amide was stabilized by a ligand, and the magnesium coordinates with the pyridine nitrogen for its base-mediated deprotonation. The deprotonated pyridine adds to another pyridine starting material coordinated with the same magnesium Lewis acid. The resulting intermediate (1) oxidized in air to form 2,2′-bipyridine products.10 | ||
| Scheme 2 C–H deprotonation followed by its reaction with an electrophile. | ||
1.3. Nucleophilic substitution
The electron-deficient nature of the pyridine ring also makes it susceptible to nucleophilic addition at C2 and C4. For an overall C–H functionalization, the hydrogen at the nucleophile-added carbon (2) must be removed with its electron pair. SNAr with hydride as the leaving group was described in 1914 by Chichibabin and co-workers for C2 amination (Scheme 3).11 The addition of sodium amide to the electrophilic C2 position is favorable, but the following hydride elimination is a high-energy step, requiring forced reaction conditions. | ||
| Scheme 3 Chichibabin reaction: C2 nucleophilic addition followed by hydride elimination. | ||
Subsequently, reactions with Grignard reagents by the Evans group and lithiated carbon nucleophiles by the Sarpong group were reported. The formation of a reactive metal hydride as a side product impedes functional group tolerance.
In 2020, Zhang, Xiong, and co-workers reported a trialkylborane-borohydride system for the alkylation of pyridines (Scheme 4). The experimental and computational mechanistic studies suggest initial hydroboration of the alkene. The resulting alkylborane 3 was added to the borane-activated pyridine (4) at the C4 position to avoid steric hindrance at C2. Borohydride removal from the corresponding intermediate 5 gives C4 alkylated pyridines. The regeneration of borohydride makes it a sub-stoichiometric reagent, while trialkylborane was used in excess, probably due to product coordination. The reaction occurred at 100 °C in THF with excellent yields and substrate scope.12
 | ||
| Scheme 4 Borohydride catalysed Chichibabin type C4 alkylation of pyridine with alkenes. | ||
In 2013, the Hartwig group reported C2 fluorination using super-stoichiometric silver difluoride (Scheme 5). The authors planned the reaction in line with the Chichibabin reaction. However, the mechanistic studies suggest that the reaction likely proceeded via the nucleophilic addition of fluoride to dihydropyridine (6), followed by oxidation with stoichiometric AgF2 to substituted pyridine with the removal of HF.13 The nucleophilic addition at C2/C4 of pyridine leading to dihydropyridine intermediate, followed by its oxidation to rearomatized substituted pyridine became a general approach for C–H functionalization of pyridines. The initial nucleophile can add to either the C2 or C4 position, leading to the possibility of mixtures of C2 and C4 regioisomers.
 | ||
| Scheme 5 AgF2 mediated C2 fluorination via silver mediated fluoride addition and subsequent oxidation. | ||
Martin and co-workers in 2019 reported site-selective C2 and C4 silylation of pyridine with Et3SiBPin and a stoichiometric KHMDS base. Their analysis with different counter-ions and solvents indicated a solvent-separated ion pair for sterically less crowded C4 functionalization. C2 silylation was also achieved through a contact ion-pair intermediate. The mechanistic proposal suggests an initial coordination of the base to the boron of silyl borane. The coordination increases the nucleophilicity of the B–Si bond, which adds to the potassium-coordinated pyridine (Scheme 6). The final product formation from the silylated dihydropyridine is not clear.14 Either air oxygen during the quench led to oxidative aromatization, or borohydride elimination can lead to product formation.
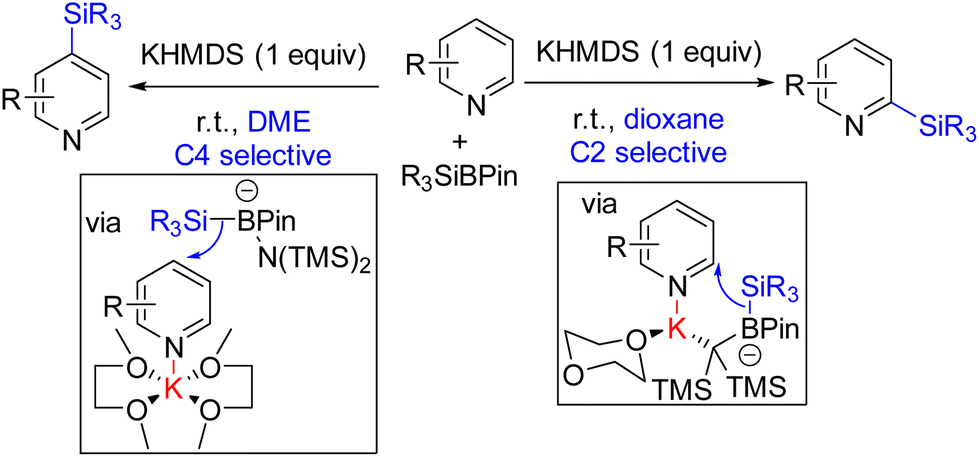 | ||
| Scheme 6 Solvent and base-mediated regio-divergent C2/C4 silylation with R3SiBPin. | ||
In 2021, List and Obradors reported a Brønsted acid catalysed C4 nucleophilic functionalization of pyridines with silyl ketene acetals (Scheme 7).15 In their reaction design, the triflimide Brønsted acid (7) acts as a precatalyst which reacts with silyl ketene acetal (8) to form the Lewis acidic silylium (Si–X). The unprecedented silylium activation of the pyridine nitrogen led to the addition of 8 to the activated pyridine. The corresponding intermediate (9) regenerated the silylium Lewis acid and dihydropyridine 10. The substituted pyridine formed upon subsequent DDQ oxidation. The large silylium led to its enhanced Lewis acidity via a longer Si–X bond, and steric hindrance at the pyridine nitrogen resulted in C4 selective nucleophilic addition.
 | ||
| Scheme 7 C4 pyridine functionalization of pyridine with silyl ketene acetals. | ||
The in situ oxidation by various oxidizing reagents compatible with a particular nucleophile became a successful approach for overall pyridine C–H functionalization. When the oxidant is not compatible, a one-pot sequential or stepwise oxidation of the dihydropyridine intermediate leads to the substituted pyridine product.16
1.4. Reaction with radicals
Oxidation of the radical intermediates from nucleophilic radical addition to pyridines became a general C–H activation strategy. In 1971, Minisci and co-workers reported that alkyl radicals generated from silver-mediated decarboxylation were added to pyridines (Scheme 8). The nucleophilic alkyl radicals add to pyridine at either C2 or C4 positions, followed by single-electron oxidation of the radical intermediate 11 to alkylated pyridine.17 They also discovered that aryl radicals could add to pyridines to give C–H arylation of pyridine.18 | ||
| Scheme 8 Minisci reaction: radical alkylation and arylation with an in situ oxidant. | ||
Unlike the anionic nucleophilic addition–oxidation sequence, nucleophilic alkyl radical generation and its addition to pyridine are generally compatible with the stoichiometric oxidant in the Minisci reaction. The advancement of mild alkyl radical generation reignites the Minisci-type C–H functionalization of pyridines. Subsequently, numerous methods for alkyl radical generation and their Minisci-type pyridine substitution were reported and reviewed.19
The Jingbo Yu group reported nucleophilic radical addition followed by a hydride elimination protocol for C4 alkylation of pyridine (Scheme 9). Alkyl halide and magnesium metal initially reacted to form an alkyl radical and radical Mg(I) species which was stabilized by the N,N′-di-tert-butylethane-1,2-diamine (DTEDA) ligand. The Mg(I) species reduces the magnesium coordinated pyridine. The radical–radical recombination led to dihydropyridine 12, and subsequent [Mg]–H elimination led to the C4 alkylated product.20
 | ||
| Scheme 9 C4 alkylation of pyridine with an alkyl halide and mechanochemically activated magnesium metal. | ||
In 2015, the MacMillan group showed that alcohol could be used as an alkylating reagent under photoredox reaction conditions. The removal of OH via a biologically relevant spin center shift (SCS) enables an inexpensive and abundant alcohol as the alkylating agent without the requirement of an oxidant (Scheme 10). The method resulted in both C2 and C4 alkylated products, which can be controlled by substrate substitution patterns.21 The reaction works via dual photoredox and HAT catalysis for the formation of an α-hydroxyalkyl radical. The addition of this radical to protonated pyridine results in the formation of the radical intermediate 13 after facile deprotonation. This radical with an α-hydroxyl group undergoes an SCS to eliminate water and generate the benzylic radical (14). The photoexcited iridium catalyst reduces this electron-deficient benzyl radical, and subsequent protonation leads to the formation of alkylpyridine.
 | ||
| Scheme 10 Alcohol as the radical alkylating reagent for pyridine alkylation without an oxidant. | ||
UV light-mediated photocatalyst-free methylation and ethylation using the corresponding alcohol were reported later under acidic conditions by Li and co-workers.22
In 2019, the Melchiorre group reported C–H hydroxyalkylation of quinolines and isoquinolines. The reaction between acyl dihydropyridine as an acyl radical generator and pyridine resulted in hydroxyalkylation products (Scheme 11).
 | ||
| Scheme 11 Acyl dihydropyridine as a dual hydroxyalkyl reagent and oxidant for pyridine C–H functionalization. | ||
Both C2 and C4 products were formed based on the substitution pattern. The reaction proceeded under 460 nm light irradiation and acidic conditions without any photocatalyst.23 The theoretical and experimental studies suggested that a light-mediated single electron reduction of acyl dihydropyridine (15) initiates the reaction. The acyl radical (16) formed was added to the protonated pyridine followed by deprotonation to form the radical intermediate 17. This intermediate undergoes an SCS to rearomatize the pyridine ring with a radical center shift to the α carbon (18). The benzylic radical undergoes single electron reduction by the pyridyl radical to form the corresponding carbanion, which protonates to yield the final product.
