
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of oxetane and azetidine ethers as ester isosteres by Brønsted acid catalysed alkylation of alcohols with 3-aryl-oxetanols and 3-aryl-azetidinols†
Peerawat
Saejong
 a,
Juan J.
Rojas
a,
Camille
Denis
ab,
Andrew J. P.
White
a,
Anne Sophie
Voisin-Chiret
b,
Chulho
Choi
c and
James A.
Bull
*a
a,
Juan J.
Rojas
a,
Camille
Denis
ab,
Andrew J. P.
White
a,
Anne Sophie
Voisin-Chiret
b,
Chulho
Choi
c and
James A.
Bull
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: j.bull@imperial.ac.uk
bNormandie Univ, UNICAEN, CERMN, 14000 Caen, France
cPfizer Global Research and Development, 445 Eastern Point Rd., Groton, CT 06340, USA
First published on 15th June 2023
Abstract
Oxetanes and azetidines continue to draw significant interest in medicinal chemistry, as small, polar and non-planar motifs. Oxetanes also represent interesting surrogates for carbonyl-containing functional groups. Here we report a synthesis of 3,3-disubstituted oxetane- and azetidine-ethers, with comparisons made to the ester functional group. The tertiary benzylic alcohols of the 4-membered rings are selectively activated using Brønsted acid catalysis and reacted with simple alcohols to form the ethers and maintain the oxetane ring intact. This approach avoids the use of strong bases and halide alkylating agents and allows alcohol libraries to be leveraged. Oxetane ethers demonstrate excellent chemical stability across a range of conditions and an improved stability vis-à-vis analogous esters under basic and reducing conditions.
Introduction
Medicinal chemistry programs can benefit from the introduction of small motifs that access new chemical- and intellectual property space, and offer advantageous physico–chemical properties. Small 4-membered heterocyclic rings, oxetanes and azetidines, present attractive motifs in this regard due to their high polarity, low molecular weight and non-planar features.1,2 In 2006, Carreira compared 3,3-disubstituted oxetanes to gem-dimethyl groups, to provide improved metabolic stability and polarity.3 Later, the comparison with alkyl-ketones was made, whereby the oxetane ring presented similar polarity, H-bonding capacity and lone pair orientation to the carbonyl group.4 Since then, oxetanes have been studied as possible replacement motifs for several carbonyl-containing functional groups such as amides, aryl-ketones, thioesters and carboxylic acids (Fig. 1a),5–8 and are increasingly employed in medicinal chemistry. Their development has been accompanied with considerable advances in the synthesis of oxetane motifs.1,3–9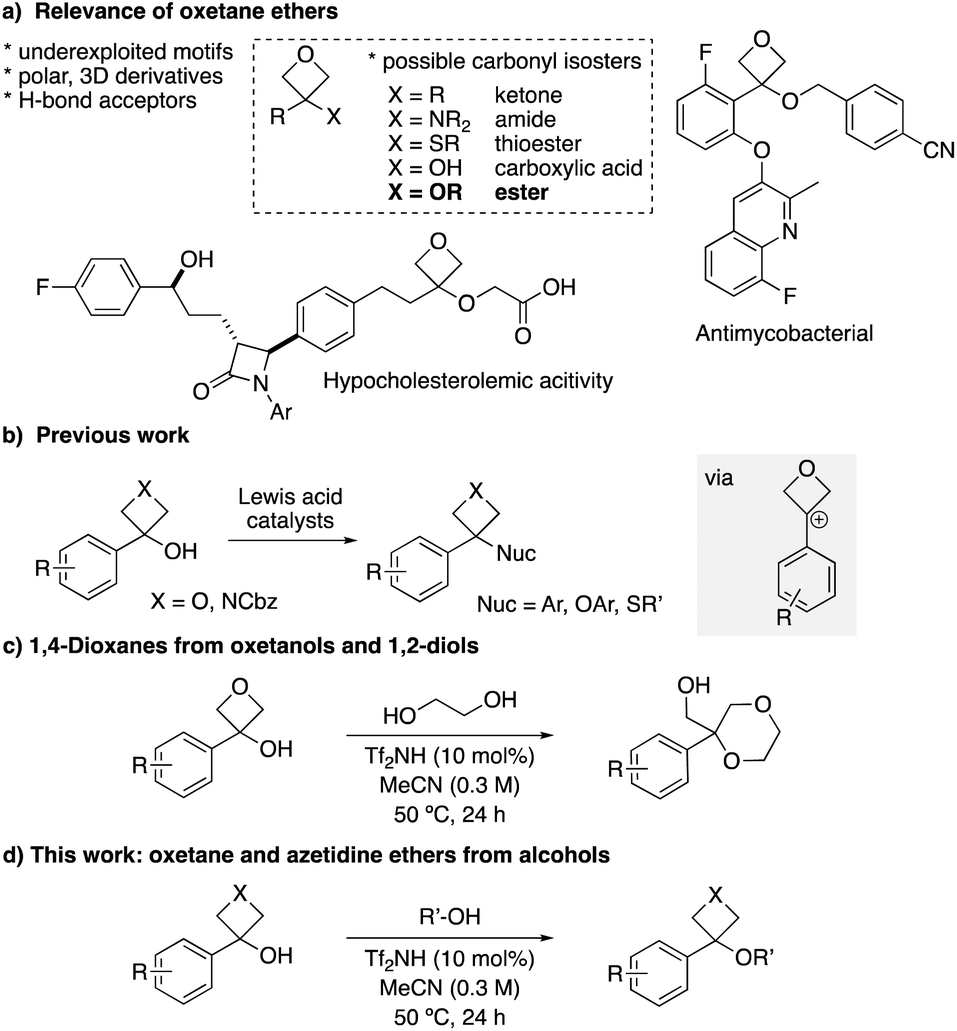 | ||
| Fig. 1 Oxetane and azetidine ethers. | ||
While the comparison of oxetane ethers with esters has been postulated,4 this remains to be exemplified. Indeed, to date there are few studies that prepare10 the oxetane ether motif and/or investigate their stability.11 Very recently, 3,3-disubstituted oxetane benzyl ethers were reported to display antimycobacterial activity,12 and have also appeared in the patent literature in compounds for the reduction of cholesterol levels (Fig. 1a).13
Azetidine ether motifs have been prepared by SN2 reaction of azetidinols with alkyl halides, or by Mitsunobu reactions.14 3,3-Disubstituted azetidine ethers showed biological activity relevant to cancer and SLE (systemic lupus erythematosus).14f,g 3,3-Disubstituted oxetane ethers have also been exploited as intermediates in the synthesis of heterocycles through intramolecular oxetane opening.15,16 In these cases, the ethers are typically prepared from oxetanols with strong bases and halide alkylating agents. We have recently developed catalytic approaches to use benzylic oxetanols and azetidinols as alkylating agents for the preparation of 3-aryl-3-functionalised-oxetanes and azetidines via carbocation intermediates (Fig. 1b).17–19,20 Lewis acid catalysts (Li+, Ca2+ and Fe3+ salts) were successful for selective alcohol activation for Friedel–Crafts alkylation reactions and reactions with thiols. Interestingly, in the Friedel–Crafts reaction with phenols, aryl oxetane ethers were identified as reversible off-cycle intermediates.17c We envisaged that the use of aliphatic alcohols as nucleophiles in a related process would provide an advantageous approach to the preparation of oxetane ethers, avoiding the use of alkyl halides. Recently, we reported the annulation of aryl-oxetanols with 1,2-diols to form 1,4-dioxanes (Fig. 1c).16,20 In this instance we proposed an intermediate oxetane ether and intramolecular oxetane opening, but this ether intermediate was neither isolated nor observed spectroscopically.
