
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free [2,3]-sigmatropic rearrangement in the reaction of sulfur ylides with allenoates†
Miguel
García-Castro
 *,
Federico
Moya-Utrera
and
Francisco
Sarabia
*,
Federico
Moya-Utrera
and
Francisco
Sarabia
Faculty of Science, University of Malaga, Campus de Teatinos s/n, 29071, Malaga, Spain. E-mail: mgcastro@uma.es
First published on 7th June 2023
Abstract
An unprecedented transition metal free [2,3]-sigmatropic rearrangement involving stabilized sulfur ylides and allenoates has been thoroughly established. The scope and utility of this reaction have been extensively studied resulting in C–C bond formation under mild conditions with greater than 20 examples reported. A highlight of the work is the simple and fully operational process that does not involve the use of carbenes or the associated hazardous and sensitive reagents. The reaction can be performed at room temperature and using an open flask. Interestingly, the new C–C bond formation reaction is gram scalable, and the obtained isomers are readily separable, affording interesting building blocks that can be used in the preparation of complex molecules.
Introduction
Since the early days of organic chemistry, the reactivity of sulfur containing compounds has played a prominent role in the discipline.1 Notably, the so-called sulfur ylides represent a very important class of chemical reagents first reported in the sixties, and the Corey–Chaykovsky reaction involving them has been extensively employed in organic synthesis.2 Since then, the development of sulfur ylide chemistry has been exponential, expanding the utility of the chemistry in new ways for their preparation, as well as discovering new reactivities exhibited by them. To date, sulfur ylides have been mainly used to synthesize a plethora of cyclopropanes, oxiranes and aziridines,3 exploiting their well-known ability to participate in the formation of three membered rings through the formation of a betaine intermediate in their reactions with α,β-unsaturated carbonyl compounds, carbonyl compounds and imines, respectively. The resulting betaine intermediate undergoes an intramolecular nucleophilic displacement, in what can be considered as a formal [2 + 1] cycloaddition reaction. On the other hand, quite more underexplored are the known [1,2]- and [2,3]-sigmatropic rearrangements, which are domino reactions involving sulfur ylide formation.1,4 Furthermore, the examples reported for these modes of reactivity usually involve metal carbenoids, such as Doyle–Kirmse and thia–Sommelet–Hauser reactions (Scheme 1).5,6 | ||
| Scheme 1 Main pathways of reactivity in sulfur ylides. | ||
However, recently the metal free version of the thia–Sommelet–Hauser reaction has been independently established by Biju and Tan groups,7 involving only aromatic rings and requiring allyl or propargyl thioethers. Mechanistically, these reactions proceed through a benzyne intermediate and in situ formation of a sulfur ylide. It is worth mentioning that in this case, the [2,3]-sigmatropic rearrangement occurs just in the ylide itself, as the allyl or propargyl moiety provides the electrophilic α-carbon (Scheme 2A). On the other hand, allenoates have emerged in the last two decades as excellent reactive partners, able to participate in a vast number of reactions, mainly cycloaddition reactions.8 Nevertheless, in the literature only one example in which their reactivities were explored with sulfur ylides is reported. In the reported example, Tong's group developed a DABCO-catalyzed [3 + 3] annulation of allenoates with sulfur ylides to deliver 9 examples of achiral 4H-pyrans.9 Additionally, Maulide's group synthesized cyclopropanes using allenamide derivatives in the presence of cationic gold complexes and doubly stabilized sulfur ylides10 (Scheme 2B).