The Overman group introduced oxalate intermediates 21 from alcohol which undergoes one-electron oxidation to generate alkyl radicals with the loss of two CO2. Reaction with the cesium salt of oxalate from a tertiary alcohol and pyridine under light and the iridium photocatalyst resulted in C2 alkylated product formation with two equivalents of ammonium persulfate as the oxidant (Scheme 12).24
 | ||
| Scheme 12 Alcohol derivative as a radical alkylating reagent of pyridine. | ||
The method was applied to many other N-heteroaromatic compounds with embedded imines. Zard's research group developed an efficient alkylation starting with different thiol derivatives, including tertiary thiols, for the formation of quaternary alkylated pyridines.25 Dilauroyl peroxide (DLP) was used for light-mediated radical initiation and subsequent oxidation of the alkyl-added pyridine intermediates.
Zhang, Yang, and co-workers, and later Poole and co-workers reported a carboxylic acid-derived hypervalent iodine(III) reagent (22) as an alkyl generation and oxidizing reagent under visible light irradiation (Scheme 13). The hypervalent iodine-carboxylate bond cleaves homolytically under light, which initiates the C–H alkylation and acylation of pyridine, quinoline, and isoquinoline.26
 | ||
| Scheme 13 Carboxylic acid and PIDA for the decarboxylating radical alkylation of pyridine. | ||
In 2023, Zhang's group discovered that tert-butyl hydroperoxide can homolytically cleave upon irradiation with 395 nm light. The alkoxy or hydroxyl radical generated undergoes HAT with carboxylic acid to form an alkyl radical via CO2 expulsion. The same alkoxy/hydroxy radical then undergoes another HAT with the radical-pyridine adduct for the synthesis of alkylated pyridines (Scheme 14).27a In the same year, Wang, Niu, and co-workers developed a desulfonative radical pyridylation with glycosyl sulfonates as the glycosyl radical precursor.27b In this case, excess tert-butyl hydroperoxide generates the tert-butyl hydroxyl radical that accepts a single electron from the sulfonate anion. The corresponding radical intermediate generates a glycosyl radical after SO2 expulsion, similar to decarboxylation. The rest of the reaction pathway runs parallel to Scheme 14.
 | ||
| Scheme 14 TBHP as a photoactive oxidant for decarboxylating alkylation. | ||
In 2017, Liu and Zhang reported the Minisci alkylation of pyridines using alkylboronic acid as an alkyl radical precursor under 1 atm oxygen (Scheme 15).28 The authors proposed aerobic oxidative alkyl radical formation from boronic acid that adds to pyridine in the DCE solvent and TFA additive. The alkyl added pyridine intermediate was subsequently oxidized by oxygen or an oxygen-derived oxidant to generate alkylpyridines. Primary and secondary alkylation was shown to work well with a variety of pyridine derivatives.
 | ||
| Scheme 15 Boronic acid under air for alkyl radical generation and oxidation for pyridine functionalization. | ||
Another approach for generating an alkyl radical is to add a hydrogen radical to an alkene. The Herzon group used Co(II)/TBHP, and the Baron group used Fe(III)/BF3·OEt2 to generate alkyl radicals from alkene and silanes. Subsequently, Liu and co-workers reported sodium borohydride and Fe(III) mediated radical alkylation of pyridines (Scheme 16). Fe(III) was proposed to oxidize hydride to a hydrogen radical that adds an alkene to form the alkyl radical. The alkyl radical added pyridine intermediate was oxidized further with Fe(III) to generate the final product.29a The reaction scope was broad, and the dialkylation product forms when multiple C2/C4 hydrogens are available for functionalization. Although inexpensive iron, borohydride, and alkenes were used for this transformation, excess reagents were necessary to obtain a good yield, and therefore the monoalkylated product was difficult to obtain.
 | ||
| Scheme 16 Radical alkene hydrogenation mediated alkyl radical generation for pyridine alkylation. | ||
Similarly, radical addition of an azide to an alkene generated alkyl radicals which were added to pyridine for the synthesis of β-azido alkylpyridines. Here, two equivalents of (diacetoxyiodo)benzene (DIB) were used as an oxidant to generate an azide radical from TMSN3.29b The reaction steps are similar to that of Zhang and Yang (Scheme 14). Later, Zhou, Li, and co-workers reacted alkene with alcohol in the presence of pyridine and persulfate to generate a β-alkoxy radical under visible light (390–400 nm LED). It is proposed that the pyridine derivative gets excited under visible light. The excited pyridine engages in a single-electron reduction reaction with the persulfate anion to form a sulfate radical anion. Alternatively, a sulfate radical anion may form via homolytic cleavage of persulfate under light irradiation. The alkene is oxidized by SO4˙− and reacts with alcohol to form a β-alkoxy radical. This radical adds to the pyridine, followed by its single electron oxidation leading to product formation.29c
Huang, Chen, and co-workers reported alkyl radical formation from bulky and inert Csp3–Csp3 bond cleavage from alcohol 23. The alcohol OH undergoes photoredox catalysed HAT to form reactive alkoxide radical 24, which forms an alkyl radical via expulsion of the carbonyl compound. The subsequent reaction of the alkyl radical with electron-deficient systems, including pyridines, led to C–H functionalization (Scheme 17).30
 | ||
| Scheme 17 Photochemical oxidative Csp3–Csp3 bond cleavage foralkyl radical generation for pyridine alkylation. | ||
Ammonium persulfate was used as a stoichiometric oxidant. Finally, alkyl radicals were also generated from C(sp3)–H, followed by their addition to pyridines for substituted pyridines.31 In line with the initial reports by the MacMillan group for alkylation from the ether, Togo reported the UV light-mediated α-alkylation of amide which needed excess amide for its success.32 Berthelot and co-workers use MacMillan conditions under visible light to improve the yield and efficiencies with a secondary amine.33 The reaction was shown to work with pyridine, quinoline, and isoquinolines, and substrates were chosen with either C2 or C4 sites blocked for selective product formation. Acyl radicals, being highly nucleophilic, are also suitable for the Minisci-type acylation of pyridines. Fluoroalkyl radicals are also compatible with the C2/C4 functionalization of pyridines.
In 2023, the Melchiorre group reported the photochemical organocatalytic C2/C4 allylation and benzylation of pyridine (Scheme 18).34 They found a suitable dithio-derivative of the phosphate anion (25−) organocatalyst that absorbs 365 nm light to generate a highly reducing excited state. The excited catalyst 25−* reduced the protonated pyridine to form a pyridinyl radical (26). The corresponding thiophosphate radical (25˙) undergoes HAT with allylic and benzylic C–H to form the corresponding radical. Hetero-coupling of these two radicals gave the alkylated dihydropyridine intermediate. The following oxidative rearomatization to product formation occurred without any oxidizing agent. The authors speculated a photochemical oxidation path in which acetone solvent acts as an oxidant.
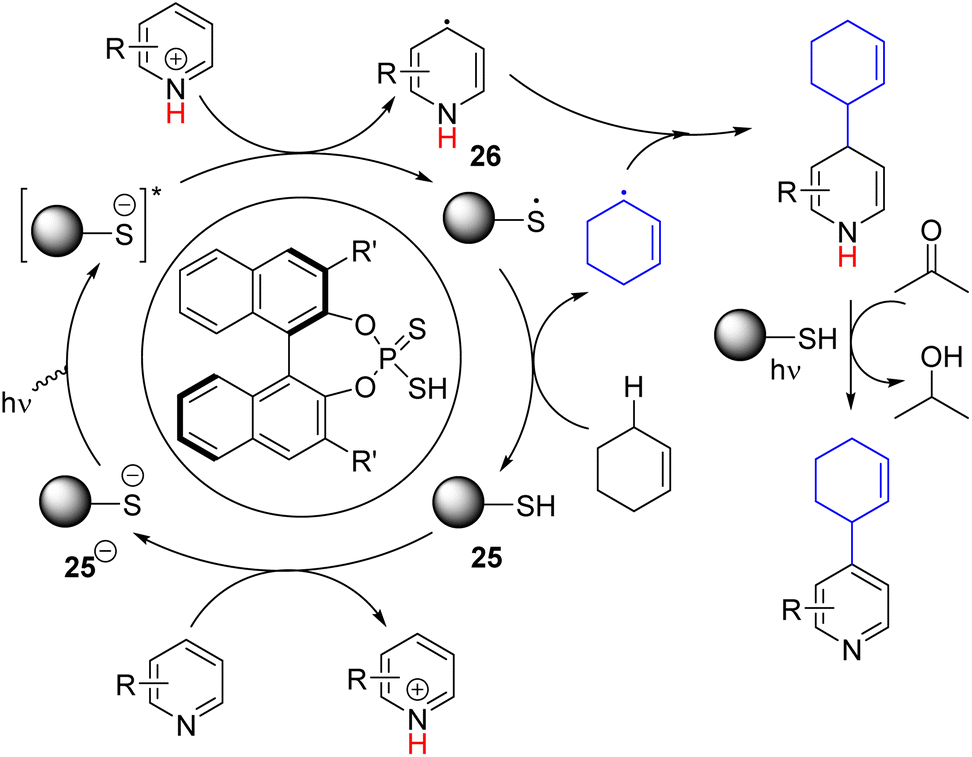 | ||
| Scheme 18 Photochemical organocatalytic allylation and benzylation of pyridine via double C–H activation. | ||
In 2021, the Leonori group utilized the Minisci-type C2 borylation of pyridines via the photocatalytic generation of nucleophilic boryl radicals (Scheme 19). The photocatalyst 4CzIPN, under blue light, gets excited and reduces ammonium persulfate to a sulfate radical anion. The radical anion undergoes HAT with B–H of borane-triethylamine (27) to form the boryl radical 28 that was added to the pyridine at the C2 position. The resulting intermediate (29) oxidized to the stable 2-borylpyridine product (30) by ammonium persulfate. The sulfate radical anion formed in the last step led to the radical chain borylation process, which was applicable to pyridine, quinoline, isoquinoline, and other electron-deficient heterocycles.35a In 2023, the Banerjee group synthesized a β-keto-enamine-based COF photocatalyst with suitable redox properties to achieve the same C2 borylation. Several catalytic cycles were performed to establish COF catalysts’ high photostability and recyclability.35b
 | ||
| Scheme 19 Photoredox C2 borylation of pyridines with ammonia-borane. | ||
Recently, Lin, Yu, and co-workers developed a tuneable C3 and C4 and electrochemical carboxylation of pyridine with CO2.36 The tuneable regioselectivity was achieved using either divided or undivided electrochemical cells (Scheme 20). The authors studied the reaction extensively via experiments and DFT calculation to suggest the reasons for this regiodivergent carboxylic acid formation. The reaction starts with the cathodic reduction of the pyridine substrate. The spin density of this radical-anion intermediate 31 is maximum at C5 carbon, and therefore it is the most nucleophilic carbon to attack electrophilic CO2. C5 nucleophilic attack to CO2 formed intermediate 32, which transformed into the dianionic 33 upon a second electron reduction. Aerobic oxidation of 33 forms the C3 carboxylate product. The spin density also suggests that the electron density at C4 is the second best, and both the C5 and C4 addition to CO2 is endothermic. Their DFT also showed that the subsequent C–H bond dissociation energy for the C4 intermediate (34) is lower than that of the C5 intermediate 32. Therefore, they speculated if tandem HAT catalysis could react faster with the C4 intermediate 34 to the C4 carboxylated product. It may shift the overall CO2 addition step in favor of C4 carboxylation. Different HAT reagents failed, presumably due to their facile reduction over the substrate pyridine at the cathode. On the other hand, running the reaction in an undivided cell serendipitously gave them the major C4 carboxylate product. They reason that iodine from the electrolyte nBu4I undergoes HAT or proton-coupled electron transfer (PCET) to abstract C4–H, leading to the regiodivergent formation of the C4 product. The iodine could also reduce at the cathode, but it can consistently regenerate at the anode oxidation of iodide, providing a constant source of iodine.