Here, we report the first preparation of oxetane and azetidine ethers via 4-membered ring carbocation intermediates, using a Brønsted acid catalyst (Fig. 1d). These small-ring ethers present an underexplored motif for medicinal chemistry, and the chemical stabilities of selected oxetane ether/ester pairs were tested under various conditions.
Results and discussion
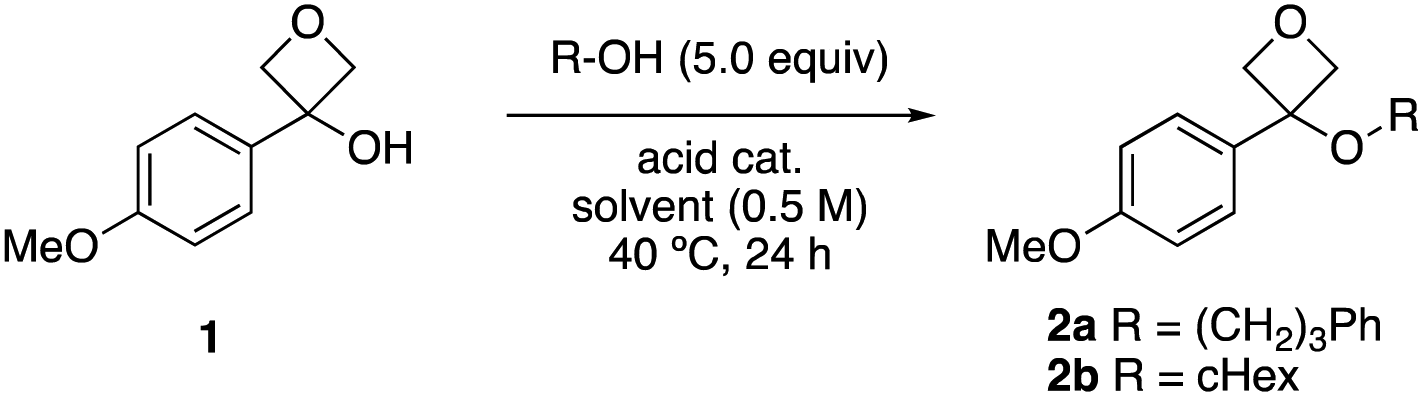
Our study began with electron rich 3-(4-methoxyphenyl)-oxetan-3-ol 1 known to be suitable to stabilise the strained ring carbocation,17 and used 3-phenyl propanol as the nucleophile. Attempts to use Lewis acid catalysts that had been previously successful for thiol and phenol nucleophiles were unsuccessful in the presence of the alcohols. On the other hand, using catalytic triflic acid (5 mol%) in chlorinated solvents (0.5 M 1) gave promising conversion with an excess of the alcohol reagent (Table 1, entries 1–3).21 Varying the solvent indicated that MeCN was optimal (entries 4–7). Varying the acid indicated an improvement using triflimidic acid over triflic or tosic acids (entries 7–9), related to the increasing acidity of the catalysts. For comparison, FeCl3 in toluene18a gave 8% of the product with a high amount of recovered oxetanol (entry 10). Using 10 mol% acid loading gave the best yields, at a reaction temperature of 50 °C (entries 9, 11–13). Unfortunately, applying these conditions directly to the secondary alcohol cyclohexanol gave relatively low yields (42%, entry 15). Reducing the concentration to 0.3 M gave an improved yield of 52% (entry 16). These conditions with the primary alcohol gave similar results (entry 14) and were selected for further study. For comparison using 1 equiv. of the alcohol gave a low yield of 17% and significant recovered oxetanol (entry 17). Alternatively, a useful yield could be obtained using excess oxetanol, with the alcohol as the limiting reagent, useful for the oxetane-functionalisation of valuable alcohol substrates (entry 18).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | –R | Acid | Acid loading (mol %) | Solvent | Yield 2a or 2ba (%) | RSM 1a,b (%) |
| Reactions performed on a 0.25 mmol scale.a Calculated by 1H NMR spectroscopy of the crude reaction mixture using 1,3,5-trimethoxybenzene as internal standard.b Recovered oxetanol 1.c Reaction performed at 30 °C.d Reaction performed at 50 °C.e Reaction performed at concentration of 0.3 M.f Using 1.0 equiv. cyclohexanol.g Using cyclohexanol as limiting reagent, with 5.0 equiv. oxetanol 1. | ||||||
| 1 | –(CH2)3Ph | TfOH | 5 | CHCl3 | 22 | 33 |
| 2 | –(CH2)3Ph | TfOH | 5 | CH2Cl2 | 22 | 28 |
| 3 | –(CH2)3Ph | TfOH | 5 | DCE | 40 | 18 |
| 4 | –(CH2)3Ph | TfOH | 5 | Toluene | 18 | 24 |
| 5 | –(CH2)3Ph | TfOH | 10 | DCE | 45 | 0 |
| 6 | –(CH2)3Ph | TfOH | 10 | DMSO | 0 | 56 |
| 7 | –(CH2)3Ph | TsOH | 10 | MeCN | 19 | 56 |
| 8 | –(CH2)3Ph | TfOH | 10 | MeCN | 64 | 4 |
| 9 | –(CH2)3Ph | Tf2NH | 10 | MeCN | 70 | 4 |
| 10 | –(CH2)3Ph | FeCl3 | 7.5 | Toluene | 6 | 87 |
| 11 | –(CH2)3Ph | Tf2NH | 5 | MeCN | 56 | 23 |
| 12 | –(CH2)3Ph | Tf2NH | 20 | MeCN | 54 | 5 |
| 13c | –(CH2)3Ph | Tf2NH | 10 | MeCN | 65 | 18 |
| 14 , | –(CH2)3Ph | Tf 2 NH | 10 | MeCN | 70 | 8 |
| 15d | –cHex | Tf2NH | 10 | MeCN | 42 | 21 |
| 16 , | –cHex | Tf 2 NH | 10 | MeCN | 52 | 33 |
| 17d,e,f | –cHex | Tf2NH | 10 | MeCN | 17 | 68 |
| 18d,e,g | –cHex | Tf2NH | 10 | MeCN | 50 | nd |
With optimised reaction conditions, the substrate scope of alcohols was investigated using primary and secondary alcohols (Scheme 2). The standard alcohol, 3-phenylpropanol, provided the oxetane ether 2a in 66% on a 2.0 mmol scale, and 61% on a 6 mmol scale (1.04 g, 2a). Ethanol gave ethyl ether 2c in 55%. Notably, bromoethanol was also successful, yielding oxetane ether 2d (45%), which would not be readily obtained under SN2 conditions with oxetanols and alkyl halides. Benzylic alcohols worked well providing bis-benzylic oxetane ethers 2e and 2f in 55% and 62% respectively. Propargylic alcohol could be employed (2g), introducing a potential click handle. Whereas ethylene glycol under these reaction conditions gives a dioxane product,16 NCbz-amino ethanol gave oxetane ether 2h in 42% yield. 1,3-Dihydroxy propane provided oxetane ether 2i as the major product in 38% yield without ring-opening of oxetane. Using serine gave the modified amino acid product 2j in 44% yield.