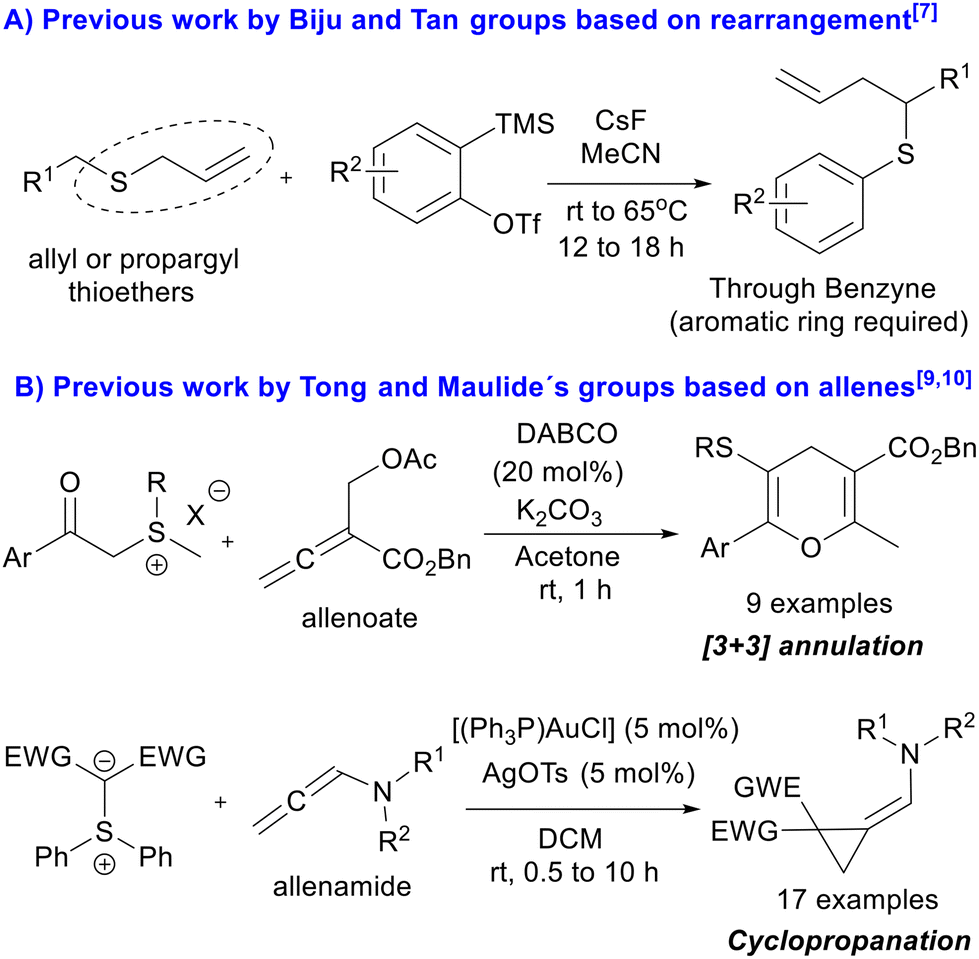 | ||
| Scheme 2 (A) Transition metal free [2,3]-sigmatropic rearrangement (thia–Sommelet–Hauser reaction) requiring special moieties. (B) Tong and Maulide's groups studied different reactions involving sulfur ylides and allenes. | ||
Herein, we wish to report the first transition metal free [2,3]-sigmatropic rearrangement with non-aromatic partners as reagents and occurring between stabilized sulfur ylides and allenoates, without the need for employing carbene transfer reactions, photochemical conditions11 or transition metals such as rhodium12 (Fig. 1).
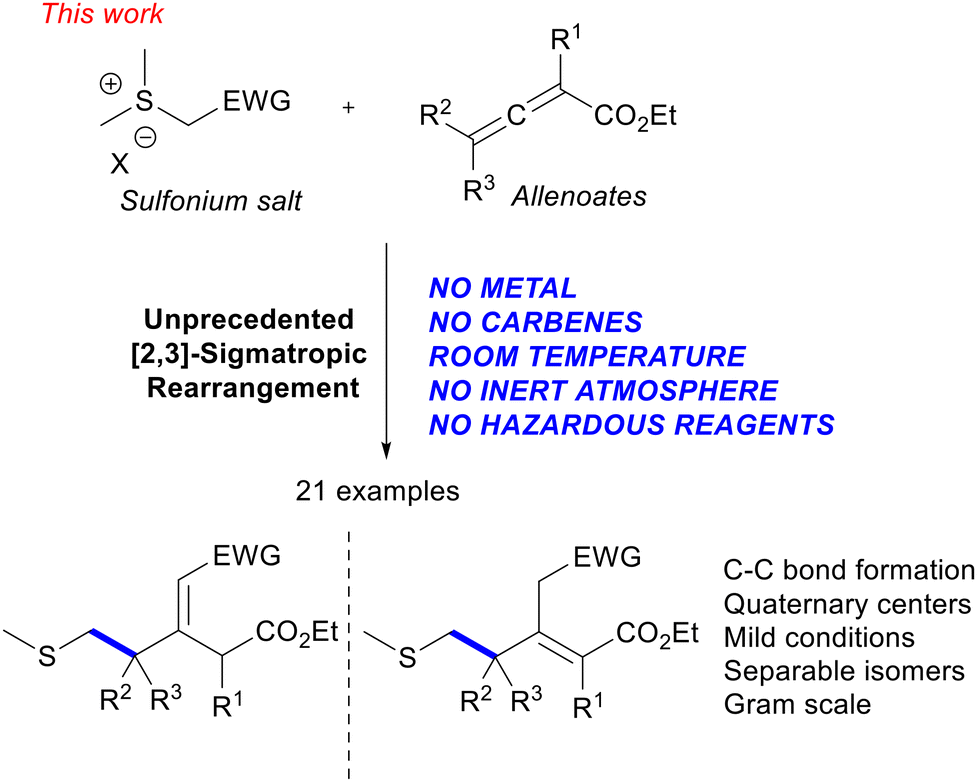 | ||
| Fig. 1 Our unprecedented transition metal free [2,3]-sigmatropic rearrangement reaction involving sulfur ylides and allenoates. | ||
Results and discussion
Our interest in the chemistry of sulfur ylides, coupled with the scarcity of studies into their reactions with allenoates, prompted us to initiate this investigation.Conceptually, the electrophilic carbon of allenoate should be attacked by the nucleophilic carbon of the sulfur ylide to obtain a betaine intermediate, which could convert to the corresponding cyclopropane derivatives. At first we studied the reaction between the in situ generated stabilized sulfur ylide 2a and simple allenoate 3a.