 | ||
| Scheme 20 Electrochemical regiodivergent C3 and C4 carboxylation with CO2. | ||
One of the challenging aspects of radical reactions is its stereocontrol. The Minisci type alkylation was mainly proposed to proceed with protonated pyridines as the activated coupling partner with alkyl radicals. In 2018, the Phipps group established that a sub-stoichiometric amount of acid was sufficient to catalyse the alkylation. Chiral phosphoric acid was tested for reductive aminoalkylation from N-acyloxyphthalimides with an iridium photocatalyst under a dual catalysis system.
The presence of NH at the aminoalkyl radical was essential to achieve significant enantioselectivity. Multiple coordination with chiral phosphoric acid was proposed for effective stereoinduction.37 Jiang and co-workers were working on the photoredox and chiral phosphoric acid dual catalysis for asymmetric radical reactions and reported an asymmetric Minisci reaction of isoquinoline and N-acyloxyphthalimides later in 2018.38 Subsequently, the Phipps group extended their work to show that alcohol is compatible under dual catalysis to yield the corresponding chiral alcohols.39 In 2023, the Wang group extended its application to β-carbolines (35) using the same redox-active ester as the alkylating agent.40 The cooperative photoredox iridium and chiral phosphoric acid catalysis gave the 2-aminoalkylation product (36) with good yields and high enantioselectivities (Scheme 21).
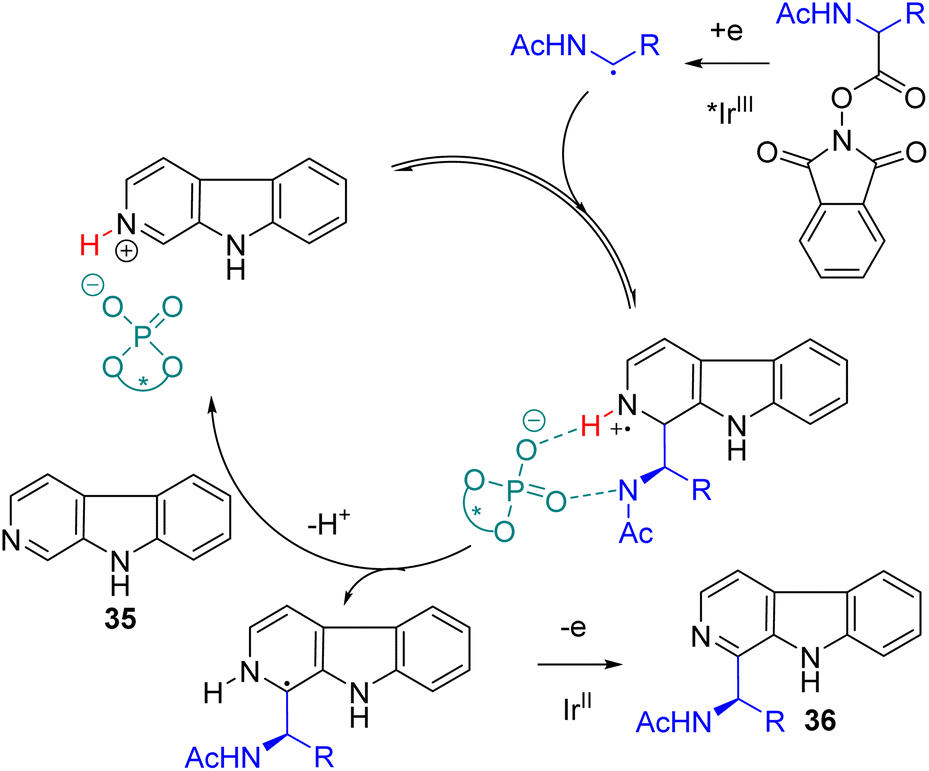 | ||
| Scheme 21 Photoredox and chiral phosphoric acid dual catalysis for asymmetric C2 aminoalkylation. | ||
1.5. Transition metal catalysis
The Jordan group reported the early examples of C–H alkylation of pyridines with alkenes using a cationic zirconium catalyst system.41 The alkylation was shown to work with 2-substituted pyridines for C6 alkylation. A follow-up report by the Jordan group and later the Hou group with a scandium catalyst reports asymmetric induction via chiral catalysts. However, those were reported with a limited substrate scope, similar to the 1989 report.42 It was speculated that the unsubstituted pyridine and quinoline nitrogen coordinate strongly with the catalyst, leading to catalyst arrest. In successful cases, the coordination power of pyridine nitrogen led to transition metal catalyst binding to the nitrogen, directing the metal center for selective C2–H activation. However, strong bonding with the substrate led to catalyst arrest, which limits the substrate scope.The acyl-directed alkylation of pyridine via a ruthenium catalyst was reported by Grigg and Savic in 1997.43 The preferential coordination of ruthenium to carbonyl over pyridine was proposed under heating conditions in toluene. The reaction failed with 2-acetyl pyridine, presumably due to double coordination mediated catalyst poisoning.
Other lanthanide complexes were reported for the C2 functionalization of pyridines. Marks, Kratish, Motta, and co-workers reported C2 borylation with HBpin (36) using a Cp*2LuCH(TMS)2 catalyst (37a).44 The catalyst coordinates with pyridine nitrogen to increase the acidity of the C2 proton, which was deprotonated by the alkyl group on the catalyst. The hydride of borane (39) coordinates with the catalyst after it loses the alkyl ligand, leading to C2 borylation (40) with catalyst–H formation (37b) (Scheme 22).
 | ||
| Scheme 22 Lanthanide catalysed C2 functionalization of pyridines. | ||
Coordination with another substrate releases the product, and Lu–H (37b) acts as an effective catalyst for the subsequent catalytic cycle. The hydride of the catalyst reacts with acidic C2 proton this time, releasing H2 as a by-product during C–H metallated intermediate formation. This protocol was shown to work with substituted pyridines, but isoquinoline led to hydroborylation product formation rather than C2 borylation.
The Xu group also reported C2 selective borylation with pinacol borane in the same year. An alkyl yttrocene catalyst undergoes a similar reaction path for C2 borylation.45
The dehydrogenative pyridine dimerization was known with RANEY® nickel or palladium on charcoal. The dimerization of 4-substituted pyridines with ruthenium or ruthenium/cobalt catalysis was also reported previously with hydrogen evolution. Those methods were generally narrow in the substrate scope. In 2019, Itami, Murakami, and co-workers reported the dehydrogenative synthesis of 2,2′-bipyridyls via C2–H activation and coupling of two pyridine molecules (Scheme 23).
 | ||
| Scheme 23 Pd(II) catalysed deprotonative C–H activation of pyridines for their reductive coupling. | ||
They used Pd(OAc)2 as the catalyst, with a stoichiometric silver salt as an oxidant with an acid additive. The reaction occurred in cyclopropyl methyl ether (CPME) at 140 °C. Pd(II) acetate was proposed to undergo C2–H activation with pyridine (41) via loss of one acetic acid, followed by a second C–H activation to bipyridyl Pd(II) intermediate 42. The reductive coupling led to the formation of the product along with a reduced Pd(0) catalyst, in which the silver salt oxidized back to the Pd(II). The reaction is effective with a variety of pyridine substitutions at different positions.46
Few other functionalizations were reported with an electron withdrawing group attached pyridines and aryl halide. Here, a transition metal undergoes oxidative addition with an aryl halide, followed by ligand exchange with C–H deprotonated pyridine and subsequent reductive coupling to product formation. A concerted metalation–deprotonation (CMD) was proposed for the catalytic process.47 Therefore, this is not technically a transition metal catalysed C–H activation, and the deprotonation of electron-deficient pyridine is crucial for this approach.
The Hiyama group used Lewis acid to coordinate the pyridine nitrogen, followed by nickel-catalyzed C–H alkenylation with alkynes. The Lewis acid activation increases the acidity of the C2 hydrogen for nickel insertion, which leads to C2 functionalization under mild reaction conditions.48 The role of Lewis acid was proposed to be more than just sacrificial coordination to pyridine for its activation, but it enhances the C–H activation step. The combination of Lewis acid and transition metal seems a general solution to tackle the pyridine C–H activation, including its asymmetric alkylations at the C2 position.
Ye, Huang, and co-workers reported a Ni–Al bimetallic catalyst system for the C2–H allylation of pyridines with 1,3-dienes (Scheme 24).49 Phosphine oxides (phosphites) with chiral diamine backbones were effective for asymmetric induction. The bimetallic catalyst synthesis was not required prior to the reaction. All reagents, metal salts, and the ligand were mixed in toluene, and heating at 135 °C resulted in a product with high enantioselectivity.