Cyclohexanol gave a moderate yield of the desired product 2b. This product displayed some instability on silica caused by the acidity of the stationary phase. Assessment of stationary phases for purification22 indicated basic alumina (activity IV) would be most suitable, which raised the isolated yield from 33% to 49%. Isopropanol afforded oxetane ether 2k in 82%, indicating lower steric demands than cyclohexanol. 2-Pentanol gave the ether 2l in 36% yield. A heterocyclic secondary alcohol, Cbz-piperidinol, provided ether 2m in 37%. However, Boc-piperidinol did not afford the desired ether. Using (S)-1-phenylethanol gave the ether product 2n in 31% yield and maintained high levels of enantioenrichment (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 er). The tertiary alcohol tert-butanol was unsuccessful.
:8 er). The tertiary alcohol tert-butanol was unsuccessful.
A competition reaction between primary and secondary alcohols was then performed, whereby ether 2a from 3-phenyl propanol dominated with 76% yield (Scheme 1). The secondary cyclohexanol gave oxetane ether 2b in only 10% yield, further indicating the selectivity based on steric grounds.
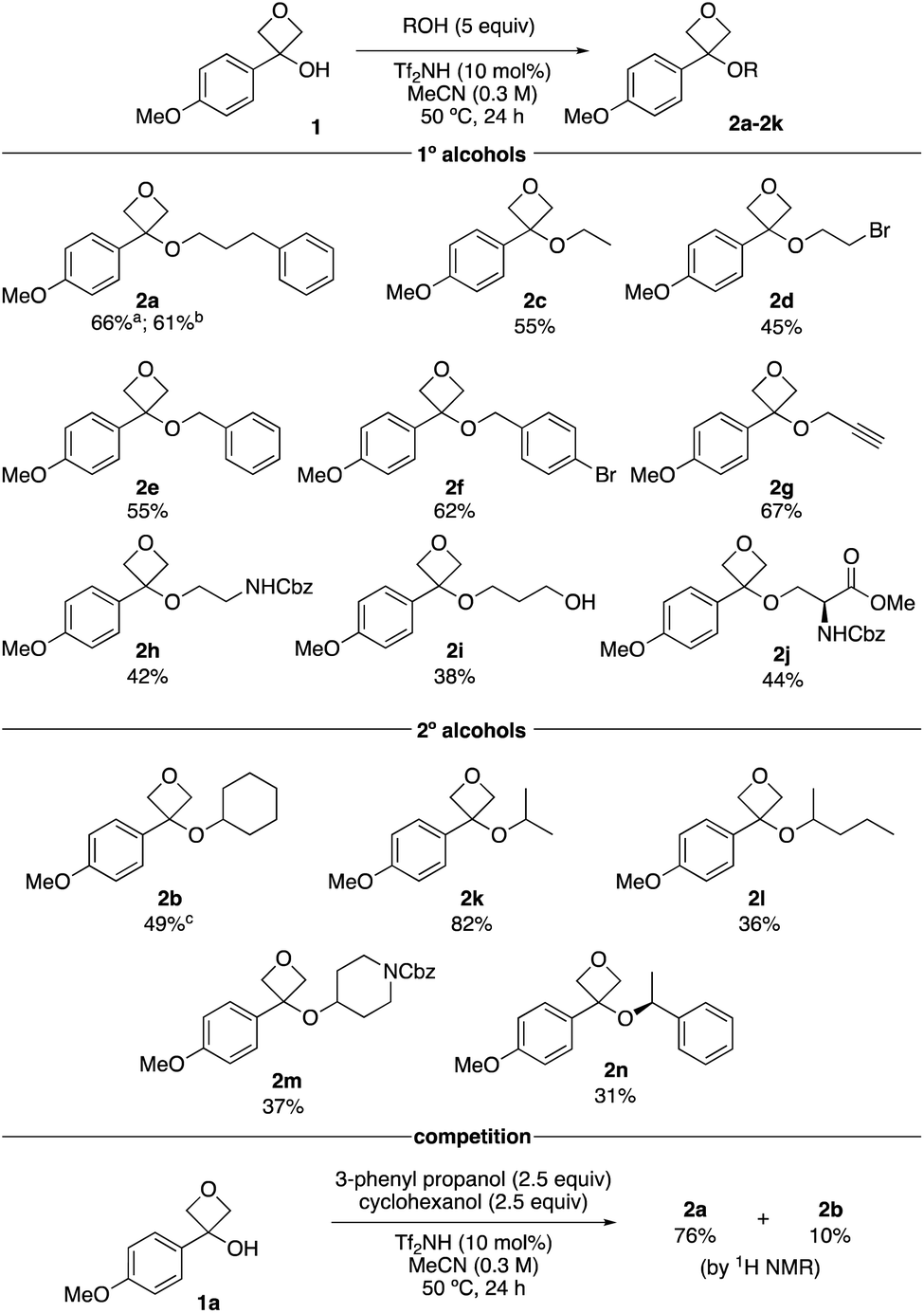 | ||
| Scheme 1 Reaction scope varying the alcohol using oxetanol 1. Reactions performed on a 0.25 mmol scale. aReaction performed on a 2.0 mmol scale. bReaction performed on a 6.0 mmol scale. cProduct isolated using chromatography on basic alumina (activity IV). | ||
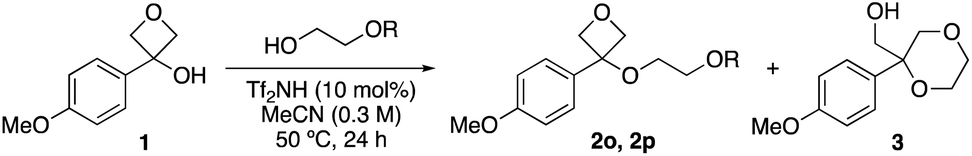
Interestingly, attempts to use methoxyethanol and ethoxy ethanol did not form the oxetane ether. Instead, dioxane 3 was formed in 71% and 43%, respectively, as a result of cyclisation comparable to our previous report using ethylene glycol (Table 2, entries 1–3).16 Using isopropoxyethanol with the bulkier iPr group gave both oxetane ether 2o (22%) and dioxane 3 (33%) products (entry 4), and with phenoxy ethanol, dioxane 3 was not observed (entry 5). Mechanistically, formation of dioxane 3 may proceed via etherification then oxetane ring opening to produce an oxonium ion followed by loss of the alkyl group either by an SN1 or SN2 mechanism. Resubjecting 2o to the reaction conditions gave dioxane 3 as the only product (25% yield by 1H NMR), confirming the viability of this route.