Importantly, we observed the formation of a mixture of two products, which, after separation and structural elucidation, were identified as 4a and 4a′. At the outset of our study, we first chose 1a and 3a as the standard substrates to identify the optimal reaction conditions, looking for reproducibility, full conversion and consistent results (Table 1). In order to optimize the reaction, we evaluated a diverse set of bases for the in situ formation of the sulfur ylide 2a from the sulfonium salt 1a.13 As a result, bases such as sodium hydride and 3 M aqueous NaOH solution were discarded due to non-reproducible results.14 In contrast, potassium tert-butoxide in tert-butanol (entries 7–9, Table 1) proved to be the base of choice, providing compounds 4a and 4a′ in a 73% combined yield.15 The optimized procedure consisted of the addition of potassium tert-butoxide to sulfonium salt 1a, and then, after stirring for 1 hour at room temperature, the addition of a solution of allenoate 3a in tert-butanol in an open flask. We optimized the required time for completion of the reaction and we observed some variations depending on the nature of allenoates;16 however, most reactions were completed after stirring for 3 hours at ambient temperature (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entrya | Baseb | Timec | Solvent | Yieldd (%) |
| a In all cases 1.0 equivalent of sulfonium salt 1a was used. b In all cases 1.0 equivalent of base was used. NaOH was a 3 M aqueous solution. c Reactions were monitored by TLC. d NR means non-reproducible results.14 Combined yield considering both formed products. e 5 M aqueous solution was used for a two phase method. f Addition of solid tBuOK to a solution of sulfonium salt in tert-butanol. g Addition of a 1 M tert-butanolic solution of tBuOK. h For scale-up: dropwise addition of allenoate for 2 h 30 min. | ||||
| 1 | NaH | 15 min | MeCN | NR |
| 2 | NaH | 1 h | MeCN | NR |
| 3 | NaH | 3 h | DMF | NR |
| 4 | NaOH | 1.5 h | t BuOH | NR |
| 5 | t BuONa | 1 h | t BuOH | 42 |
| 6e | NaOH | 1 h | DCM/H2O | NR |
| 7f | t BuOK | 1 h | t BuOH | 69 |
| 8g | t BuOK | 1 h | t BuOH | 73 |
| 9h | t BuOK | 1 h | t BuOH | 74 |

Furthermore, we also evaluated a set of reactions where the sulfur ylide 2a was isolated,17 and reactions with allenoates were evaluated in four different solvents: dichloromethane, acetonitrile, tetrahydrofuran and tert-butanol, in order to answer two main questions: first, will avoiding the basic media generated with the in situ formation of sulfur ylide prevent the formation of two isomers, and second, will the nature of the solvents influence the course of the main reaction. Gratifyingly, first we were able to demonstrate that when using isolated sulfur ylide 2a, the main reaction results in the formation of two separable isomers in the same proportion, and second, and quite interestingly, a variety of solvents can be used for the [2,3]-sigmatropic rearrangement, empowering the newly discovered methodology.
To rationalize the formation of these products, we propose a [2,3]-sigmatropic rearrangement from a transient sulfur ylide, which could be generated from the starting betaine intermediate (pathway A). It is somewhat expected that the outcome of the reaction was mainly represented by cyclopropanes (pathway B), but surprisingly and to our delight, we discovered that we obtained an easily separable mixture of compounds 4a and 4a′ in an overall yield of 73% and in a ratio of 1:2, whose molecular structures were established by careful and extensive NMR and GC-MS analyses.18 These structures reveal that a [2,3]-sigmatropic rearrangement could be operating and serves as a mechanistic explanation for the formation of the products. Accordingly, the nucleophilic carbon of the ylide attacks the electrophilic carbon of the allenoate, which stabilizes the negative charge due to the presence of the electron-withdrawing alkoxycarbonyl group in the α-position. Furthermore, this carbanion abstracts a proton from the methyl group attached to the sulfur atom, generating an unstable sulfur ylide that quickly triggers the formation of a new C–C bond and the breaking of C–S bond, representing a [2,3]-sigmatropic rearrangement, which results in the formation of both isomers due to the ability of the molecule to isomerize, which is facilitated by the acidic protons in the α-position of the ester (Scheme 3A).
 | ||
| Scheme 3 (A) Proposed mechanism for the formation of isomers 4a and 4a′ through a novel [2,3]-sigmatropic rearrangement. (B) Experiments designed to prove the proposed mechanism. | ||
In order to demonstrate the proposed mechanism we devised two new sulfur ylides, derived from sulfonium salts 1c and 1d, respectively. Sulfonium salt 1c contains two phenyl groups attached to the sulfur atom and lacks acidic protons, preventing the formation of a type II intermediate. Using 1c in the standard reaction resulted in no reaction. On the other hand, the synthesis of deuterated sulfonium salt 1d allowed us to realize the first isotopic studies by analyzing the 1H-NMR spectra of the reaction products. In this case, we obtained deuterated compound 4e′, supporting the formation of a transient ylide by deuterium abstraction of the betaine intermediate19 (Scheme 3B).
While the formation of these products could be rationalized through a [2,3] sigmatropic rearrangement, the geometry of the resulting trisubstituted double bond, found in 4a as in 4a′, was justified according to Scheme 4. Thus, assuming a cisoid arrangement between the reactants as the preferred approach, in which the two polar groups are in an almost eclipsed orientation, the resulting betaine intermediate A should evolve to the transient sulfur ylide B by intramolecular deprotonation of the sulfonium salt by the basic carbanion located at the α-position of the ester. Intermediate B then should undergo a [2,3]-sigmatropic rearrangement to product 4a′ after rotation of the C–C bond allowing for the spatial approach of the nucleophilic carbon of the sulfur ylide to the double bond carbon. Two possible conformers (B′ and B′′) could participate in the [2,3]-sigmatropic rearrangement, with the Z-isomer being the only possible product (4a′) in both cases. Finally, the basic conditions of the reaction mixture should promote an equilibrium isomerization of 4a′ to the thermodynamically favored 4a, through intermediate C. In contrast, cyclopropane formation would require a high-energy conformer, whose carbanion and sulfonium groups are oriented in an antiperiplanar arrangement. The high barrier for ring closure establishes this process as the rate-determining step and, considering that betaine formation is reversible, makes this pathway non-productive.