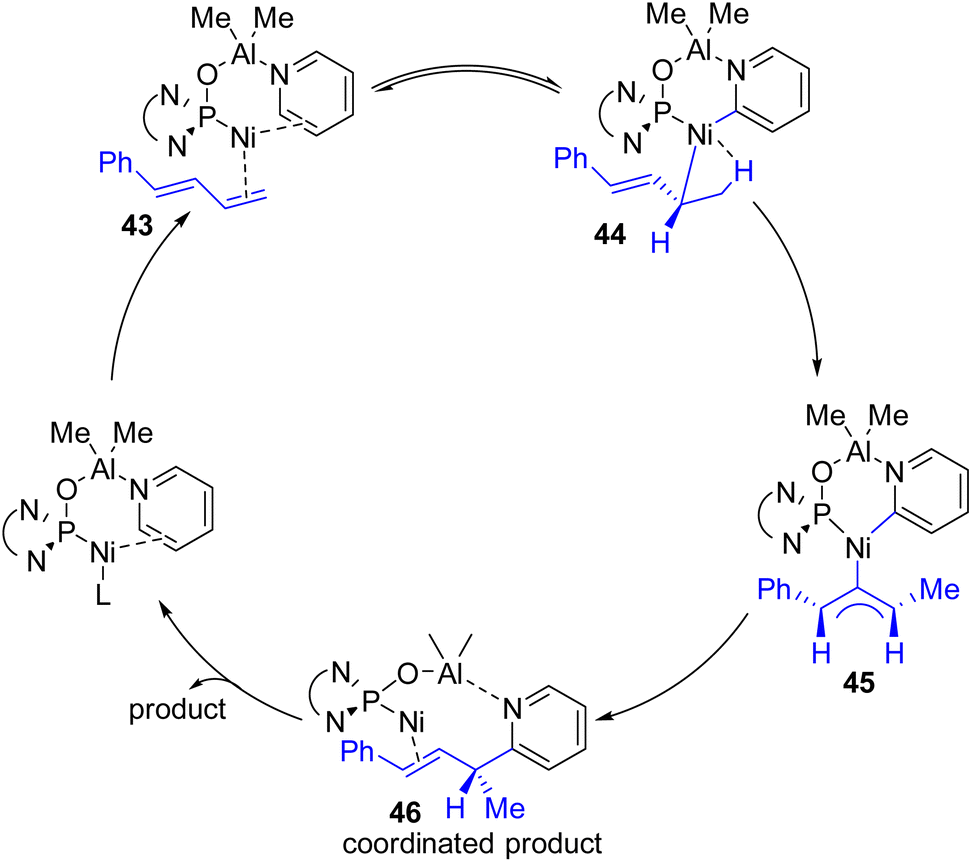 | ||
| Scheme 24 Ni–Al bimetallic complex with the chiral phosphine oxide ligand on nickel for asymmetric C2 allylation. | ||
The reaction was shown to work with aryl-substituted dienes (43), whereas an alkyl-substituted diene resulted in the formation of a major linear product. This bimetallic catalyst system does not need C2 alkylated sterically suitable pyridines for its success. Simple unsubstituted pyridines and a variety of substituents at different positions were successful substrates. The method was successfully applied to quinoline and quinoxaline with moderate yields and enantioselectivities. However, heterocycles such as bipyridines, terpyridines, pyridazines, and imidazoles did not work. It was reasoned that the stronger coordination of Lewis acidic aluminium with these substrates probably inhibited the bimetallic catalyst regeneration.
Shi and co-workers synthesized a bulky N-heterocyclic carbene ligand for Ni(0) (47), which undergoes C–H activation of pyridine and its insertion into an intramolecular alkene (Scheme 25). The combination of stoichiometric bulky Lewis acid and the bulky ligand on nickel led to C–H activation far from the pyridinic nitrogen to give excellent regiocontrol. The C2-symmetric chiral NHC ligand also resulted in high enantioselectivity.50a
 | ||
| Scheme 25 Lewis acid-transition metal bimetallic complex for pyridine C–H functionalization. | ||
Shi, Zhang, and co-workers followed up with C4 selective alkylation with intermolecular substituted styrene. Pyridine, 2-substituted pyridine, and quinolines were functionalized with moderate yields and high enantioselectivities. The reaction with a terminal styrene was less efficient and mainly generated a linear product.50b The computational mechanistic studies suggest that pyridine C–H activation and hydrometallation of alkene occur simultaneously without a distinct metal-hydride intermediate formation. The strong π–π stacking of styrene and aryl fragments of the NHC ligand is crucial for reaction efficiencies and enantioselectivity.
The Nakao group developed C2 selective silylation with silyl hydride as a reagent using a rhodium–aluminum bimetallic complex catalyst.51 The Lewis acidic aluminum coordinates to the pyridine nitrogen, bringing the rhodium catalyst in close proximity to the C2–H. The C–H-activated Rh–H intermediate cannot react with Si–H, and therefore a sacrificial alkene is needed. The alkene coordinates with Rh–H for its hydrometallation and reacts with Si–H to release alkane. The reductive coupling from the pyridine and silyl coordinated Rh intermediate generated the C2 silylpyridine product with the catalyst regeneration.
Tayama and co-workers reported a base-mediated Sommelet Hauser rearrangement of N-(pyridinylmethyl) tetraalkylammonium salts (Scheme 26). The ester stabilized acidic proton of ammonium substitution at C2 of pyridine (50) was deprotonated with a tert-butoxide base, which led to a [2,3] rearrangement. The electron-deficient pyridine ring facilitates the carbanion attack at the C3 position to the non-aromatic intermediate 52. A proton shift to aromatization gave the final C3 substituted product 53 in good yields.52
 | ||
| Scheme 26 [2,3] rearrangement from 2-pyridine ammonium salt for C3 functionalization. | ||
In 2021, the Doria group developed an enzyme-like supramolecular iridium catalyst system for C3 borylation (Scheme 27).53 The designed catalyst geometry of the supramolecular ligand 54 binds both zinc and iridium. The coordination of the pyridine substrate to zinc brings the C3–H in close proximity to the iridium catalyst for its activation and borylation. The substrate scope is relatively limited, with iodide, electron-deficient acid, and amide at the C3 position of pyridine did not give a borylation product. The C4 alkylated substrates were also unsuccessful.
 | ||
| Scheme 27 Bimetallic catalyst with an enzyme-like ligand to coordinate both the Lewis acid and iridium for C3 borylation. | ||
The Yu group focused on the transition metal catalysed direct C3 functionalization of pyridines. The direct C3–H metalation generally proceeds via an electrophilic activation pathway, and the nitrogen coordination of electron-deficient pyridine makes it even harder to achieve. In 2011, they developed a strongly coordinating bidentate ligand for palladium which prevents the metal from being poisoned by pyridine coordination. However, this approach only works with a large excess of pyridines and at a higher concentration.54 Conversely, C2/C4–H functionalization via Lewis acid-transition metal catalysis was developed via an oxidative addition pathway. In 2021, the Yu group utilized the oxidative addition pathway rather than the electrophilic activation mode to achieve C3 alkenylation efficiently (Scheme 28).55
 | ||
| Scheme 28 Bimetallic ligand-directed C3 alkenylation with alkynes. | ||
The inherent C2/C4 reactivity was circumvented via a bifunctional ligand design which coordinated both the transition metal and Lewis acid simultaneously. The aluminum Lewis acid coordination to pyridine directs the nickel (0) to C3–H for metalation (intermediate 55). The bifunctional ligand complex formation was achieved in situ to generate alkenylation products in good yields.
The Ackermann group in 2021 reported a reusable heterogeneous manganese catalyst system for C3 arylation and alkylation of C2-amide pyridines. The bipyridine ligand on the manganese catalyst was anchored on the solid support SBA-15. Both alkylation and arylation were achieved with the corresponding Grignard reagent at 70 °C.56
2. Functionalization via a dual activator and reactive partner
Although pyridine is electron deficient, the loss of aromaticity during nucleophilic addition makes it relatively challenging. As a result, a strong nucleophile is needed for successful addition at an elevated temperature. The nitrogen lone pair of pyridine is not part of the aromaticity and therefore acts as a strong electron donor. Donating nitrogen lone pairs to an activator makes the ring more electron deficient, and nucleophilic addition becomes facile.N-Activated pyridines for radical reactions were well developed mainly due to their dual roles. Stephenson and co-workers treated pyridine N-oxide with acetyl chloride to generate an N-acyloxypyridinium salt. The in situ generated salt undergoes single electron reduction and decarboxylation to generate an alkyl radical that couples with the pyridine ring for the C2 alkylated product.57 Starting with oxidized pyridine makes this alkylation a redox neutral process.
In 2021, the Hong group discovered that an O-aryl oxime pyridinium salt with a photoactive aryloxy group (59) homolytically cleaves under visible light. The generated imine radical (60) undergoes 1,6-intramolecular HAT to form the carbon radical 62, which was added intramolecularly to the pyridinium. The resulting intermediate (63) loses a proton and subsequently oxidizes with an aryloxy radical (61) to form the C2 alkylated pyridinium intermediate 64. Finally, the base mediated C–N bond cleavage led to the alkyl pyridine product (Scheme 29).58
 | ||
| Scheme 29 Intramolecular C2 alkylation of the photoactive O-aryl oxime pyridinium salt. | ||
Hong, Baik, and co-workers later utilized the N-aminopyridinium salt 65 in which the amino group acts as both the activator of pyridine and a radical reagent for β-aminoalkylation of pyridines (Scheme 30). The reaction starts with an electron-deficient aminopyridinium salt (65) undergoing single electron reduction via a photoredox catalyst under visible light.
 | ||
| Scheme 30 N-Aminopyridinium as a pyridine activator and radical generator for photoredox C4 alkylation. | ||
The resulting intermediate gives rise to pyridine after the expulsion of the amine radical 66. The electrophilic amine radical adds to an electron rich alkene, and the resulting nucleophilic alkyl radical (67) attacks the electrophilic C4 position of another pyridinium salt. This radical cation adduct (68) led to the product after deprotonation and expulsion of the amine radical for a radical chain reaction.59 Alternatively, intermediate 68 could be oxidized by Ir(IV) to form intermediate 69. Photocatalytic reduction of 69 by excited Ir(III) led to product formation with the regeneration of an amine radical for reaction progression. The C4 selectivity observed was proposed on the basis of the steric hindrance at C2.