Variation of the oxetanol substrate was investigated using 3-phenyl propanol as a nucleophile (Scheme 2). 4-Isopropoxy derivative (13a) was formed in a similar yield to the methoxy-example. Protected phenolic derivatives bearing OTIPS and OBn groups at the 4-position were also successful (14a and 15a). TIPS protected product 14a displayed instability on acidic silica and was obtained in 47% using basic alumina (activity IV) for chromatography. The free phenol could also be applied and 16a was obtained in 51% yield. Di-substituted phenyl oxetanols also underwent etherification, providing the ethers in moderate yield (17a–20a). The lower yield observed with dimethoxy phenyl (17a) was ascribed to rapid formation of the carbocation, followed by degradation, since the dimethoxy substitution pattern is expected to best stabilize the transition state to form the oxetane carbocation.7a 3,4,5-Trimethoxy-substituted derivative gave the product (21a) in 23% yield, presumably due to the deactivating electronic effect from the 3- and 5-positions which prevent effective planarization and conjugation from the 4-methoxy group.7a The lower yield observed with both the carbocation-stabilising dimethoxy- (17a) and the carbocation-destabilising trimethoxy substitution pattern (21a) highlight the requirement of the etherification reaction for a defined stability window of the oxetane carbocation to prevent its degradation due to too rapid formation (dimethoxy) or insufficient stability (trimethoxy). Less electron rich aromatic groups were unreactive: the desired oxetane ether was not obtained in the reaction with 3-phenyl oxetan-3-ol, nor ortho-/meta-methoxy phenyl derivatives.
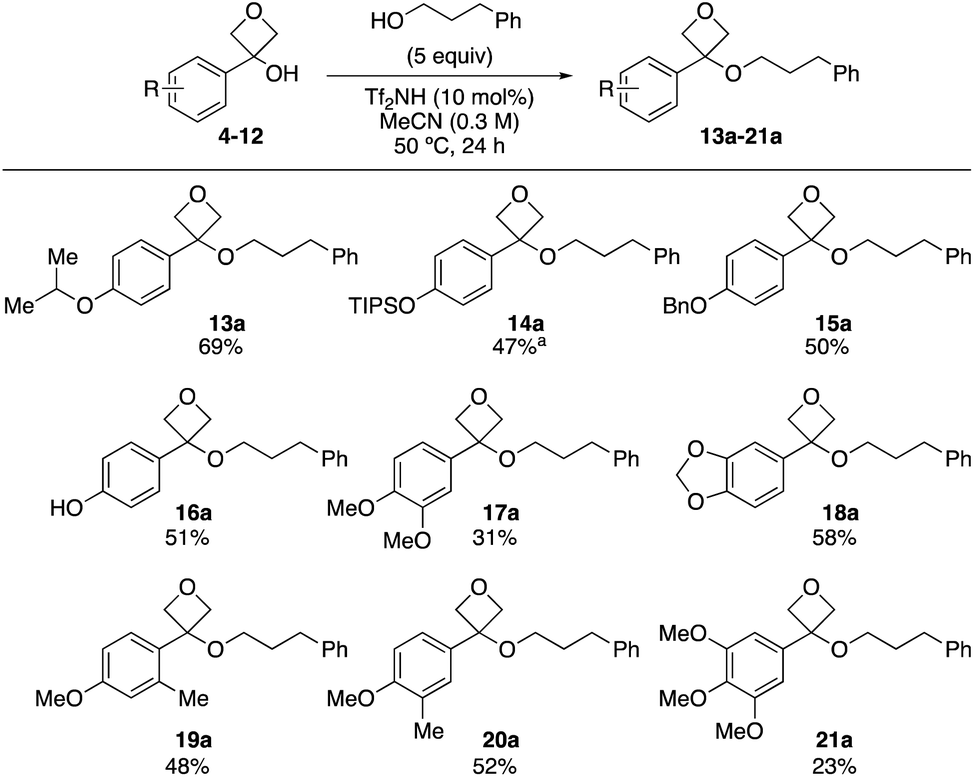 | ||
| Scheme 2 Reaction scope varying oxetan-3-ol substrate. Reactions performed on a 0.25 mmol scale. aProduct isolated using chromatography on basic alumina (activity IV). | ||
The conformation of oxetane ethers 2a and 2b, determined by X-ray crystallography, differs to that of related esters (Fig. 2). The absence of the π-carbonyl-pO conjugated system in the oxetane structure confers oxetane ethers with increased 3-dimensionality. The aryl ring is twisted at ca. 80° out of the O3–C1–CAr plane (CAr = C15 in 2a and C12 in 2b). In benzoates on the other hand, the aryl ring is only ca. 3° out of plane to maximize π-conjugation resulting in a flat conformation. As observed in our previous analysis on 3-aryl-3-alkyl-oxetanes,23 the increased steric requirement of the oxetane ring provokes a switch in conformational preference of the ethereal substituent which lies on the aryl side in oxetane ethers in a quasi-staggered conformation, but on the carbonyl side in benzoate structures.
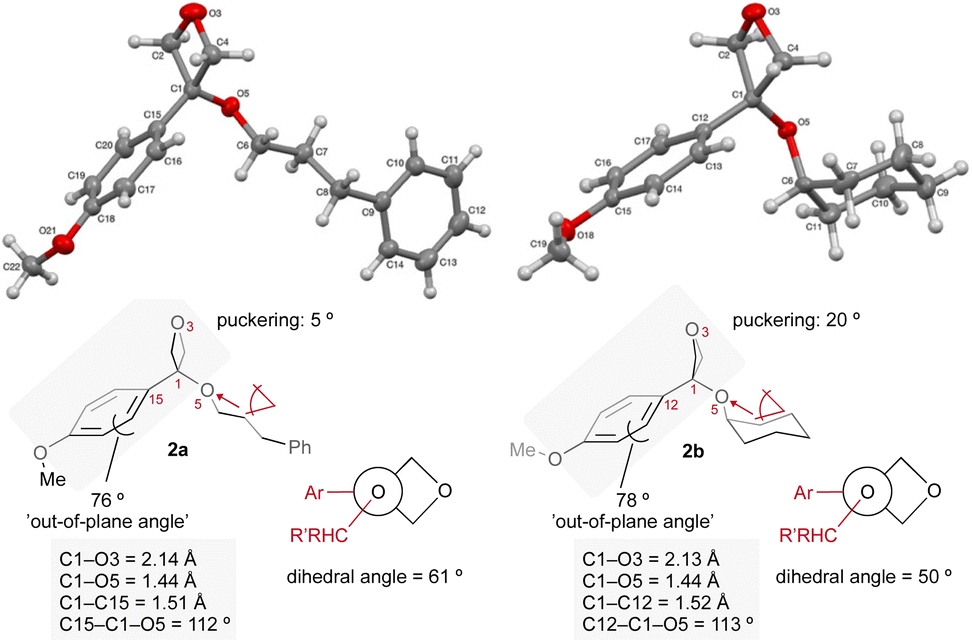 | ||
| Fig. 2 X-ray structures (50% probability ellipsoids) of oxetane ethers 2a and 2b. | ||
Similar optimisation was conducted on N-Cbz 3-PMP-azetidin-3-ol 22. Using very similar conditions, at 0.5 M concentration, slightly improved yields were obtained for the azetidine ether derivatives (Scheme 3). The reaction with primary alcohols yielded the desired products in moderate to high yields (24a, 24c–24j). Interestingly, methoxy ethanol and ethylene glycol gave azetidine ethers 24g and 24h in 52% and 79% yield without ring-opening. The secondary alcohols, cyclohexanol, 3-hydroxytetrahydrofuran and isopropanol, gave the azetidine ether products in 47%, 49%, and 57%, respectively. With OTIPS-azetidinol 23, alkylation and TIPS deprotection were observed, which was not seen in the oxetane derivatives, though the yield was similar to that obtained with the methoxyphenyl derivative 22.
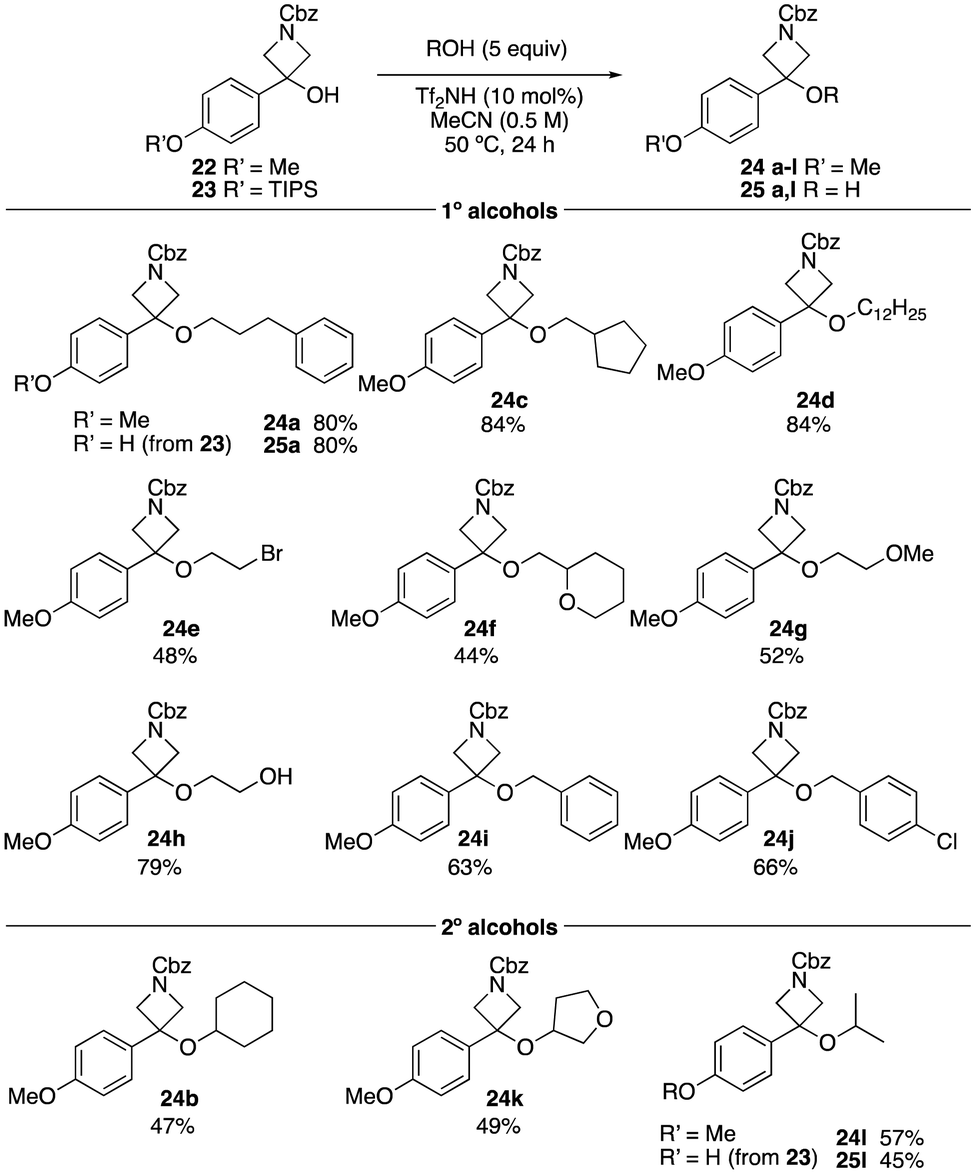 | ||
| Scheme 3 Reaction scope varying the alcohol using azetidinols 22 and 23. Reactions performed on a 0.5 mmol scale. | ||
Throughout the study, the requirement for excess nucleophile suggested the reaction may be reversible. This was confirmed by resubmitting oxetane and azetidine ethers to the reaction conditions in the presence of water, which led to formation of the corresponding alcohols (Scheme 4). The oxetane ether was recovered in 22% with oxetanol and cyclohexanol formed in 58% and 78%, in similar proportions to the forward reaction. Oxetane ether 2b was resubmitted to the reaction conditions in the presence of 3-phenylpropanol which yielded the trans-etherification product (2a) in 82%. Azetidine ether 24b gave back azetidin-3-ol 22 in 99% yield, consistent with a more rapid carbocation formation and preferential trapping by the smaller nucleophile (H2O) potentially due to increased steric hindrance around the azetidine carbocation caused by the Cbz group.18b,24
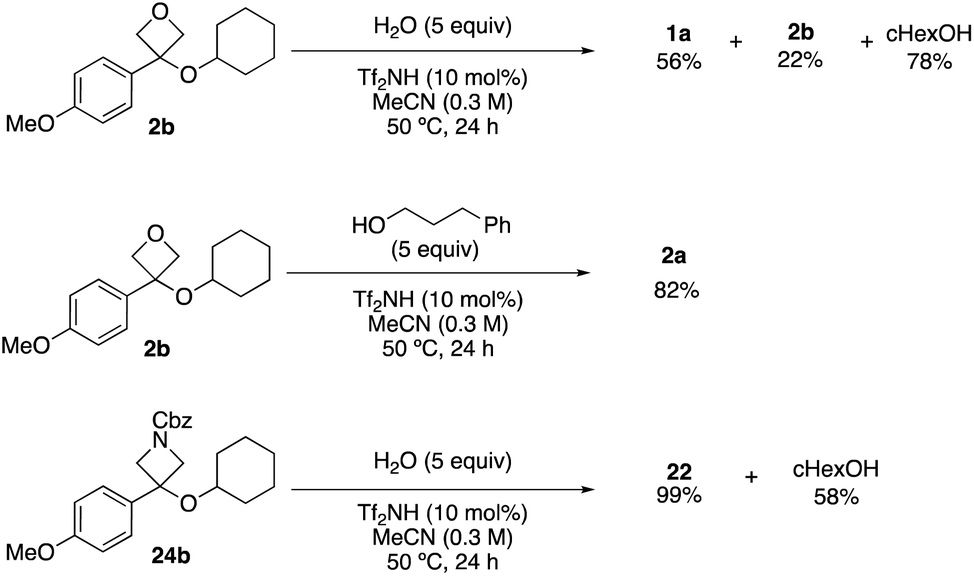 | ||
| Scheme 4 Control experiments indicating reaction reversibility. | ||
As the reaction was limited to electron-rich aryl groups, we next examined the potential to diversify the arene through cross-coupling reactions. TIPSO-phenyl oxetane ether 14a was readily deprotected with TBAF to form phenol 16a, and triflate 26a was formed on reaction with triflic anhydride. This proved to be a versatile intermediate for palladium-catalysed cross coupling processes. Reduction to the unsubstituted phenyl ring (27a) was achieved using formic acid, triethylamine with palladium acetate and 1,1′-bis(diphenylphosphino)ferrocene (dppf) ligand at 60 °C (Scheme 5).25 Applying triflate 26a to Suzuki–Miyaura reactions with pinacol boronates gave biaryls 28a and 29a in 99% and 81% yield respectively, including the incorporation of the (trifluoromethyl)pyrimidine motif (Scheme 5). These reactions occurred without degradation of the oxetane ether motif and demonstrated the potential for incorporation into more complex molecules.