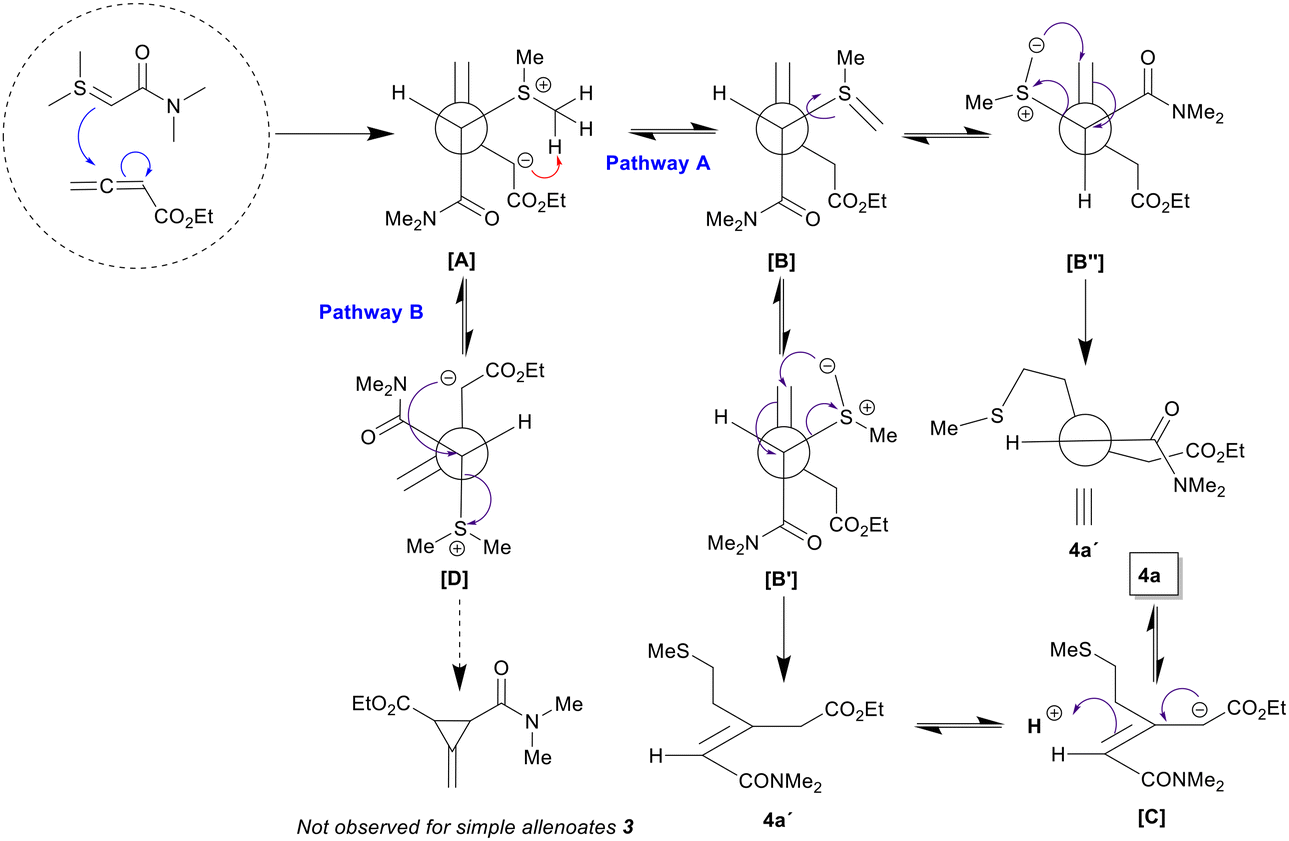 | ||
| Scheme 4 Theoretical rationale of the reaction of 2a with 3a and justification of the stereochemical outcome. | ||
Having determined the course of the reaction, we were prompted to study the scope of the reaction, in the first instance, by exploration of simple γ-substituted allenoates.
To our delight, compounds 4 and 4′ were obtained in moderate to good overall yields and as separable isomers (Table 2). Particularly relevant is the case of allenoate 3g, which bears a geminal dimethyl group, in which compound 4g′ (entry 6, Table 2) was obtained in 57% yield, demonstrating that the rearrangement is possible despite the steric hindrance present at the allenoate moiety. The remaining examples show that there is no significant difference between the employment of aliphatic substituted allenoates 3b–f (entries 1–5, Table 2) and aromatic substituted allenoates 3h–k (entries 7–10, Table 2). The use of a benzyl group in the γ-position of the allenoate, compound 3l (entry 11, Table 2), affords a mixture of compounds 4l and 4l′ in a combined 71% yield, depicting the generality and robustness of the newly discovered synthetic methodology. In addition, compound 4f could be isolated in the form of monocrystals and the subsequent X-ray analysis allowed for the unambiguous confirmation of the proposed structure of the product (Fig. 2).
 | ||
| Fig. 2 X-ray structure of compound 4f. | ||
|
|
|||
|---|---|---|---|
| Entrya | R3b |
Timec | Products (% yield)d [isomer ratio] |
| a In all cases 1.0 equivalent of sulfonium salt 1a and 1.0 equivalent of freshly prepared allenoate 3 were used. b In all cases R2 = H, except for entry 6, where R2 = methyl. c Stirring after the addition of allenoate. d Yield is given for pure compounds after separation using flash column chromatography. The overall yield is considered by taking both pure compounds together. e Only one isomer can be formed. | |||
| 1b | Methyl (3b) | 1 h |
4b, 4b′ (68%) [1:1.3] |
| 2 | Ethyl (3c) | 1 h |
4c, 4c′ (65%) [1:1] |
| 3 | Propyl (3d) | 1 h |
4d, 4d′ (67%) [1.4:1] |
| 4 | Isopropyl (3e) | 3 h |
4e, 4e′ (54%) [1.3:1] |
| 5 | t Butyl (3f) | 1 h |
4f, 4f′ (54%) [1:1.5] |
| 6b | Methyl (3g) | 12 h | 4g′ (57%)e |
| 7 | Phenyl (3h) | 3 h |
4h, 4h′ (40%) [2.3:1] |
| 8 | p-Chlorophenyl (3i) | 1 h |
4i, 4i′ (63%) [1:2.1] |
| 9 | m-Bromophenyl (3j) | 3 h |
4j, 4j′ (40%) [1.8:1] |
| 10 | m-Methoxyphenyl (3k) | 3 h |
4k, 4k′ (55%) [1:2] |
| 11 | Benzyl (3l) | 2 h |
4l, 4l′ (71%) [1:3] |

In order to further explore the scope and limitations of the reaction, we decided to explore two important structural factors for the two reactants, the role of the electron-withdrawing group in the sulfonium salt and the use of more complex allenoates, such as the α,γ-substituted variants.20
To this aim, we tested the reaction employing the sulfur ylide 2b, derived from sulfonium salt 1b, where the electron-withdrawing group is an alkoxycarbonyl instead of an aminocarbonyl group, and allenoates 3(a, f and h). In these cases, the reactions proceeded in lower yields and the formation of product 4 dominated, which was ascribed to the lower reactivity of the sulfur ylide 2b with respect to the amide-stabilized sulfur ylide (Scheme 5). For example, compounds 4m–o were obtained in 20–30% versus the 50–70% yield obtained when sulfur ylide 2a was used.
 | ||
| Scheme 5 Changing the EWG on sulfur ylides affects dramatically the yield of the reaction. | ||
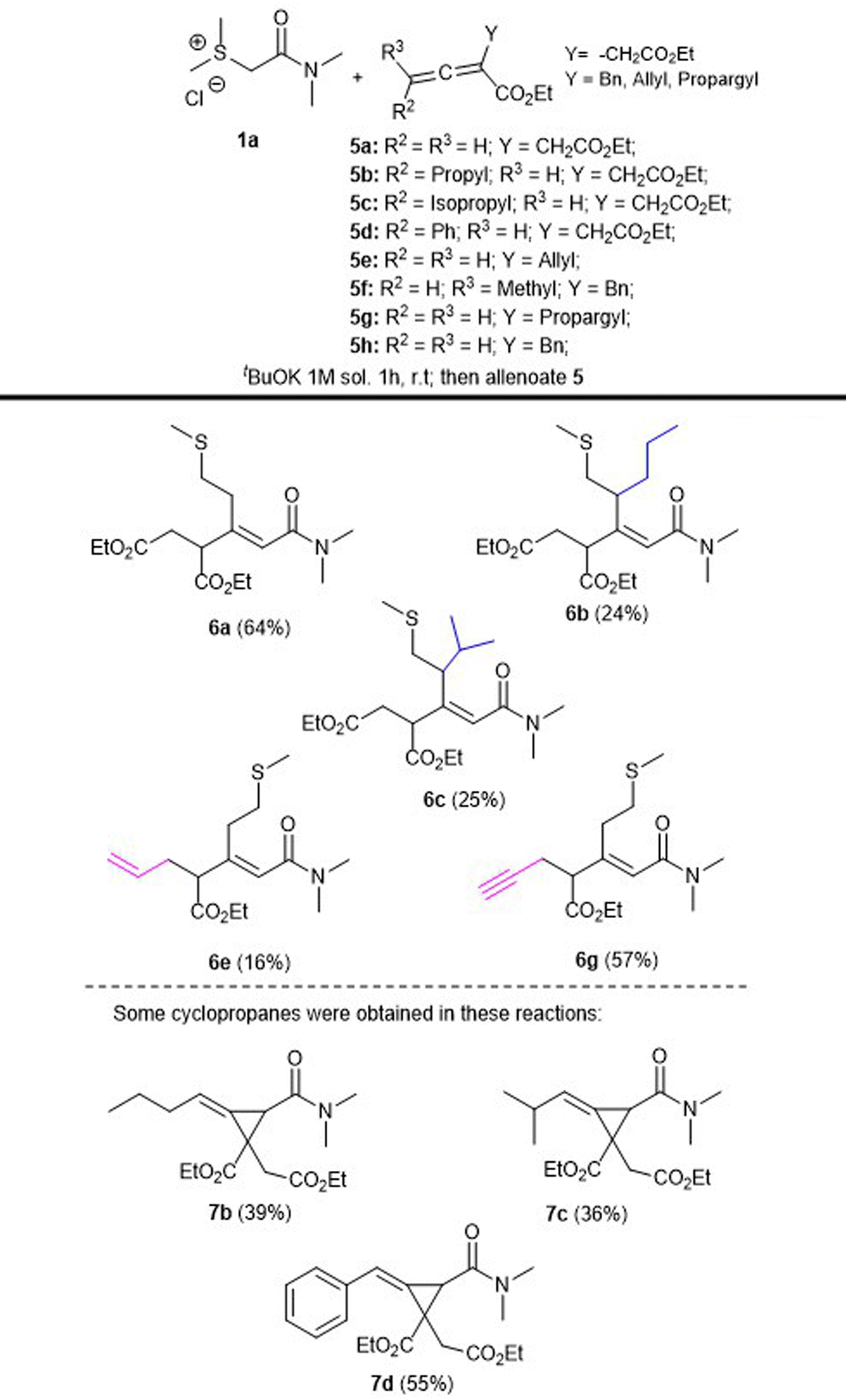
Finally, more complex allenoates 5 were tested in order to expand the reaction scope and to afford structurally complex products. Thus, we explored the reaction between sulfonium salt 1a and allenoates 5(a–h) under the previously optimized conditions, to afford products 6(a–c, e, g) in moderate to good yields as separable mixtures of isomers (Table 3).
|
Particularly interesting is the study of this reaction using allenoates bearing different groups in the α-position, with the consideration that they should exhibit higher reactivities, for example, the allyl (5e) and propargyl (5g) groups. However, the corresponding products 6e and 6g were obtained in low to moderate yields (16 and 57%, respectively) and also as a separable mixture of isomers in the case of the allyl derivative, and in the propargyl case, surprisingly only one isomer was afforded opening up the possibility of generating any desired carbon skeleton with different patterns of substitution using the appropriate corresponding allenoates.
However, it is important to point out that in several reactions using complex allenoates, we isolated in moderate to good yields the corresponding cyclopropane derivatives as the main products; for example, compounds 7b, 7c and 7d were obtained in 39, 36 and 55% yields, respectively, along with the corresponding rearrangement products (6b and 6c in 24 and 25% yields, respectively). These results demonstrate that the initially expected outcome of these reactions resulting in the formation of cyclopropyl derivatives is operative when additional groups are included in the allenoate moiety. These observations offer the opportunity for further exploration of complex allenoates with sulfur ylides. Furthermore, we could isolate other reaction products in specific reactions employing allenoate 5f and 5h, affording diverse complex products21 (Table 3).
We envision that the present methodology can be further applied to the total synthesis of natural products and complex molecules. In fact, during the preparation of this manuscript, a related reaction based on Baldwin's work4 was proposed by Fürstner's group for the total synthesis of the marine natural product scabrolide A.22
Conclusions
In conclusion, we have systematically explored the reactivity between stabilized sulfur ylides and allenoates, discovering a novel and simple method to synthesize interesting scaffolds through a new carbon–carbon bond formation via a [2,3]-sigmatropic rearrangement which does not require transition metals or carbene formation, a very important feature of this reaction. Currently, the synthesized compounds are being tested in biological screenings. Further exploitation of this new methodology in the synthesis of natural products is currently in progress.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Malaga for its support and funding. F. M.-U. and M. G.-C. thank the University of Malaga for research contracts corresponding to the research programme of the university (I Plan Propio UMA). This article was supported by project B1-2021_08 funding from the University of Malaga (I Plan Propio UMA. Main researcher is Dr. M. García-Castro). M. G.-C and F. S thank Dr Iván Cheng from ETH, Zurich (Switzerland), for his assistance with the X-ray structure. The authors thank Dr J. I. Trujillo from Pfizer (Groton, CT) for assistance in the preparation of this manuscript.References
- (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867–868 CrossRef CAS.
- (a) J. B. Sweeney and A. E. J. Walsh, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Ireland, 2014, vol. 1, pp. 609–652 Search PubMed; (b) R. Appel, N. Hartmann and H. Mayr, J. Am. Chem. Soc., 2010, 132, 17894–17900 CrossRef CAS PubMed.
- J. E. Baldwin, R. E. Hackler and D. P. Kelley, J. Am. Chem. Soc., 1968, 90, 4758–4759 CrossRef CAS.
- (a) M. P. Doyle, J. H. Griffin, M. S. Chinn and D. Van Leusen, J. Org. Chem., 1984, 49, 1917–1925 CrossRef CAS; (b) W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994–1003 CrossRef CAS; (c) T. Fukuda and T. Katsuki, Tetrahedron Lett., 1997, 38, 3435–3438 CrossRef CAS; (d) P. W. Davies, S. J. C. Albrecht and G. Assanelli, Org. Biomol. Chem., 2009, 7, 1276–1279 RSC; (e) P. Jia and Y. Huang, Adv. Synth. Catal., 2018, 360, 3044–3048 CrossRef CAS; (f) S.-J. Shen, X.-L. Du, X.-L. Xu, Y.-H. Wu, M.-G. Zhao and J.-Y. Liang, RSC Adv., 2019, 9, 34912–34925 RSC; (g) C. L. Makitalo, A. Yoshimura, G. T. Rohde, I. A. Mironova, R. Y. Yusubova, M. S. Yushubov, V. V. Zhdankin and A. Saito, Eur. J. Org. Chem., 2020, 6433–6439 CrossRef CAS.
- (a) M. Liao, L. Peng and J. Wang, Org. Lett., 2008, 10, 693–696 CrossRef CAS PubMed; (b) Y. Li, Y. Shi, Z. Huang, X. Wu, P. Xu, J. Wang and Y. Zhang, Org. Lett., 2011, 13, 1210–1213 CrossRef CAS PubMed.
- (a) M. Thangaraj, R. N. Gaykar, T. Roy and A. T. Biju, J. Org. Chem., 2017, 82, 4470–4476 CrossRef CAS PubMed; (b) J. Tan, T. Zheng, K. Xu and C. Liu, Org. Biomol. Chem., 2017, 15, 4946–4950 RSC; (c) R. N. Gaykar, M. George, A. Guin, S. Bhattacharjee and A. T. Biju, Org. Lett., 2021, 23, 3447–3452 CrossRef CAS PubMed.
- For representative reviews on allene reactivity and cycloaddition: (a) N. De and E. J. Yoo, ACS Catal., 2018, 8(1), 48–58 CrossRef CAS; (b) E.-Q. Li and Y. Huang, Chem. Commun., 2020, 56, 680–694 RSC; (c) S. M. Gillbard and H. W. Lam, Chem. – Eur. J., 2022, 28, e202104230 CAS; (d) H. Hopf and M. S. Sherburn, Synthesis, 2022, 864–886 CrossRef CAS; (e) P. Matton, S. Huvelle, M. Haddad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2022, 4–32 CAS; (f) M. Song, J. Zhao and E.-Q. Li, Chin. Chem. Lett., 2022, 33(5), 2372–2382 CrossRef CAS.
- K. Li, J. Hu, H. Liu and X. Tong, Chem. Commun., 2012, 48, 2900–2902 RSC.
- J. Sabbatani, X. Huang, L. F. Veiros and N. Maulide, Chem. – Eur. J., 2014, 20, 10636–10639 CrossRef CAS PubMed.
- Z. Yang, Y. Guo and R. M. Koenigs, Chem. – Eur. J., 2019, 25, 6703–6706 CrossRef CAS PubMed.
- (a) A. C. S. Reddy, K. Ramachandran, P. M. Reddy and P. Anbarasan, Chem. Commun., 2020, 56, 5649–5652 RSC; (b) S. Yan, J. Rao and C.-Y. Zhou, Org. Lett., 2020, 22, 9091–9096 CrossRef CAS PubMed.
- M. Valpuesta-Fernández, P. Durante-Lanes and F. J. López-Herrera, Tetrahedron, 1990, 46, 7911–7922 CrossRef.
- See the ESI† for unexpected results due to highly observed reactivity on allenoate patterns.
- Either adding solid tBuOK over a tert-butanolic solution of sulfonium salt or adding a commercially available 1 M tert-butanolic solution of tBuOK.
- For allenoates 3 preparation: Z. Huang, X. Yang, F. Yang, T. Lu and Q. Zhou, Org. Lett., 2017, 19, 3524–3527 CrossRef CAS PubMed.
- We used an earlier modified protocol previously used in our labs, consisting of reacting the sulfonium salt with NaH in acetonitrile at room temperature followed by stirring for 3 hours, in the presence of two drops of H2O. The reaction mixture was filtered and concentrated in a rotavapor, affording a freshly prepared sulfur ylide. F. Sarabia, F. Martín-Gálvez, M. García-Castro, S. Chammaa, A. Sánchez-Ruiz and J. F. Tejón-Blanco, J. Org. Chem., 2008, 73, 8979–8986 CrossRef CAS PubMed.
- Structural determination was realized by a combination of GC-MS and deep NMR analysis. And finally, X-ray diffraction was employed for crystalline products.
- See the ESI† for additional information regarding mechanistic studies.
- M. G. Sankar, M. García-Castro, C. Golz, C. Strohmann and K. Kumar, Angew. Chem., Int. Ed., 2016, 55, 9709–9713 CrossRef CAS PubMed.
- Those structures were not clearly defined, and they are currently under further elucidation studies.
- Z. Meng and A. Fürstner, J. Am. Chem. Soc., 2022, 144, 1528–1533 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, including synthesis and characterization of all products reported in this study, NMR spectra of all products, High Resolution Mass spectrometry (HRMS) and elemental analysis. CCDC 2253009. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00657c |
| This journal is © The Royal Society of Chemistry 2023 |