The Hong group continued their dual activation and alkylating reagent strategy with other activators for photoredox functionalization. In 2020, they reported C2 and C4 alkylation using N-alkenoxypyridinium salts and alkenes (Scheme 31).60 The pyridinium salts were synthesized from pyridine N-oxide and terminal alkyne, which acts as a bifunctional reagent.
 | ||
| Scheme 31 N-Alkenoxypyridinium as a pyridine activator and acyl radical generator for photoredox C2 alkylation. | ||
In 2020, the Hong group used a similar strategy for radical three-component coupling to obtain C4 β-trifluoromethyl alkyl pyridines and quinolines. Trifluoromethanesulfonic anhydride was used to activate pyridine, which undergoes single electron reduction by photoexcited eosin Y. The reduced form generates a trifluoromethane radical after the removal of pyridine and SO2. The CF3 radical was added to the alkene, and the following nucleophilic radical was added to another pyridinium triflate.61
In 2022, the Hong group utilized the N-aminopyridinium salt 65 to generate the amine radical 66 that undergoes HAT with alcohol 75 to generate an alkyl radical coupling partner (76). The generated alkyl radical was added to another pyridinium salt. The resulting intermediate (77) was proposed to oxidize by the oxidized photocatalyst (IrIV) for the formation of a C4 alkylated pyridinium salt (78). This salt undergoes reductive elimination to form the amine radical 66 and the final product.62 The scope of the reaction is broad, with a variety of functional groups shown to be tolerated (Scheme 32).
 | ||
| Scheme 32 N-Aminopyridinium as a pyridine activator and HAT reagent for photoredox C4 alkylation. | ||
Maity's research group, in 2019, developed a phosphite catalysed C2 allylation strategy starting with an N-allylpyridinium salt (79).63 The N-allylation activated the pyridine ring for phosphite catalyst (80) addition at C2 to form adduct 81. The subsequent base mediated [2,3] aza-Wittig rearrangement of anionic 81 transferred the allyl group from nitrogen to C2 (82).
The anionic nitrogen intermediate 82 releases the catalyst to form branched allyl pyridines. The use of chital TADDOL-based phosphite led to high enantioselective product formation. Using this methodology, isoquinoline, quinoline, and 4-substituted pyridine substrates successfully formed branched allylation products (Scheme 33). The enantioselectivities were very good for most of the substrates, including fully substituted terminal alkene substrates to all-carbon quaternary centers.
 | ||
| Scheme 33 N-Allylpyridinium as a pyridine activator and branched C2-allylation reagent. | ||
In 2021, they followed up with the phosphite catalysed C–H alkylation of pyridines (Scheme 34). The N-alkylpyridiniums (83) were synthesized from the Zinke salt and amine, and a base-mediated protocol similar to their allylation method gave alkylpyridines. The tandem [1,2] aza-Wittig rearrangement proceeded via solvent-caged radical pair formation and predominantly intramolecular recombination to product formation. As a result, the reaction starting with a chiral amine led to the chiral product with high stereoretention. The stereoretention is solvent dependent, and a racemic starting material with a chiral phosphite catalyst in THF led to the establishment of a catalytic enantioselective version.64 Pyridines with both C2 and C4 positions free for catalyst addition led to minor C4 alkylated products along with major C2 alkylpyridines.
 | ||
| Scheme 34 N-Alkylpyridinium as a pyridine activator and d C2 and C4 alkylation reagent. | ||
The Biju group developed a unique three-component strategy using reactive aryne to functionalize C2–H of pyridine (Scheme 35). The pyridine nitrogen donates electron pairs to aryne (84), and the following anion of the zwitterion (85) was proposed to abstract C2–H to generate carbene species 86. The nucleophilic carbene reacts with the carbonyl compounds, followed by intramolecular SNAr for overall C2 functionalization.65 The method was shown to be applicable to pyridines and electron-rich pyridines.
 | ||
| Scheme 35 Pyridine C–H functionalization via aryne three-component coupling. | ||
3. Functionalization via a sacrificial activator
The dual role of the pyridine activator in C–H activation is elegant but not applicable to all functionalization types. A mild and general approach is to quaternize the pyridine nitrogen to make it a better electrophile. After nucleophile addition, the C–H and the group on nitrogen have to leave to regenerate the pyridine ring. If the activator becomes a good leaving group upon the addition of nucleophiles, then base-mediated E2 elimination reinstalls the pyridine core. The success of this approach depends on selective nucleophile addition to C2 or C4 carbon rather than the electrophilic center of the activator. In the case of certain nucleophiles, such as amines, ring-opening product formation could dominate over the C–H functionalization path. The oxidation of pyridine to its N-oxide, followed by its activation with an activator enjoys a better outcome due to its less electrophilicity and better leaveability, but suffers from step and atom economy. Additionally, functional groups on pyridine prone to oxidative decomposition are not compatible. A variety of functionalization dependent activators starting from pyridine N-oxide have been developed and reviewed recently.66The Corey group reported triflic anhydride as a direct activator, but the efficiencies highly depend on the nature of the nucleophile. For example, electron-rich aromatics as nucleophiles gave moderate to good yields for intermediate formation, but cyanide resulted in a poor yield. The nucleophile-added intermediate to product formation was achieved via a tBuOK base in DMSO with quantitative yields.67
Following Corey's lead, Tan, Li, and co-workers used triflic anhydride as the activator and β-naphthol (89) as a nucleophile to obtain C2 arylation of isoquinolines (Scheme 36). The tautomerization of β-naphthol after nucleophilic addition and in situ K2CO3 mediated triflic acid removal gave the final product in good yields.68
 | ||
| Scheme 36 Triflic anhydride activation for nucleophilic arylation of isoquinoline with β-naphthols. | ||
The Anders group in 1987 reported that pyridine was converted to 4-phosphonium or 2,4-diphosphonium salts upon treatment with triflic anhydride and triphenylphosphine in the presence of trimethylamine base.69 In 2016, the McNally group re-examined the triphenylphosphonium pyridine (93) synthesis.70 They found that the N-triflyl salt undergoes a selective C4 nucleophilic addition to differently substituted pyridines. The choice of the solvent and organic base for subsequent base-mediated triflic acid removal was important to obtain good results. The phosphonium substitution occurred at C2 when the C4 position was blocked. McNally and co-workers studied the subsequent transformations of this phosphonium pyridine 93 for various C–C and C–heteroatom bond formations at the pyridine C4 position (94). In their 2016 report, they treated the phosphonium salt with alcohols in the presence of sodium hydride, which led to the formation of C4 alkoxypyridines. The substitution of other nucleophiles, such as sulfur, nitrogen, and halides, was reported subsequently (Scheme 37).71
 | ||
| Scheme 37 Triflic anhydride activation C4 phosphonium salts for their subsequent functionalization. | ||
The mechanistic examination of the nucleophilic substitution reactions of phosphonium pyridine revealed that the reaction proceeds via the P(V) intermediate 96 rather than direct SNAr substitution for most nucleophiles.71d,e Cross-coupling from P(V) was proposed for product formation, similar to transition metal cross-coupling reactions. With that knowledge, they continued their exploration with phosphonium containing a nucleophile (95) for intramolecular coupling. The suitable second nucleophile, less prone to cross-coupling, assisted the intramolecular cross-coupling product via the P(V) intermediate. Heteroaryl and fluoroalkyl nucleophiles were shown to be effective (Scheme 38).72
 | ||
| Scheme 38 Nucleophilic substitution of pyridine phosphonium via phosphorus ligand couplings. | ||
In 2018, they reported the carbonate assisted incorporation of deuterium and tritium at C4 of pyridine via the intermediate of C4 phosphonium 93 (Scheme 39).73 The carbonate is a poor nucleophile for cross-coupling, and the expulsion of CO2 from carbonate-bound phosphonium 98 led to strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O mediated electrophilic substitution.
O mediated electrophilic substitution.
 | ||
| Scheme 39 Carbonate assisted electrophilic substitution of pyridine phosphonium with deuterium and tritium. | ||
The Manolikakes group used triflic anhydride as the pyridine activating group, followed by the addition of sulfinate salt and base-mediated elimination of triflic acid for C–H sulfonylation. In their first publication, they achieved moderate selectivity for C4 functionalization over C2 with DABCO as the base. In a subsequent report, N-methypiperidine as a base led to improved regiocontrol for C4 sulfonylation. They proposed that a base lone pair interacts with the electrophilic pyridinium intermediate and therefore has an additional role in controlling the regioselectivity of nucleophilic addition.74
In 2017, Fier developed chlorooxime 99 as an in situ activator for C–H functionalization with nucleophiles at C2.75 The chlorooxime is generally good with the cyanide nucleophile but gave poor results with oxygen and carbon nucleophiles (Scheme 40). Amine as a nucleophile did not produce the required product.
 | ||
| Scheme 40 Chlorooxime activating group for direct pyridine C2–H functionalization with nucleophiles. | ||
The Fier group came up with an explanation for the poor yields or no product formation with certain nucleophiles. They subsequently developed a bifunctional activator 102 that contains the amine nucleophile in it for effective C2 amination (Scheme 41).76
 | ||
| Scheme 41 A multifunctional pyridine activating group with a nucleophile for C2–H amination of pyridines. | ||
The Phil Baran group in 2021 reported C4 selective Minisci-type alkylation with a stable N-alkyl salt (106) derived from fumaric acid. Its subsequent Minisci reaction with a carboxylic acid in the presence of a silver nitrate catalyst and ammonium persulfate oxidant gave the C4 alkylated pyridinium salt 107. The N-alkyl group was removed under ambient conditions by treating it with excess DBU in dichloromethane to form the final product (Scheme 42).77a The already activated pyridinium salt (106) does not require typical acid activation for the Minisci reaction.
 | ||
| Scheme 42 A Minisci-type C4 alkylation from fumaric acid derived N-alkylpyridinium salt. | ||
Following the lead from Baran's work, the Noel group utilized the same activator for the radical C4 alkylation of pyridines with alkane as the radical source.77b Benzophenone under 365 nm light irradiation acts as a HAT catalyst for alkane C–H abstraction to form the alkyl radical. The rest of the catalytic cycle is identical to that of Baran's work. The Noel group developed a continuous-flow process for the synthesis of alkylated pyridine from the pyridinium salt 106 in a single set-up. In 2022, Zhang, Yang, and co-workers used redox active N-(acyloxy)phthalimides as the radical coupling partner for the alkylation of 106 using photoredox catalysis under oxidant free conditions.77c
Chen, Fu, and co-workers reported the mono- and di-arylation of N-methylated pyridinium using copper and palladium catalysis. All the reagents, including methylating reagents, aryl bromide coupling partners, and catalysts, were added together with the pyridine and carbonate base for a one-pot process. The reaction was carried out in dimethylacetamide solvent at 150 °C.