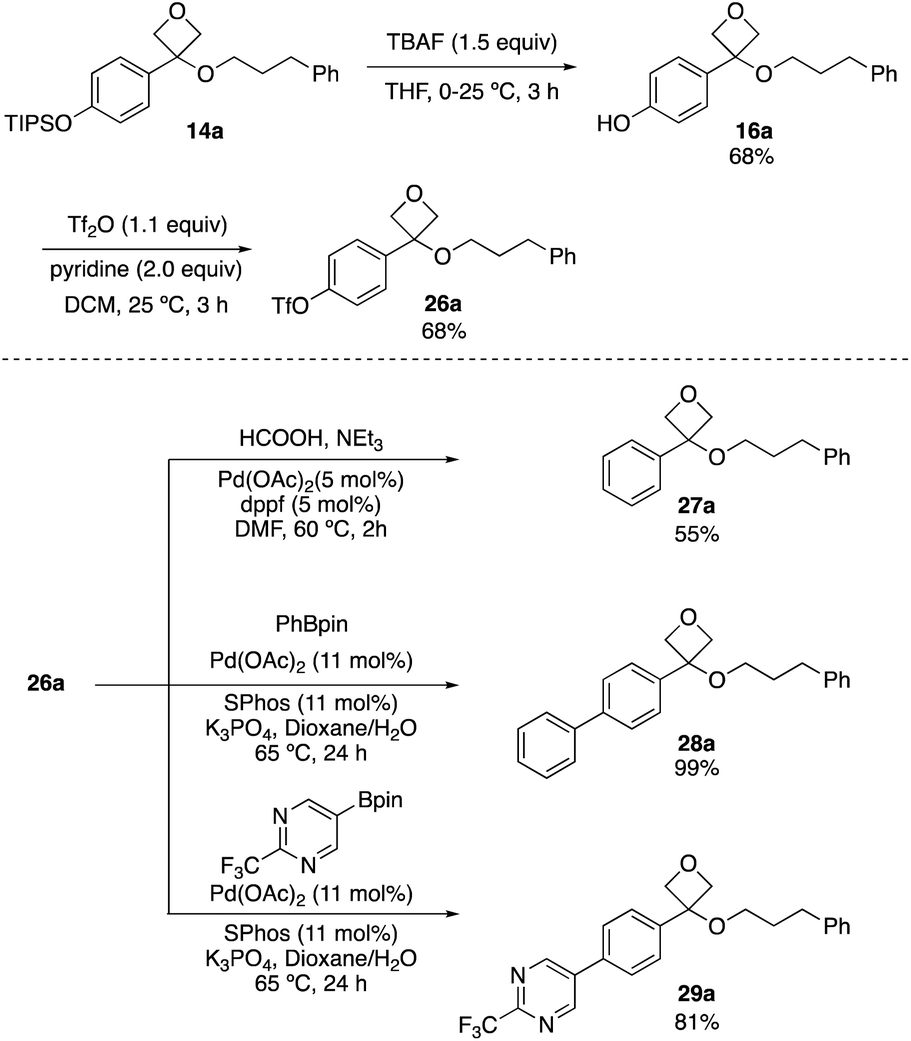 | ||
| Scheme 5 Cross-couplings of oxetane ether triflate 26a. | ||
The oxetane and azetidine ethers present potentially interesting motifs for application in drug discovery programmes, to provide an unusual 3D motif. Also of interest is the comparison with the corresponding esters. Esters typically hydrolyse rapidly under physiological conditions, catalysed by esterase enzymes, and so are extensively employed as pro-drugs which can improve solubility.26
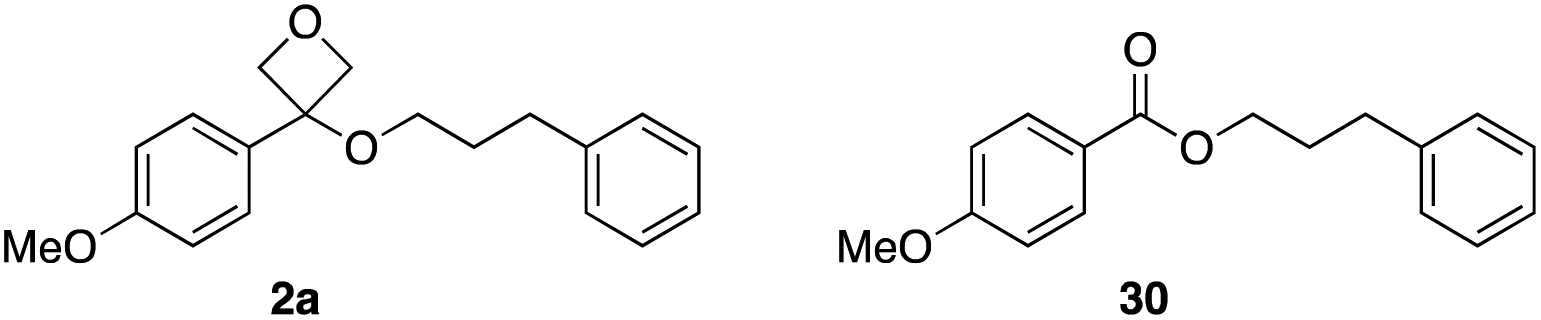
The stability of oxetane ethers in comparison with analogous esters was investigated by subjecting oxetane ether 2a and ester ‘pair’ 30 to different reaction conditions to assess compound recovery (Table 3). Initially, heating at 80 °C in DMSO and toluene for 24 h gave excellent recovery for both compounds (entries 1 and 2). Generally, esters are easily hydrolysed under acidic or basic aqueous conditions. The oxetane ether/ester pairs were treated with 1 M HCl at 37 °C (entry 3), related to average body temperature, though neither substrate was lost through hydrolysis. On the other hand, 1 M NaOH at rt gave significant loss of the ester (36% recovery), but full recovery of the oxetane (entry 4). Nucleophilic reagents NaI/acetone gave good recovery for both. The oxetane ether was also stable to LiBH4 whereas the ester underwent reduction, giving only 13% recovery (entry 6). The oxetane ether was treated with MeO-cysteine to mimic glutathione and was unreactive. Taken together, these results demonstrate high stability for the oxetane ether 2a under acidic, basic and nucleophilic conditions. Secondary alcohol oxetane ether 2b showed some instability in acid conditions in 1 M aqueous HCl solution (94% recovery 1 h at rt; 77% recovery 1 h, 37 °C, 31% recovery 24 h, 37 °C), presumed to proceed through the re-formation of the oxetane carbocation. This further indicated a steric influence on compound stability.
|
|
|||
|---|---|---|---|
| Entry | Conditions | Recovery (%)c | |
| Oxetane ether 2a | Ester 30 | ||
| a Reactions performed on a 0.05 mmol scale. b Reactions performed on a 0.1 mmol scale. c Amount of recovered compound was calculated by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. | |||
| 1a | Toluene 80 °C for 24 h | 99 | 100 |
| 2a | DMSO 80 °C for 24 h | 99 | 100 |
| 3a | 1 M aq. HCl, 37 °C, 24 h | 96 | 96 |
| 4a | 1 M aq. NaOH, rt, 24 h | 99 | 36 |
| 5b | 10 equiv. NaI in acetone, 50 °C, 1 h | 96 | 97 |
| 6b | 5 equiv. LiBH4 in THF, 65 °C, 1 h | 99 | 13 |
| 7b | 1 equiv. MeO-cysteine, DMF/H2O, rt, 12 h | 99 | 99 |
Conclusions
A protocol for the preparation of oxetane and azetidine ethers using oxetanols and azetidinols as electrophiles with alcohol nucleophiles has been developed. This avoids the requirement for alkyl halide reagents, and produces water as the only by-product. The condensation reaction requires a Brønsted acid catalyst and proceeds through an oxetane carbocation intermediate. The reaction was shown to be reversible under the reaction conditions. Oxetane ether products were diversified on a reactive phenol handle to access more complex aromatic structures. Oxetane ethers showed a significantly higher stability to basic and reducing conditions in comparison to the corresponding ester, as well as stability to high temperatures, acidic aqueous conditions, and thiols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge The Royal Society (University Research Fellowship UF140161 and URF\R\201019 to J. A. B.), Pfizer and Imperial College London for a studentship (J. J. R.). P. S. is grateful for funding support provided by the Development and Promotion of Science and Technology Talents Project (DPST) and the Institute for the Promotion of Teaching Science and Technology (IPST). C. D. is grateful for funding support provided by the Normandy County Council and Cancéropôle Nord-Ouest.References
- (a) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed; (b) J. J. Rojas and J. A. Bull, in Comprehensive Heterocyclic Chemistry IV, ed. D. StC Black, J. Cossy and C. V. Stevens, Elsevier Ltd, 2022, pp. 212–256 Search PubMed; (c) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS PubMed.
- (a) J. A. Burkhard, C. Guérot, H. Knust, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2010, 12, 1944–1947 CrossRef CAS PubMed; (b) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed; (c) A. A. Kirichok, I. Shton, M. Kliachyna, I. Pishel and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2017, 56, 8865–8869 CrossRef CAS PubMed; (d) D. R. Parmar, J. Y. Soni, R. Guduru, R. H. Rayani, R. V. Kusurkar and A. G. Vala, Arch. Pharm., 2021, 354, 1–33 CrossRef PubMed.
- G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736–7739 CrossRef CAS PubMed.
- G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512–4515 CrossRef CAS PubMed.
- (a) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227–3246 CrossRef CAS PubMed; (b) M. A. J. Dubois, R. A. Croft, Y. Ding, C. Choi, D. R. Owen, J. A. Bull and J. J. Mousseau, RSC Med. Chem., 2021, 12, 2045–2052 RSC.
- For studies on peptides: (a) M. McLaughlin, R. Yazaki, T. C. Fessard and E. M. Carreira, Org. Lett., 2014, 16, 4070–4073 CrossRef CAS PubMed; (b) N. H. Powell, G. J. Clarkson, R. Notman, P. Raubo, N. G. Martin and M. Shipman, Chem. Commun., 2014, 50, 8797 RSC; (c) G. P. Möller, S. Müller, B. T. Wolfstädter, S. Wolfrum, D. Schepmann, B. Wünsch and E. M. Carreira, Org. Lett., 2017, 19, 2510–2513 CrossRef PubMed; (d) S. Roesner, G. J. Saunders, I. Wilkening, E. Jayawant, J. V. Geden, P. Kerby, A. M. Dixon, R. Notman and M. Shipman, Chem. Sci., 2019, 10, 2465–2472 RSC.
- (a) For synthesis of aminooxetanes in comparison to amides: J. J. Rojas, R. A. Croft, A. J. Sterling, E. L. Briggs, D. Antermite, D. C. Schmitt, L. Blagojevic, P. Haycock, A. J. P. White, F. Duarte, C. Choi, J. J. Mousseau and J. A. Bull, Nat. Chem., 2022, 14, 160–169 CrossRef CAS PubMed; (b) For synthesis of oxetane sulfides in comparison to thioesters: R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Chem. – Eur. J., 2018, 24, 818–821 CrossRef CAS PubMed.
- P. Lassalas, K. Oukoloff, V. Makani, M. James, V. Tran, Y. Yao, L. Huang, K. Vijayendran, L. Monti, J. Q. Trojanowski, V. M. Y. Lee, M. C. Kozlowski, A. B. Smith, K. R. Brunden and C. Ballatore, ACS Med. Chem. Lett., 2017, 8, 864–868 CrossRef CAS PubMed.
- For leading examples also see: (a) E. D. Butova, A. V. Barabash, A. A. Petrova, C. M. Kleiner, P. R. Schreiner and A. A. Fokin, J. Org. Chem., 2010, 75, 6229–6235 CrossRef CAS PubMed; (b) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550–8551 CrossRef CAS PubMed; (c) O. A. Davis and J. A. Bull, Angew. Chem., Int. Ed., 2014, 53, 14230–14234 CrossRef CAS PubMed; (d) O. A. Davis, R. A. Croft and J. A. Bull, Chem. Commun., 2015, 51, 15446–15449 RSC; (e) O. A. Davis and J. A. Bull, Synlett, 2015, 26, 1283–1288 CrossRef CAS; (f) K. Rykaczewski and C. Schindler, Org. Lett., 2020, 22, 6516–6519 CrossRef CAS PubMed; (g) P. R. D. Murray, W. M. M. Bussink, G. H. M. Davies, F. W. van der Mei, A. H. Antropow, J. T. Edwards, L. A. D'Agostino, J. M. Ellis, L. G. Hamann, F. Romanov-Michailidis and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 4055–4063 CrossRef CAS PubMed; (h) D. M. Flores and V. A. Schmidt, J. Am. Chem. Soc., 2019, 141, 8741–8745 CrossRef CAS PubMed.
- (a) A.-C. Nassoy, P. Raubo and J. P. A. Harrity, Tetrahedron Lett., 2013, 54, 3094–3096 CrossRef CAS; (b) P. Li, J. R. Zbieg and J. A. Terrett, ACS Catal., 2021, 11, 10997–11004 CrossRef CAS; (c) M. A. Argiriadi, E. C. Breinlinger, E. Y. T. Chien, M. D. Coward, K. E. Frank, M. M. Friedman, D. J. Hardee, J. M. Herold, H. Liu, W. Qiu, M. J. Scanio, M. R. Schrimpf, T. R. Vargo, S. A. V. Epps, M. P. Webster, A. J. Little, T. A. Dunstan, M. H. Katcher and D. A. Schiedler, Inhibitors of tyrosine kinase 2 mediated signaling, International PatentWO2019178079A1, 2019 Search PubMed; (d) M. Härter, H. Beck, P. Ellinghaus, K. Berhörster, S. Greschat, K. Thierauch and F. Süssmeier, Heteroaromatic compounds for use as HIF inhibitors, United States PatentUS20110301122A1, 2011 Search PubMed.
- (a) T. P. Heffron, L. Salphati, B. Alicke, J. Cheong, J. Dotson, K. Edgar, R. Goldsmith, S. E. Gould, L. B. Lee, J. D. Lesnick, C. Lewis, C. Ndubaku, J. Nonomiya, A. G. Olivero, J. Pang, E. G. Plise, S. Sideris, S. Trapp, J. Wallin, L. Wang and X. Zhang, J. Med. Chem., 2012, 55, 8007–8020 CrossRef CAS PubMed; (b) S. Leth-Petersen, I. N. Petersen, A. A. Jensen, C. Bundgaard, M. Bæk, J. Kehler and J. L. Kristensen, ACS Chem. Neurosci., 2016, 7, 1614–1619 CrossRef CAS PubMed.
- A. Shinde, S. R. Ugale, Y. Nandurkar, M. Modak, A. P. Chavan and P. C. Mhaske, ACS Omega, 2022, 7, 47096–47107 CrossRef CAS PubMed.
- T. Fessard, D.-B. Li, D. Barbaras, S. Wolfrum and E. M. Carreira, (Lipideon Biotechnology AG) Preparation of azetidinone-containing compounds for pharmaceutical hypocholesterolemic compositions, International Patent WO2010100255A1, 2010 Search PubMed.
- (a) J. E. P. Davidson, J. M. Bentley, C. E. Dawson, K. Harrison, H. L. Mansell, A. L. Pither, R. M. Pratt, J. R. A. Roffey and V. J. Ruston, Azetidine carboxamide derivatives and their use in the treatment of CB1 receptor mediated disorders, International PatentWO2004096794A1, 2004 Search PubMed; (b) M. C. T. Fyfe, W. Gattrell and C. M. Rasamison, Azetidine derivatives as G-protein coupled teceptor (GPR119) agonist, InternationalWO20090281076A1, 2009 Search PubMed; (c) M. S. Malamas, S. I. Farah, M. Lamani, D. N. Pelekoudas, N. T. Perry, G. Rajarshi, C. Y. Miyabe, H. Chandrashekhar, J. West, S. Pavlopoulos and A. Makriyannis, Bioorg. Med. Chem., 2020, 28, 115195 CrossRef CAS PubMed; (d) M. Asselin, J. W. Ellingboe and R. E. Mewshaw, [(indol-3-yl)-cycloalkyl]-3-substituted azetidines for the treatment of central nervous system disorders, United State PatentUS6245799 B1, 2001 Search PubMed; (e) E. Isabel, D. A. Powell, W. C. Black, C. C. Chan, S. Crane, R. Gordon, J. Guay, S. Guiral, Z. Huang, J. Robichaud, K. Skorey, P. Tawa, L. Xu, L. Zhang and R. Oballa, Bioorg. Med. Chem., 2011, 21, 479–483 CrossRef CAS PubMed; (f) M. Zhang, A. Aguilar, S. Xu, L. Huang, K. Chinnaswamy, T. Sleger, B. Wong, S. Gross, B. N. Nicolay, S. Ronseaux, K. Harvey, Y. Wang, D. McEachern, P. D. Kirchhoff, Z. Liu, J. Stuckey, A. E. Tron, T. Liu and S. Wang, J. Med. Chem., 2021, 64, 10333–10349 CrossRef CAS PubMed; (g) T. Tsuji, A. Nakao, Y. Nakamura, Z. Zhang, M. Yokoyama and M. Iwanami, Azetidine sulfonamide compound, International PatentWO2021112251A1, 2021 Search PubMed.
- (a) W. Yang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 1868–1871 CrossRef CAS PubMed; (b) Z. Wang, Z. Chen and J. Sun, Org. Biomol. Chem., 2014, 12, 6028–6032 RSC; (c) C. A. Malapit and A. R. Howell, J. Org. Chem., 2015, 80, 8489–8495 CrossRef CAS PubMed; (d) D. A. Strassfeld, Z. K. Wickens, E. Picazo and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 9175–9180 CrossRef CAS PubMed.
- J. J. Rojas, E. Torrisi, M. A. J. Dubois, R. Hossain, A. J. P. White, G. Zappia, J. J. Mousseau, C. Choi and J. A. Bull, Org. Lett., 2022, 24, 2365–2370 CrossRef CAS PubMed.
- (a) R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Chem. – Eur. J., 2016, 22, 16271–16276 CrossRef CAS PubMed; (b) see ref. 7b. ; (c) R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Tetrahedron, 2018, 74, 5427–5435 CrossRef CAS; (d) M. A. J. Dubois, M. A. Smith, A. J. P. White, A. L. W. Jie, J. J. Mousseau, C. Choi and J. A. Bull, Org. Lett., 2020, 22, 5279–5283 CrossRef CAS PubMed.
- (a) M. A. J. Dubois, A. Lazaridou, C. Choi, J. J. Mousseau and J. A. Bull, J. Org. Chem., 2019, 84, 5943–5956 CrossRef CAS PubMed; (b) C. Denis, M. A. J. Dubois, A. S. Voisin-Chiret, R. Bureau, C. Choi, J. J. Mousseau and J. A. Bull, Org. Lett., 2019, 21, 300–304 CrossRef CAS PubMed; (c) R. A. Croft, M. A. J. Dubois, A. J. Boddy, C. Denis, A. Lazaridou, A. S. Voisin-Chiret, R. Bureau, C. Choi, J. J. Mousseau and J. A. Bull, Eur. J. Org. Chem., 2019, 5385–5395 CrossRef CAS . For a review on benzylic oxetane- and azetidine carbocations, see: J. J. Rojas and J. A. Bull, Chimia, 2023, 77, 192–195 Search PubMed.
- Also see: M. Das, A. Weissenfluh, N. Ly and M. L. Trudell, J. Org. Chem., 2020, 85, 8209–8213 CrossRef CAS PubMed.
- For a related process, proposing a para-quinone methide intermediate with phenolic derivatives, see: S. Liu, Y. Zang, H. Huang and J. Sun, Org. Lett., 2020, 22, 8219–8223 CrossRef CAS PubMed.
- For a review of Brønsted acid catalysed substitution, see: S. Estopiñá-Durán and J. E. Taylor, Chem. – Eur. J., 2021, 27, 106–120 CrossRef PubMed.
- T. Boultwood, D. P. Affron, A. D. Trowbridge and J. A. Bull, J. Org. Chem., 2013, 78, 6632–6647 CrossRef CAS PubMed.
- M. A. J. Dubois, J. J. Rojas, A. J. Sterling, H. C. Broderick, M. A. Smith, A. J. P. White, P. W. Miller, C. Choi, J. Mousseau, F. Duarte and J. A. Bull, J. Org. Chem., 2023, 88, 6476–6488 CrossRef CAS PubMed.
- As a comparison of reactivity, 2-phenylpropan-2-ol was examined under the reaction conditions with 3-phenyl propanol as the nucleophile. This gave only 10% yield of the corresponding ether accompanied by degradation of the substrate.
- S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 5541–5544 CrossRef CAS.
- (a) G. L. Patrick, An introduction to Medicinal Chemistry, Oxford University Press, Oxford, 2013 Search PubMed; (b) L. D. Lavis, ACS Chem. Biol., 2008, 3, 203–206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data; additional optimization; compound stability to stationary phases; experimental procedures; characterization data and copies NMR spectra. CCDC 2250896 and 2250897. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00731f |
| This journal is © The Royal Society of Chemistry 2023 |