In a subsequent report, Fu, Zheng, and co-workers conducted a detailed computational and experimental study to provide a plausible mechanism (Scheme 43).78 Initial N-methylation of pyridine activates it for C2–H insertion by the copper catalyst (108). In parallel, the oxidative insertion of palladium into aryl bromide generated an arylpalladium(II) intermediate. trans-Metallation of pyridyl from copper to palladium to 109, followed by its reductive elimination formed the C2 arylated N-methylpyridinium intermediate. The demethylation also occurred under the reaction conditions via SN2 demethylation with a methyl sulfonate counter anion. The stoichiometry of aryl halide led to either mono or 2,6-diaryl pyridine formation.
 | ||
| Scheme 43 Copper and palladium catalysed mono- and di-arylation of methylpyridinium salt. | ||
Hong and co-workers, in 2022, started with a sacrificial amine activator on pyridine (65) for its decarboxylative alkylation with carboxylic acid.79a The photoexcited acridinium oxidized the carboxylic acid to generate an alkyl radical. The alkyl radical reacts with activated aminopyridinium salt (65). The corresponding intermediate 110 loses a proton and amine radical (66) to yield the 4-alkylated pyridine product. The amine radical 66 undergoes single electron reduction with the reduced photocatalyst to regenerate acridinium (Scheme 44).
 | ||
| Scheme 44 Photocatalytic decarboxylative alkylation of N-aminopyridinium salt. | ||
Zhang, Neu, and co-workers found that N-methoxy pyridinium salts can form an EDA complex with alkyl sulfoxide in the presence of potassium fluoride. The 455 nm light irradiation of LED light to this EDA complex led to methoxy radical formation, which reacts with an alkyl sulfoxide to form an alkyl radical and sulfinate by-product. The following reaction of the alkyl radical with the N-methoxy pyridinium salt gave C2 alkylated pyridine with methoxy radical generation for the radical chain reaction.79b In 2021, Zipse, Renaud, and co-workers reported alkyl radical addition to N-methoxy pyridinium salts for C2 alkylated pyridine formation.79c
In 2022, the Hong group utilized the same sacrificial activator for asymmetric C4 alkylation with chiral NHC catalysis under blue light irradiation (Scheme 45).80 Sodium pivalate hydrate was used as an additive, and forms an EDA complex with the aminopyridinium salt (65) that could generate an amine radical (66). The NHC added intermediate of enal (111) undergoes single electron oxidation with 65 to form radical intermediate 112. The reaction of 112 with activated pyridine 65 formed 103, and the elimination of the proton and amine radical from it resulted in the final product. The chiral NHC catalyst determines the stereochemistry.
 | ||
| Scheme 45 Enantioselective C4 functionalization of N-aminopyridinium salts via NHC catalysis. | ||
When the N-activating group is not a good leaving group, oxidation of the corresponding N-substituted dihydropyridine is required. For example, the acyl activation of pyridine allows soft organocuprate and silylcuprate to add under mild conditions with C4 selectivity. The corresponding intermediate is stable, and oxidation with elemental sulfur in boiling toluene was required for the oxidation and removal of N-acyl to C4 functionalized pyridines.81
Kanai, Kuninobu, and co-workers showed that a stoichiometric amount of triarylborane Lewis acid could activate the pyridines for nucleophilic addition by trifluoroalkylsilanes. The corresponding intermediate was shown to be oxidized with different oxidants for trifluoroalkylpyridines. In 2023, the Lakhdar group reported that BF3·OEt2 can be a compatible stoichiometric activator for C2 and C4 phosphonation of pyridines with phosphites. Chloronil was used as the subsequent oxidant.82
The Wang group, in 2022, disclosed an overall double and triple nucleophile substitution in a stepwise fashion (Scheme 46).83
 | ||
| Scheme 46 Double and triple functionalization of pyridine via sequential functionalization and final oxidative aromatization. | ||
Initial activation of pyridine with stoichiometric BF3·OEt2 and Grignard addition led to the formation of 4-substituted dihydropyridine. Then dihydropyridine was treated with trifluoroacetic acid for the formation of enamine-iminium, and trapping the iminium with an N-substituted indole for the formation of C2 added tetrahydropyridine intermediate (114). The authors established that DDQ oxidation of tetrahydropyridine could double oxidize it back to 2,4-substituted pyridines. With excess TFA and indole, the other enamine can also react similarly to form a 2,4,6-trifunctionalized piperidine intermediate (115). The piperidine intermediate was also oxidized with DDQ to generate trisubstituted pyridines.
The C2/C4 C–H functionalization of pyridine predominates over C3, which remains difficult to achieve. The Lewis acid catalysed hydroboration and hydrosilylation of pyridines were known and generally functionalized further for substituted tetrahydropyridine or piperidine synthesis.84 Wang and co-workers utilized the nucleophilic reactivity of 1,4-dihydropyridine to react it with an electrophile and subsequent oxidative aromatization for the formation of C3 functionalized pyridines (Scheme 47).85 The one-pot tandem reaction sequence was conducted sequentially to avoid the reduction of the electrophile rather than the pyridine itself and the oxidation of dihydropyridine in the last step rather than borane. The mechanistic studies reveal that the triarylborane Lewis acid catalyses both the hydroboration of pyridine and its subsequent reaction with the imine and ketone electrophile. The initial hydroboration is reversible, and thermodynamic equilibrium was reached between 1,2- and 1,4-dihydropyridines. Since 1,2-dihydropyridines are also nucleophilic at C3 and C5 positions, they can also react with the electrophile to generate C3 functionalized pyridines. Two equivalent electrophiles were used, where the second equivalent oxidized the C3 functionalized dihydropyridine 117 to the product. C4 substituted pyridines did not work with imines and ketones, although initial hydroboration led to 1,2-dihydropyridine. The authors suspect that steric hindrance might be the reason for this failure, and a smaller and stronger iminium electrophile led to successful C3 substitution.
 | ||
| Scheme 47 Lewis acid catalysed HBpin mediated C3 functionalization of pyridines with CX electrophiles. | ||
The Kuninobu group developed the C3 trifluoromethylation of pyridines using reduction to dihydropyridine 118, followed by reacting it with an electrophilic trifluoromethylating reagent for C3 functionalization (119). Finally, oxidation of the C3 functionalized dihydropyridine 119 led to the formation of the product.86 The whole sequence was achieved in a single-pot reaction with a single solvent (Scheme 48).
 | ||
| Scheme 48 Lewis acid catalysed silane mediated C3 trifluoromethylation of pyridines with a DDQ oxidant. | ||
Wang's group reported a conceptually similar approach for C3 fluorothiolation. The reaction is versatile for mono- and difunctionalization of a variety of pyridine, quinoline, and isoquinoline derivatives (Scheme 49).87
 | ||
| Scheme 49 Lewis acid catalysed hydroborane mediated C3 fluorothiolation of pyridines under aerobic oxidation. | ||
In 2023, Wang, Xu, and co-workers reported C3 cyanation with an electrophilic cyanation reagent following similar tandem processes.88a In this case, most of the electrophilic cyanation reagents failed to give good yields. The authors reasoned that the oxidizing power of the cyanation reagent could oxidize the dihydropyridine intermediate rather than provide “CN+”. To circumvent this problem, they screened a variety of reagents and the reaction with 2,2-bis-(4-cyanatophenyl)propane with the lowest oxidation potential led to good results. The final oxidation of cyano-substituted dihydropyridine was achieved with DIAD. A large variety of 3-cyano products were formed from substituted pyridine, quinoline, and isoquinolines.
Very recently, the Wang group developed a dual catalysis mode to form asymmetric C3 allylation of pyridines.88b Their BAr3 catalysed hydroboration forms 116, as reported earlier. In this report, they found that the nucleophilic 116 can react with an Ir–π–allyl electrophile to form C3 allylated dihydropyridine 117. The crude reaction mixture was stirred under air at room temperature, which oxidized the intermediate to the pyridine product. The well-developed chiral BINOL derived phosphoramide ligand on iridium induces high enantioselectivity in the product.
In 2022, both the Studer group and McNally, Paton, and co-workers separately came up with their redox neutral strategies, which avoid the reductive dearomatiozation–oxidative rearomatization protocol. The Studer group used the three-component coupling of pyridines with dimethyl acetylenedicarboxylate (DMAD) and methyl pyruvate (MP) to form an oxazinopyridine intermediate (120).89 This intermediate, 120, behaves as a dienamine and reacts with ionic and radical electrophiles. The C3 functionalized intermediate 121 could be converted back to the C3 functionalized pyridine under acidic removal of activating groups (Scheme 50). This protocol used fluoroalkyl iodides for fluoroalkylation under blue light irradiation. The electron-rich oxazinopyridine (120) forms an EDA complex with electron-deficient iodide, which initiates radical functionalization.
 | ||
| Scheme 50 Pyridine activation and its radical and ionic C3 functionalization. | ||
Additionally, ionic electrophilic halogenation with NCS, NBS, and NIS led to the C3 halogenation product. Functionalization at C3 and C5 of the dienamine-type intermediate is feasible, and the sequential addition of different electrophiles led to the formation of a regioselective difunctionalization product.
McNally and co-workers utilized the Zincke salt ring opening type reactivity of pyridinium with an amine to generate an electron-rich acyclic intermediate.90 They reacted pyridine with triflic anhydride to pyridinium salt and treated it with a secondary amine to the ring opening intermediate (122). The ring-opening intermediate is electron-rich and reacts with NCS, NBS, and NIS for its halogenation at C3 and/or C5 positions. The removal of triflate from nitrogen and ring-closure were achieved under acidic conditions to reinstall the pyridine ring with 3 and 3,5-disubstitution (Scheme 51). Like the Studer approach, sequential electrophilic substitution led to the formation of regioisomeric dihalogenation product.
 | ||
| Scheme 51 Triflic anhydride mediated C3 halogenation of pyridines via a Zincke imine intermediate. | ||
4. Conclusions
In this review, we classified various types of pyridine C–H activation modes. Owing to its electro-deficient nature and coordination ability, SEAr at C3 occurred only at higher temperatures with a limited scope. SNAr for C–H activation requires hydride as a leaving group and, although discovered a century ago, it did not find widespread application. Transition metal catalysed C–H functionalization of pyridine is generally difficult compared to other aryl C–H functionalizations. Lanthanide complexes turned out to be very effective for pyridine C2–H activation. A combination of dual Lewis acid-transition metal catalysis was recently developed for effective and regioselective C–H activation. Catalytic or stoichiometric pyridine nitrogen activation is the most common and efficient approach for its C–H reactivity. The choice of ligands and metals proved effective in functionalizing all 2, 3, and 4 positions regioselectively. The radical reaction in line with the Minisci reaction, or with activated pyridinium substrates, became one of the most frequently used techniques for various C2 and C4 functionalizations. Many pyridine activating functional groups were chosen that act as dual reagents for activation, followed by subsequent functionalization. Very recently, the conversion of pyridine to dihydropyridine, followed by its C3 functionalization and rearomatization, led to overall C3 functionalization. Significant progress has been made in the last five years, and it is expected to grow faster owing to the importance of functionalized pyridines.Author contributions
Susmita Maity, Asish Bera, and Ayantika Bhattacharjya researched the relevant papers. Susmita drafted the direct functionalization part, Asish drafted the functionalization via the dual activator and reactive partner part, and Ayantika drafted the part on functionalization via sacrificial activators. Pradip Maity: conceptualization, resources, supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the SERB (CRG/2019/001065 and SCP/2022/000465). Susmita and Ayantika thank the SERB for a fellowship and Asish thanks the CSIR for a fellowship. We greatly acknowledge the CSIR-NCL for providing infrastructure facilities.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–11027 CrossRef CAS PubMed.
- S. Pal, Pyridine, 2018, ch. 5, ISBN 978-1-78923-423-7. DOI:10.5772/intechopen.76986.
- (a) P. Patil, S. P. Sethy, T. Sameera and K. Shailaja, Asian J. Res. Chem., 2013, 6, 888–899 Search PubMed; (b) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC.
- R. Gujjarappa, N. Vodnala and C. C. Malakar, ChemistrySelect, 2020, 5, 8745–8758 CrossRef CAS.
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita and H. Ichimura, Pyridine and Pyridine Derivatives, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113(5), 3084–3213 CrossRef CAS PubMed; (b) C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114(21), 10829–10868 CrossRef CAS PubMed; (c) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed; (d) X. L. Liu, L. B. Jiang, M. P. Luo, Z. Ren and S. G. Wang, Org. Chem. Front., 2022, 9, 265–280 RSC.
- (a) H. M. L. Davies, J. Du Bois and J. Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC; (b) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- (a) D. E. Pearson, W. W. H. Judith, K. T. Chaw and B. R. Suthers, J. Org. Chem., 1961, 26, 789–792 CrossRef CAS; (b) L. I. Belen'kii, Chem. Heterocycl. Compd., 1986, 22, 587–608 CrossRef.
- S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7685 CrossRef CAS PubMed.
- (a) L. Davin, W. Clegg, A. R. Kennedy, M. R. Probert, R. McLellan and E. Hevia, Chem. – Eur. J., 2018, 24, 14830–14835 CrossRef CAS PubMed; (b) M. Balkenhohl, H. Jangra, I. S. Makarov, S.-M. Yang, H. Zipse and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 14992–14999 CrossRef CAS PubMed.
- A. E. Chichibabin and O. A. Zeide, J. Russ. Phys.-Chem. Soc., 1914, 46, 1216–1236 CAS.
- Y. Wang, R. Li, W. Guan, Y. Li, X. Li, J. Yin, G. Zhang, Q. Zhang, T. Xiong and Q. Zhang, Chem. Sci., 2020, 11, 11554–11561 RSC.
- P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956–960 CrossRef CAS PubMed.
- Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef CAS PubMed.
- C. Obradors and B. List, J. Am. Chem. Soc., 2021, 143, 6817–6822 CrossRef CAS PubMed.
- Q. Chen, X. M. Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958–4961 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 4411–4416 CrossRef CAS.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- C. Wu, T. Ying, H. Fan, C. Hu, W. Su and J. Yu, Org. Lett., 2023, 25, 2531–2536 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- W. Liu, X. Yang, Z. Z. Zhou and C. J. Li, Chem, 2017, 2, 688–702 CAS.
- B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878–16883 CrossRef CAS PubMed.
- S. P. Pitre, M. Muuronen, D. A. Fishman and L. E. Overman, ACS Catal., 2019, 9, 3413–3418 CrossRef CAS.
- (a) M. G. Braun, G. Castanedo, L. Qin, P. Salvo and S. Z. Zard, Org. Lett., 2017, 19(15), 4090–4093 CrossRef CAS PubMed; (b) V. L. Revil-Baudard, J. P. Vors and S. Z. Zard, Org. Lett., 2018, 20(12), 3531–3535 CrossRef CAS PubMed.
- (a) X. Y. Zhang, W. Z. Weng, H. Liang, H. Yang and B. Zhang, Org. Lett., 2018, 20, 4686–4690 CrossRef CAS PubMed; (b) J. M. Anderson, N. D. Measom, J. A. Murphy and D. L. Poole, Org. Lett., 2023, 25, 2053–2057 CrossRef CAS PubMed.
- (a) J. Xu, C. Liang, J. Shen, Q. Chen, W. Li and P. Zhang, Green Chem., 2023, 25, 1975–1981 RSC; (b) S. Xu, W. Zhang, C. Li, Y. Li, H. Zeng, Y. Wang, Y. Zhang and D. Niu, Angew. Chem., Int. Ed., 2023, 62, e202218303 CrossRef CAS PubMed.
- L. Zhang and Z. Q. Liu, Org. Lett., 2017, 19, 6594–6597 CrossRef CAS PubMed.
- (a) B. Liang, Q. Wang and Z. Q. Liu, Org. Lett., 2017, 19, 6463–6465 CrossRef CAS PubMed; (b) Z. Liu and Z. Q. Liu, Org. Lett., 2017, 19, 5649–5652 CrossRef CAS PubMed; (c) C. Wang, S. Song, Z. Chen, D. Shen, Z. Wang, J. Zhou, J. Guo and J. Li, J. Org. Chem., 2022, 87, 16794–16806 CrossRef CAS PubMed.
- K. Liao, F. Wu, J. Chen and Y. Huang, Cell Rep. Phys. Sci., 2022, 3, 100763 CrossRef CAS.
- A. Ghosh, P. Pyne, S. Ghosh, D. Ghosh, S. Majumdar and A. Hazra, Green Chem., 2022, 24, 3056–3080 RSC.
- (a) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed; (b) N. Okugawa, K. Moriyama and H. Togo, J. Org. Chem., 2017, 82, 170–178 CrossRef CAS PubMed.
- C. Bosset, H. Beucher, G. Bretel, E. Pasquier, L. Queguiner, C. Henry, A. Vos, J. P. Edwards, L. Meerpoel and D. Berthelot, Org. Lett., 2018, 20, 6003–6006 CrossRef CAS PubMed.
- E. L. Saux, E. Georgiou, I. A. Dmitriev, W. C. Hartley and P. Melchiorre, J. Am. Chem. Soc., 2023, 145, 47–52 CrossRef PubMed.
- (a) K. J. Kim, T. Constantin, M. Simonetti, J. Llaveria, N. S. Sheikh and D. Leonori, Nature, 2021, 595, 677–683 CrossRef PubMed; (b) A. Basak, S. Karak and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 7592–7599 CrossRef CAS PubMed.
- G. Q. Sun, P. Yu, W. Zhang, Y. Wang, L. L. Liao, Z. Zhang, L. Li, Z. Lu, D. G. Yu and S. Lin, Nature, 2023, 615, 67–73 CrossRef CAS PubMed.
- (a) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS PubMed; (b) R. S. J. Proctor, P. Chuentragool, A. C. Colgan and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 4928–4934 CrossRef CAS PubMed.
- X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Org. Lett., 2018, 20, 6298–6301 CrossRef CAS PubMed.
- A. C. Colgan, R. S. J. Proctor, D. C. Gibson, P. Chuentragool, A. S. K. Lahdenpera, K. Ermanis and R. J. Phipps, Angew. Chem., Int. Ed., 2022, 61, e202200266 CrossRef CAS PubMed.
- M. P. Luo, Y. J. Gu and S. G. Wang, Chem. Sci., 2023, 14, 251–256 RSC.
- R. F. Jordan and D. F. Taylor, J. Am. Chem. Soc., 1989, 111, 778–779 CrossRef CAS.
- (a) S. Rodewald and R. F. Jordan, J. Am. Chem. Soc., 1994, 116, 4491–4492 CrossRef CAS; (b) B. T. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086–18089 CrossRef CAS PubMed; (c) G. Song, W. N. O. Wylie and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209–12212 CrossRef CAS PubMed.
- R. Grigg and V. Savic, Tetrahedron Lett., 1997, 38, 5737–5740 CrossRef CAS.
- J. O. Rothbaum, A. Motta, Y. Kratish and T. J. Marks, J. Am. Chem. Soc., 2022, 144, 17086–17096 CrossRef CAS PubMed.
- Y. Luo, S. Jiang and X. Xu, Angew. Chem., Int. Ed., 2022, 61, e202117750 CAS.
- S. Yamada, T. Kaneda, P. Steib, K. Murakami and K. Itami, Angew. Chem., Int. Ed., 2019, 58, 8341–8345 CrossRef CAS PubMed.
- (a) H. Q. Do and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 1128–1129 CrossRef CAS PubMed; (b) X. Zhang, S. Fan, C. Y. He, X. Wan, Q. Q. Min, J. Yang and X. Z. Jiang, J. Am. Chem. Soc., 2010, 132, 4506–4507 CrossRef CAS PubMed; (c) P. Guo, J. M. Joo, S. Rakshit and D. Sames, J. Am. Chem. Soc., 2011, 133, 16338–16341 CrossRef CAS PubMed; (d) O. Rene and K. Fagnou, Org. Lett., 2010, 12, 2116–2119 CrossRef CAS PubMed; (e) C. Z. Wu, C. Y. He, Y. Huang and X. Zhang, Org. Lett., 2013, 15, 5266–5269 CrossRef CAS PubMed.
- Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 CrossRef CAS PubMed.
- J.-F. Li, D. Pan, H.-R. Wang, T. Zhang, Y. Li, G. Huang and M. Ye, J. Am. Chem. Soc., 2022, 144, 18810–18816 CrossRef CAS PubMed.
- (a) W. B. Zhang, X. T. Yang, J. B. Ma, Z. M. Su and S. L. Shi, J. Am. Chem. Soc., 2019, 141, 5628–5634 CrossRef CAS PubMed; (b) J. B. Ma, X. Zhao, D. Zhang and S. L. Shi, J. Am. Chem. Soc., 2022, 144(30), 13643–13651 CrossRef CAS PubMed.
- N. Hara, N. Uemura and Y. Nakao, Chem. Commun., 2021, 57, 5957–5960 RSC.
- E. Tayama, G. Shimizu and R. Nakao, Tetrahedron, 2022, 111, 132721 CrossRef CAS.
- J. Trouve, P. Zardi, S. Al-Shehimy, T. Roisnel and R. G. Doria, Angew. Chem., Int. Ed., 2021, 60, 18006–18013 CrossRef CAS PubMed.
- M. Ye, G.-L. Gao and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 6964–6967 CrossRef CAS PubMed.
- T. Zhang, Y.-X. Luan, N. Y. S. Lam, J.-F. Li, Y. Li, M. Ye and J.-Q. Yu, Nat. Chem., 2021, 13, 1207–1213 CrossRef CAS PubMed.
- I. Choi, Z. Shen, E. Ronge, V. Karius, C. Jooss and L. Ackermann, Chem. – Eur. J., 2021, 27, 12737–12741 CrossRef CAS PubMed.
- A. C. Sun, E. J. McClain, J. W. Beatty and C. R. J. Stephenson, Org. Lett., 2018, 20, 3487–3490 CrossRef CAS PubMed.
- B. Kweon, C. Kim, S. Kim and S. Hong, Angew. Chem., Int. Ed., 2021, 60, 26813–26821 CrossRef CAS PubMed.
- Y. Moon, B. Park, I. Kim, G. Kang, S. Shin, D. Kang, M.-H. Baik and S. Hong, Nat. Commun., 2019, 10, 4117 CrossRef PubMed.
- G. R. mathi, Y. Jeong, Y. Moon and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 2049–2054 CrossRef CAS PubMed.
- K. Lee, S. Lee, N. Kim, S. Kim and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 13379–13384 CrossRef CAS PubMed.
- M. Vellakkaran, T. Kim and S. Hong, Angew. Chem., Int. Ed., 2022, 61, e202113658 CrossRef CAS PubMed.
- A. Motaleb, S. Rani, T. Das, R. G. Gonnade and P. Maity, Angew. Chem., Int. Ed., 2019, 58, 14104–14109 CrossRef CAS PubMed.
- S. Rani, S. R. Dash, A. Bera, M. N. Alam, K. Vanka and P. Maity, Chem. Sci., 2021, 12, 8996–9003 RSC.
- A. Guin, S. Bhattacharjee and A. T. Biju, Chem. – Eur. J., 2021, 27, 13864–13869 CrossRef CAS PubMed.
- D. Wang, L. Desaubry, G. Li, M. Huang and S. Zheng, Adv. Synth. Catal., 2021, 363, 2–39 CrossRef CAS.
- E. J. Corey and Y. Tian, Org. Lett., 2005, 7(24), 5535–5537 CrossRef CAS PubMed.
- P.-Y. Jiang, K.-F. Fan, S. Li, S.-H. Xiang and B. Tan, Nat. Commun., 2021, 2384 CrossRef CAS PubMed.
- E. Anders and F. Markus, Tetrahedron Lett., 1987, 28, 2675–2676 CrossRef CAS.
- M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806 CrossRef CAS PubMed.
- (a) R. D. Dolewski, M. C. Hilton and A. Mcnally, Synlett, 2018, 29, 8–14 CrossRef CAS; (b) R. G. Anderson, B. M. Jett and A. McNally, Tetrahedron, 2018, 74, 3129–3136 CrossRef CAS PubMed; (c) C. Patel, M. Mohnike, M. C. Hilton and A. McNally, Org. Lett., 2018, 20, 2607–2610 CrossRef CAS PubMed; (d) R. G. Anderson, B. M. Jett and A. McNally, Angew. Chem., Int. Ed., 2018, 57, 12514 CrossRef CAS PubMed; (e) J. N. Levy, J. V. Alegre-Requena, R. Liu, R. S. Paton and A. McNally, J. Am. Chem. Soc., 2020, 142, 11295 CrossRef CAS PubMed; (f) R. D. Dolewski, P. J. Fricke and A. McNally, J. Am. Chem. Soc., 2018, 140, 8020 CrossRef CAS PubMed.
- (a) M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Science, 2018, 362, 799 CrossRef CAS PubMed; (b) B. T. Boyle, M. C. Hilton and A. McNally, J. Am. Chem. Soc., 2019, 141, 15441 CrossRef CAS PubMed; (c) X. Zhang, K. G. Nottingham, C. Patel, J. V. Alegre-Requena, J. N. Levy, R. S. Paton and A. McNally, Nature, 2021, 594, 217 CrossRef CAS PubMed.
- J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS PubMed.
- (a) M. Friedrich and G. Manolikakes, Eur. J. Org. Chem., 2022, e202200915 CAS; (b) M. Friedrich, L. Schluz, K. Hofman, R. Zangl, N. Morgner, S. Shaaban and G. Manolikakes, Tetrahedron Chem., 2022, 1, 100003 CrossRef.
- P. S. Fier, J. Am. Chem. Soc., 2017, 139, 9499–9502 CrossRef CAS PubMed.
- P. S. Fier, S. Kim and R. D. Cohen, J. Am. Chem. Soc., 2020, 142, 8614–8618 CrossRef CAS PubMed.
- (a) J. Choi, G. Laudadio, E. Godineau and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 11927–11933 CrossRef CAS PubMed; (b) J. S. Orduna, R. C. Silva, F. Raymenants, B. Reus, J. Thaens, K. T. de Oliveira and T. Noel, Chem. Sci., 2022, 13, 12527–12532 RSC; (c) Z. Zhang, Q. He, X. Zhang and C. Yang, Org. Biomol. Chem., 2022, 20, 1969–1973 RSC.
- (a) C. Lin, D. Li, B. Wang, J. Yao and Y. Zhang, Org. Lett., 2017, 19, 1970–1973 CrossRef PubMed; (b) F. Jia, C. Yin, Y. Zheng, R. Sun, Y.-C. Ge, D. Xu, R. Li, H. Chen, C. Zheng and H. Fu, J. Org. Chem., 2018, 83, 10389–10397 CrossRef CAS PubMed.
- (a) C. Kim, J. Jeong, M. Vellakkaran and S. Hong, ACS Catal., 2022, 12, 13225–13233 CrossRef CAS; (b) D. Xie, Y. Wang, X. Zhang, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2022, 61, e202204922 CAS; (c) S. Rieder, C. Melendez, F. Denes, H. Jangra, K. Mulliri, H. Zipse and P. Renaud, Chem. Sci., 2021, 12, 15362–15373 RSC.
- H. Choi, G. R. Mathi, S. Hong and S. Hong, Nat. Commun., 2022, 13, 1776 CrossRef CAS PubMed.
- D. L. Comins, L. S. King, E. D. Smith and F. C. Fevrier, Org. Lett., 2005, 7, 5059–5062 CrossRef CAS PubMed.
- (a) M. Nagase, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 6103–6106 CrossRef CAS PubMed; (b) V. Quint, T. H. V. Nguyen, G. Mathieu, S. Chelli, M. Breugst, J.-F. Lohier, A.-C. Gaumont and S. Lakhdar, ACS Org. Inorg. Au, 2023, 3, 151–157 CrossRef CAS PubMed.
- D. Wang, L. Xu, S. Zheng and X. Yang, Adv. Synth. Catal., 2022, 364, 2720–2728 CrossRef CAS.
- (a) X. Fan, J. Zheng, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2015, 137, 4916–4919 CrossRef CAS PubMed; (b) N. Gandhamsetty, S. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 15176–15184 CrossRef CAS PubMed; (c) Z.-Y. Liu, Z.-H. Wen and X.-C. Wang, Angew. Chem., Int. Ed., 2017, 56, 5817–5820 CrossRef CAS PubMed; (d) J.-J. Tian, Z.-Y. Yang, X.-S. Liang, N. Liu, C.-Y. Hu, X.-S. Tu, X. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2020, 59, 18452–18456 CrossRef CAS PubMed.
- Z. Liu, J.-H. He, M. Zhang, Z.-J. Shi, H. Tang, X.-Y. Zhou, J.-J. Tian and X.-C. Wang, J. Am. Chem. Soc., 2022, 144, 4810–4818 CrossRef CAS PubMed.
- R. Muta, T. Torigoe and Y. Kuninobu, Org. Lett., 2022, 24, 8218–8222 CrossRef CAS PubMed.
- X.-Y. Zhou, M. Zhang, Z. Liu, J.-H. He and X.-C. Wang, J. Am. Chem. Soc., 2022, 144, 14463–14470 CrossRef CAS PubMed.
- (a) M. Zhang, Q. Zhou, H. Luo, Z.-L. Tang, X. Xu and X.-C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216894 CrossRef CAS PubMed; (b) Z. Liu, Z.-J. Shi, L. Liu, M.-C. Zhang, H.-Y. Guo and X.-C. Wang, J. Am. Chem. Soc., 2023, 145, 11789–11797 CrossRef CAS PubMed.
- H. Cao, Q. Cheng and A. Studer, Science, 2022, 378, 779–785 CrossRef CAS PubMed.
- B. T. Boyle, J. N. Levy, L. de Lescure, R. S. Paton and A. McNally, Science, 2022, 378, 773–779 CrossRef CAS PubMed.
Footnote |
| † All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |