
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis and biological evaluation of histone deacetylase inhibitor WF-3161†
Michael
Kohr
 a,
Niklas
Papenkordt
b,
Manfred
Jung
b and
Uli
Kazmaier
*a
a,
Niklas
Papenkordt
b,
Manfred
Jung
b and
Uli
Kazmaier
*a
aOrganic Chemistry, Saarland University, D-66123 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
bInstitute of Pharmaceutical Sciences, University of Freiburg, Albertstr. 25, 79104 Freiburg, Germany
First published on 16th May 2023
Abstract
A novel synthesis of the naturally occurring HDAC inhibitor WF-3161 is described. Key steps include the Matteson homologation to generate the stereogenic centres in the side chain, and Pd-catalysed C–H functionalisation to connect the side chain to the peptide backbone. WF-3161 was found to be highly selective for HDAC1, whereas no activity was observed towards HDAC6. High activity was also found against the cancer cell line HL-60.
Introduction
Pharmaceuticals that target histone deacetylase (HDAC) enzymes have gained much attention in recent decades.1 The overexpression of HDACs correlates with numerous prevalent diseases, such as Alzheimer's disease2 and cancer,3 which makes these enzymes potential targets for drug development. Several acyclic HDAC inhibitors (HDACi), such as the naturally occurring trichostatin A (TSA)4 or the synthetic compound vorinostat (SAHA), have already been evaluated in clinical trials, with vorinostat the first HDACi to be approved by the FDA in the United States.5 More complex HDACi have been isolated from fungi, including chlamydocin,6 Cyl-1 and Cyl-2 (PMP: p-methoxyphenyl),7 the trapoxins,8 and WF-3161,9 among others (Fig. 1). All of these compounds have three major functional groups in common: (a) a zinc-binding motif, e.g. a hydroxamic acid or an epoxyketone; (b) a linker (spacer), which simulates the lysine side chain of the natural substrate; and (c) a cap region, which interacts with the protein surface surrounding the active site.10 | ||
| Fig. 1 Natural and synthetic HDAC inhibitors. | ||
Prototypical HDAC inhibitors, such as the natural product trichostatin A (TSA) or the approved drug vorinostat (SAHA), lack HDAC isoform selectivity, which leads to undesirable interactions with the ‘wrong’ isoforms (Tables 1 and 2).11 In contrast, macrocyclic HDACi contain a more complex cap region and are capable of differentiating among HDAC isoforms, enabling the investigation of their individual functions and association with cancer progression.12 Macrocyclic HDACi containing (2S,9S)-2-amino-9,10-epoxy-8-oxodecanoic acid (Aeo), an amino acid bearing an epoxyketone group in the side chain, highly differentiate between HDAC1 and HDAC6, for example, which belong to different HDAC classes (Table 1).13a The presence of the 8-ketogroup of Aeo has been proven to be essential for the interaction of HDACi with the protein's active site. Replacements of the keto-group or its reduction leads to a significantly lower bioactivity, as shown by Kim et al.13b
| HDACi | IC50, [nM] | IC50, [nM] | HDAC6/HDAC1 |
|---|---|---|---|
| HDAC1 | HDAC6 | ||
| TSA | 6.0 ± 2.5 | 8.6 ± 1.4 | 1.4 |
| Trapoxin A | 0.82 ± 0.29 | 524 ± 240 | 640 |
| Trapoxin B | 0.11 ± 0.01 | 360 ± 160 | 3300 |
| Chlamydocin | 0.15 ± 0.03 | 1100 ± 430 | 7300 |
| Cyl-2 | 0.70 ± 0.45 | 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 11000 000 ± 11000 |
57000 |
| HDACi | IC50, [nM] | IC50, [nM] | GI50, [nM] |
|---|---|---|---|
| HDAC1 | HDAC6 | HL-60 | |
| WF-3161 | 0.7 ± 0.1 | n.i. @ 25000 |
13 ± 1.1 |
| Vorinostat | 126 ± 7 | 58 ± 8 | 846 ± 48 |
| Mocetinostat | 110 ± 8 | n.i. | 592 ± 63 |
While these results provide deep insights into the behaviour of the HDACi investigated, to the best of our knowledge, other important representatives of this class of compounds, such as WF-3161 and Cyl-1, have not been thoroughly investigated to date.
The interesting biological activities of these tetrapeptide-based HDACi and their derivatives have initiated wide-ranging efforts towards a synthesis of these compounds.14 A key challenge in this approach is the stereoselective synthesis of the unusual amino acid Aeo, which contains an epoxyketone as a warhead and a zinc-binding motif. Several synthetic approaches have been developed in recent years, including alkylation of imino glycinate,15 phosphonate condensation of glycine derivatives,16 and the use of enantiomerically pure amino acid precursors,17 among others. Since our group is also involved in the synthesis of unusual amino acids18 and peptidic natural products,19 we became interested in the synthesis of this type of HDACi and began to develop several independent routes towards protected Aeo-precursors. An asymmetric chelate-Claisen rearrangement20 was the key step in the synthesis of chlamydocin21 and Cyl-1,22 while a Pd-catalysed peptide allylation23 gave access to trapoxin A.24 We subsequently became interested in developing a third independent route based on C–H functionalisation, with the goal of obtaining access to WF-3161. This natural product was first isolated from a strain of fungus, Petriella guttulata, by Umehara et al., who amassed a large body of structural and biological data on its activity, including anti-cancer activity against blood cancer cells in mice and antifungal activity against Trichophyton asteroides.9a In 2008, Proksch et al. obtained similar results with lymphoma cells from mice.25 However, no investigations on the HDAC activity of WF-3161 have been carried out to date, although the first synthesis of this compound was already reported in 1989 by Schmidt et al.26
Transition metal-catalysed C–H functionalisation has become an extremely popular tool in modern organic synthesis,27 and its widespread applications have been described in a series of recent reviews.28 Pd-catalysed functionalisation of amino acids and peptides, in particular, should offer an ideal method of synthesising and modifying peptidic natural products.29 Suitable directing groups (DGs) are required to address a specific Csp3–H bond in an amino acid in the presence of others.30 This protocol is also suitable for the modification of peptides, as long as no acidic N–H-bond is present in the reacting amino acid.31 To date, the best results have been obtained with amides of 8-aminoquinoline (AQ)32 or 2-(methylthio)aniline (MTA),33 with both directing groups developed by Daugulis et al.34 In general, (het)aryliodides or -bromides are employed for the functionalisation of cyclic or N-methylated amino acids at the β-position, whereas the introduction of alkyl groups is relatively rare. We were therefore inspired to determine if we could introduce a protected, highly functionalised side chain in a single step into a suitably protected alanine derivative, e.g. an N-phthaloylated aminoquinoline ester (Scheme 1). This particular approach is highly attractive, as it should enable the introduction of various side chains as potential zinc-binding motifs, regardless of the stereogenic centre at the α-position, as long as it does not epimerise under the reaction conditions used.
 | ||
| Scheme 1 Planned synthesis of protected Aeo via C–H-functionalization. | ||
Results and discussion
Based on the total synthesis of WF-3161 by Schmidt et al., who carefully investigated the four different macrocyclisation positions, we chose to focus on the position between Aeo and D-phenylalanine as the most promising. Before developing an entirely new route, we initially decided to verify the applicability of C–H functionalisation in this presumably challenging approach. Therefore, we synthesised iodide 5 in a similar manner as described by Schreiber et al.35 In this approach, we started with threitol-derived triisopropylsilyl (TIPS)-ether 1 (Scheme 2), which we previously used as a starting material in Pd-catalysed allylic alkylations.36 Swern oxidation and subsequent Wittig olefination using the p-methoxybenzyl (PMB)-protected phosphonium salt 237 provided (Z)-alkene 3 as a single stereoisomer. The olefin geometry does not play a role in this case, because the double bond was hydrogenated before the PMB was removed. It should be noted, however, that the order of the reaction steps is important, because hydrogenation after PMB-cleavage preferentially affords the deoxygenated product. The primary alcohol 4 was converted into iodide 5via the Appel reaction.38 With the required building block in hand, we subsequently investigated the key step of our synthesis: the C–H functionalisation of N-phthaloyl-protected alanine 8-aminoquinoline ester 6.39 The first experiments were carried out in t-butanol/dichloroethane (1:1), which afforded the desired product 7 in an acceptable yield of 44%. As a major side product, the direct substitution of the iodine by t-butanol was observed. Switching to the more sterically hindered t-amylalcohol solved this problem and afforded 7 in excellent yield for an alkyl coupling.
 | ||
| Scheme 2 Synthesis of an Aoe-precursor 7 from threitol derivative 1via C–H-functionalization. Reagents and conditions: (a) (COCl)2 (1.6 equiv.), DMSO (3.0 equiv.), NEt3 (5.0 equiv.), CH2Cl2, 0 °C, 1 h. (b) KHMDS (1.1 equiv.), Wittig salt 2 (1.2 equiv.), THF, 0 °C, 75 min (c) H2 (1 atm), Pd/C, MeOH, rt, 16 h. (d) DDQ (1.1 equiv.), CH2Cl2:H2O (20:1), rt, 2 h. (e) PPh3 (1.25 equiv.), imidazole (1.25 equiv.), I2 (1.24 equiv.), CH2Cl2, 1. 0 °C, 25 min; 2. rt, 3.5 h. (f) 6, Ag2CO3 (0.8 equiv.), Pd(OAc)2 (10 mol-%), dibenzyl phosphate (30 mol%), tert-amyl-OH:C2H4Cl2 (1:1), 60 °C, 24 h. | ||
Following the successful optimisation of the C–H functionalisation step, we developed a new synthetic approach by employing the Matteson homologation for the generation of the stereogenic centres in the side chain (Scheme 3).40 We recently utilised this highly flexible concept during the total synthesis of several natural products.41 Because we were unable to homologate benzyloxy-substituted methyl boronic esters, we started our synthesis with the known O-trityl-protected boronic ester 8,42 which had already been used by Matteson for a similar purpose (Scheme 3).43 Deprotonated benzyl alcohol was chosen as a nucleophile under standard reaction conditions, which afforded boronic ester 9 in good yield and in enantiomerically pure form. The second stereogenic centre and the linear spacer unit were generated by converting the O-silylated alkyl halide 10 into the corresponding Grignard reagent, which was subsequently used as a nucleophile in the second homologation step. The prolongated boronic ester formed was directly oxidised to the corresponding alcohol 11, which was obtained as a single stereoisomer. In the next step, we aimed to remove the trityl protecting group and protect the two OH-functionalities as benzyl esters, which would allow us to remove all protecting groups at once as the final step in the synthesis. Although, we had already replaced the previously used TIPS group with the more acid-stable tert-butyldiphenylsilyl (TBDPS) group, the selective cleavage of the trityl group was far from trivial. With HCl in dioxane or Amberlyst 15, partial or complete cleavage of the silyl protecting group was observed. With acetic acid, we were able to cleave the trityl group selectively, but by far the best results were obtained using anhydrous ZnBr2 in the presence of exactly one equivalent of H2O per equivalent ZnBr2. Under these conditions, only traces of the silyl cleavage product were observed and 7% of 11 could be recovered. The resulting diol 12 was double benzyl-protected (13) and following cleavage of the silyl protecting group, the terminal primary alcohol 14 was converted into the desired iodide 15 in an analogous manner to 5.
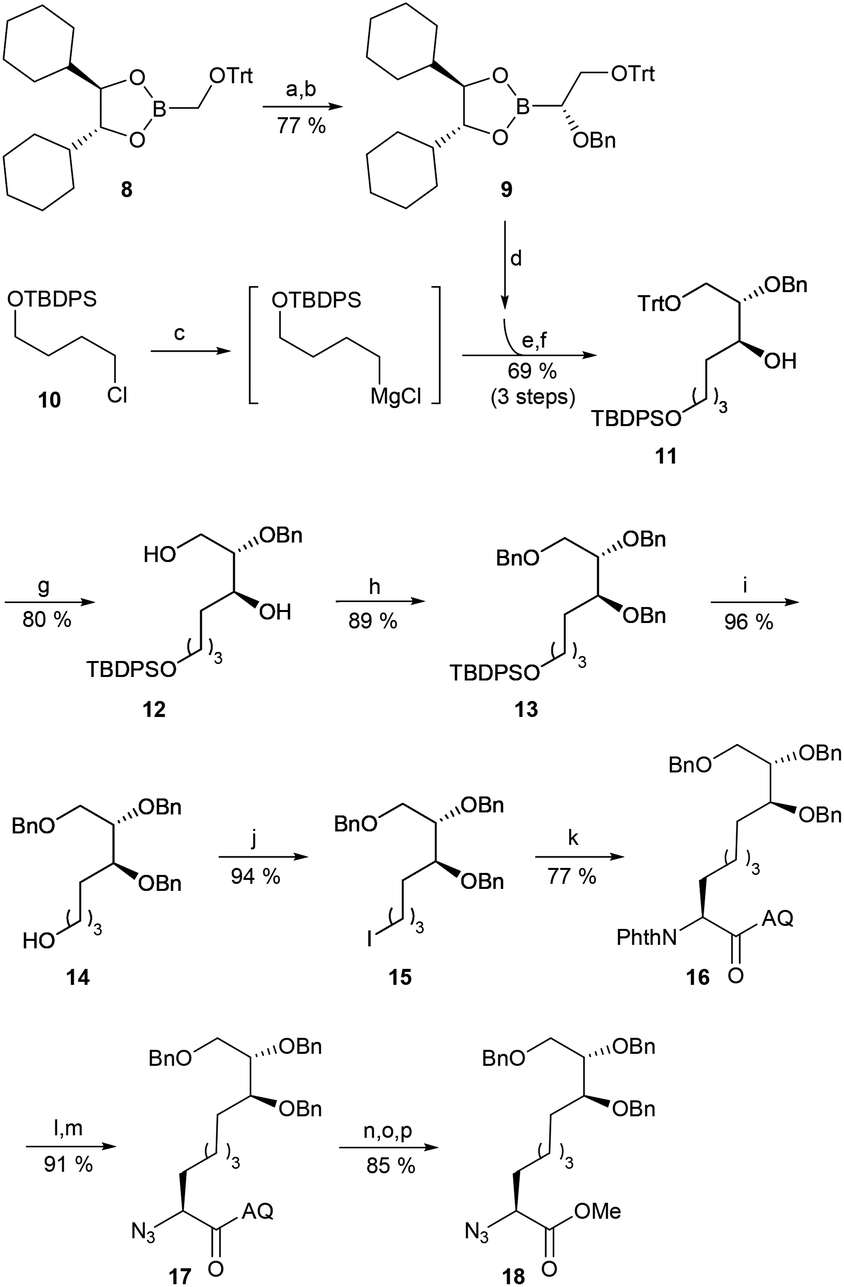 | ||
| Scheme 3 Synthesis of an Aoe-precursor 18via Matteson homologation and C–H functionalization. Reagents and conditions: (a) (1) CH2Cl2 (3.0 equiv.), LDA (1.35 equiv.), −40 °C; (2) ZnCl2 (3.0 equiv.), THF, rt. (b) BnOH (2.3 equiv.), NaH (2.0 equiv.), THF/DMSO, 0 °C to rt, 16 h. (c) Mg (2.7 equiv.), 1,2-dibromoethane (13 mol%), THF, Δ, 2 h. (d) (1) CH2Cl2 (3.0 equiv.), LDA (1.35 equiv.), −40 °C: (2) ZnCl2 (4.0 equiv.), THF, rt. (e) 0 °C to rt, 16 h. (f) NaOH (2.25 equiv.), H2O2 (2.25 equiv.), THF, rt, 90 min (g) ZnBr2, (4.0 equiv.), H2O (4.0 equiv.), CH2Cl2, rt, 3.5 h. (h) KOtBu (3.0 equiv.), BnBr (4.0 equiv.), THF, 0 °C, 100 min (i) TBAF (1.25 equiv.), THF, rt, 16 h. (j) PPh3 (1.25 equiv.), imidazole (1.25 equiv.), I2 (1.24 equiv.), CH2Cl2, rt, 3.5 h. (k) 6, Ag2CO3 (0.8 equiv.), Pd(OAc)2 (10 mol-%), dibenzyl phosphate (30 mol-%), tert-amyl-OH:C2H4Cl2 (1:1), 60 °C, 22 h. (l) hydrazine (1.2 equiv.), DIPEA (1.0 equiv.), EtOH, 80 °C, 20 h. (m) TfN3 (3.1 equiv.) in toluene, NaHCO3 (4.0 equiv.), CuSO4·5 H2O (4.3 mol%), MeOH/H2O, rt, 2 h. (n) Boc2O (4.0 equiv.), DMAP (60 mol%), MeCN, rt, 16 h. (o) LiOH (2.0 equiv.), H2O2 (5.0 equiv.), THF, 0 °C, 1 h. (p) K2CO3 (2.5 equiv.), MeI (3.0 equiv.), DMF, 0 °C to rt, 3 h. | ||
The subsequent C–H functionalisation under the previously optimised conditions afforded comparable results as previously. Protected amino acid 16 was obtained in high yield and as a single stereoisomer. The phthaloyl protecting group was removed under standard conditions in excellent yield, and the free amine was converted into the corresponding azide 17. Subsequent cleavage of the aminoquinoline, followed by O-methylation, provided methylester 18, which was used in the subsequent peptide couplings.
In our first attempt, we tried to couple our new amino acid with a preformed tripeptide; unfortunately, however, significant epimerisation in the peptide fragment was observed. Therefore, we decided to build up the peptide step by step (Scheme 4). Staudinger reduction of the azide of 18 afforded the free amine, which was coupled with Boc-protected pipecolic acid to 19. Boc-cleavage under standard conditions, using HCl in dioxane, and coupling with Boc-Leu afforded the desired tripeptide 20 in an overall high yield. Interestingly, a partial peptide bond cleavage was observed when reacting tripeptide 20 with HCl in dioxane, as in the previous step. After some optimisations, however, high yields were achieved with TFA/triisopropylsilane (TIPS-H),44 in which no decomposition was observed. Following peptide coupling to tetrapeptide 21 and saponification, the free carboxylic acid was obtained, and was first activated with pentafluorophenol in analogy to the Schmidt synthesis. Unfortunately, no synthetically useful yield was obtained from our attempts to cyclise this active ester. Instead, macrocyclisation with PyAOP and HOAt afforded the desired product 22 in high yields and without any epimerisation. The O-benzyl protecting groups were easily removed at ambient pressure in the presence of small amounts of HCl. Under these conditions, however, the formation of peptide cleavage products was an issue, as WF-3161 has a relatively high ring tension compared to other tetrapeptidic HDACi.45 This problem was solved by carrying out the hydrogenation reaction under neutral conditions and at higher pressure. The triol obtained was subsequently converted into the corresponding epoxide, which was not a trivial step. Attempts to tosylate the primary alcohol led to low conversions and poor selectivity. In contrast, a selective mesylation at low temperature was possible. Full conversion was still not achieved, though it was possible to suppress double mesylation using 1.2 equiv. of mesyl chloride. Fortunately, unreacted triol could be recovered from the reaction mixture. The best results in the epoxide formation step were obtained using the non-nucleophilic base DBU to afford epoxy alcohol 23, which was subsequently oxidised with Dess–Martin periodinane (DMP) to afford the desired WF-3161.
 | ||
| Scheme 4 Synthesis of WF-3161. Reagents and conditions: (a) PPh3 (3.0 equiv.), THF/H2O, 50 °C, 3 h. (b) Boc-L-Pip-OH (1.1 equiv.), TBTU (1.1 equiv.), DIPEA (1.2 equiv.), MeCN, 0 °C to rt, 16 h. (c) HCl/1,4-dioxane (10 equiv.), rt, 60 min (d) Boc-L-Leu-OH (1.2 equiv.), PyAOP (1.2 equiv.), DIPEA (2.5 equiv.), DMF, 0 °C to rt, 16 h. (e) TFA/TIPS-H/H2O (92.5:5:2.5), CH2Cl2, 0 °C, 60 min (f) Boc-D-Phe-OH (1.1 equiv.), PyAOP (1.1 equiv.), DIPEA (2.5 equiv.), DMF, 0 °C to rt, 16 h. (g) LiOH (1.2 equiv.), 1,4-dioxane/H2O, 0 °C to rt, 16 h. (h) TFA/TIPS-H/H2O (92.5:5:2.5), CH2Cl2, 0 °C, 40 min (i) PyAOP (10 equiv.), HOAt (10 equiv.), DIPEA (10 equiv.), DMF, high dilution (1 mM), 0 °C to rt, 16 h. (j) H2 (20 bar), Pd/C, MeOH, 16 h. (k) MsCl (1.2 equiv.), 2,4,6-collidine (10 equiv.), CH2Cl2, 0 °C, 22 h. (l) DBU (5.0 equiv.), MeOH, 0 °C, 5.5 h. (m) DMP (2.0 equiv.), CH2Cl2, rt, 2 h. | ||
Next, WF-3161 was initially screened in vitro for HDAC inhibitory activity, followed by an assessment of its ability to reduce cancer cell viability. A fluorescence-based assay was used to determine the activity of WF-3161 on the class I nuclear isoforms HDAC1, as well as the class II cytoplasmatic isoform HDAC6. The data are presented as IC50 values in Table 2. The already approved HDAC inhibitor vorinostat and the HDAC1 selective inhibitor mocetinostat were included as positive controls. WF-3161 inhibited HDAC activity in a dose-dependent manner, with IC50 values of 0.7 nM ± 0.1 nM for HDAC1. Interestingly, no inhibition of HDAC6 was observed at 25 μM, suggesting a strong selectivity towards class I HDACs, similar to that observed with Cyl-2, but different from that of the trapoxins.
To further measure the activity of the synthesised WF-3161, we examined its growth-inhibiting effects on an acute myeloid leukaemia cell line, based on the MTS assay (Promega), with vorinostat and mocetinostat as reference controls. The results are shown in Table 2. As indicated by the data, the macrocyclic WF-3161, which contains an epoxyketone, inhibits the cell viability of HL-60 in the low nanomolar range, with a GI50 value of 13 ± 1.1 nM. The low GI50 value obtained is also in accordance with the low IC50 value observed in the in vitro tests.
The increased potency of WF-3161 compared to that of vorinostat or mocetinostat in vitro and in the MTS assay could be related to the epoxyketone as a zinc-binding group. Interestingly, the mechanism of epoxyketone inhibition of HDACs is not entirely clear. In 1993, M. Kijima and colleagues hypothesised that trapoxin A, which also contains the epoxyketone, might act as either a tight noncovalent binder or covalently bind to the HDAC.46 This observation suggests that WF-3161 inhibits class I HDACs in a similar fashion and leads to increased potency compared to the hydroxamic acid and benzamide zinc-binding groups.
Conclusions
We have developed a novel synthesis for the HDAC inhibitor WF-3161, based on Matteson homologations and C–H functionalisation. The Matteson homologation was used to generate the stereogenic centre in the side chain and should be a suitable method for introducing other comparable zinc-binding motifs. The side chain was subsequently coupled to an alanine derivative via palladium-catalysed C–H-functionalisation. The WF-3161 obtained was screened in vitro for HDAC inhibitory activity and for its ability to reduce cancer-cell viability. Highly selective inhibition of HDAC1 compared to HDAC6 was observed, in addition to low nM activity towards the myeloid leukaemia cell line HL-60.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University and the Deutsche Forschungsgemeinschaft (DFG,Bruker Neo 500-447298507 to U. K.; SFB 992 (Project-ID: 192904750) to M. J. is gratefully acknowledged.References
- (a) H. Rajak, A. Singh, P. K. Dewangan, V. Patel, D. K. Jain, S. K. Tiwari, R. Veerasamy and P. C. Sharma, Curr. Med. Chem., 2013, 20, 1887–1903 CrossRef CAS PubMed; (b) S. Tan and Z.-P. Liu, ChemMedChem, 2015, 10, 441–450 CrossRef CAS PubMed; (c) X. Peng, Z. Sun, P. Kuang and J. Chen, Eur. J. Med. Chem., 2020, 208, 112831 CrossRef CAS PubMed; (d) T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed; (e) M. Daśko, B. de Pascual-Teresa, I. Ortín and A. Ramos, Molecules, 2022, 27, 715 CrossRef PubMed.
- M. Kilgore, C. A. Miller, D. M. Fass, K. M. Hennig, S. J. Haggarty, J. D. Sweatt and G. Rumbaugh, Neuropsychopharmacology, 2010, 35, 870–880 CrossRef CAS PubMed.
- (a) D. Marchion and P. Münster, Expert Rev. Anticancer Ther., 2007, 7, 583–598 CrossRef CAS PubMed; (b) J. M. Wagner, B. Hackanson, M. Lübbert and M. Jung, Clin. Epigenet., 2010, 1, 117–136 CrossRef CAS PubMed; (c) G. Shanmugam, S. Rakshit and K. Sarka, Transl. Oncol., 2022, 16, 101312 CrossRef CAS PubMed.
- (a) M. Yoshida, M. Kijima, M. Akita and T. Beppu, J. Biol. Chem., 1990, 265, 17174–17179 CrossRef CAS PubMed; (b) A. G. Kazantsev and L. M. Thompson, Nat. Rev. Drug Discovery, 2008, 7, 854–868 CrossRef CAS PubMed; (c) M. Jakovcevski and S. Akbarian, Nat. Med., 2012, 18, 1194–1204 CrossRef CAS PubMed; (d) F. F. Wagner, M. Weiwer, M. C. Lewis and E. B. Holson, Neurotherapeutics, 2013, 10, 589–604 CrossRef CAS PubMed; (e) A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- A. Closse and R. Huguenin, Helv. Chim. Acta, 1974, 57, 533–545 CrossRef CAS PubMed.
- (a) A. Hirota, A. Suzuki, H. Suzuki and S. Tamura, Agric. Biol. Chem., 1973, 37, 643–647 CrossRef CAS; (b) S. Takayama, A. Isogai, M. Nakata, H. Suzuki and A. Suzuki, Agric. Biol. Chem., 1984, 48, 839–842 CAS.
- H. Itazaki, K. Nagashima, K. Sugita, H. Yoshida, Y. Kawamura, Y. Yasuda, K. Matsumoto, K. Ishii, N. Uotani, H. Nakai, A. Terui, S. Yoshimatsu, Y. Ikenishi and Y. Nakagawa, J. Antibiot., 1990, 43, 1524–1532 CrossRef CAS PubMed.
- (a) K. Umehara, K. Nakahara, S. Kiyoto, M. Iwami, M. Okamoto, H. Tanaka, M. Kohsaka, H. Aoki and H. Imanaka, J. Antibiot., 1983, 36, 478–483 CrossRef CAS PubMed; (b) M. Kawai, R. Pottorf and D. H. Rich, J. Med. Chem., 1986, 29, 2409–2411 CrossRef CAS PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 188–193 CrossRef CAS PubMed.
- (a) R. Furumai, Y. Komatsu, N. Nishino, S. Khochbin, M. Yoshida and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 87–92 CrossRef CAS PubMed; (b) P. Gupta, R. C. Reid, A. Iyer, M. J. Sweet and D. P. Fairlie, Curr. Top. Med. Chem., 2012, 12, 1479–1499 CrossRef CAS PubMed.
- K. Cheng, S. Li and C. Liao, Curr. Med. Chem., 2017, 24, 4166–4179 CrossRef CAS PubMed.
- (a) A. V. Bieliauskas and M. K. H. Pflum, Chem. Soc. Rev., 2008, 37, 1402–1413 RSC; (b) S.-D. Kim, H. W. Knoche and L. D. Dunkle, Physiol. Mol. Plant Pathol., 1987, 30, 433–440 CrossRef CAS.
- (a) T. L. Newkirk, A. A. Bowers and R. M. Williams, Nat. Prod. Rep., 2009, 26, 1293–1320 RSC; (b) A. R. Maolanon, H. M. E. Kristensen, L. J. Leman, M. R. Ghadiri and C. A. Olsen, ChemBioChem, 2017, 18, 5–49 CrossRef CAS PubMed.
- D. H. Rich, J. Singh and J. H. Gardner, J. Org. Chem., 1983, 48, 432–434 CrossRef CAS.
- U. Schmidt, A. Lieberknecht, H. Griesser and F. Bartkowiak, Angew. Chem., Int. Ed. Engl., 1984, 23, 318–320 CrossRef.
- (a) J. E. Baldwin, R. M. Adlington, C. R. A. Godfrey and V. K. Patel, J. Chem. Soc., Chem. Commun., 1991, 1277 RSC; (b) J. E. Baldwin, R. M. Adlington, C. R. A. Godfrey and V. K. Patel, Tetrahedron, 1993, 49, 7837–7856 CrossRef CAS.
- (a) U. Schmidt, A. Lieberknecht, U. Kazmaier, H. Griesser, G. Jung and J. Metzger, Synthesis, 1991, 49–55 CrossRef CAS; (b) A. Krebs and U. Kazmaier, Tetrahedron Lett., 1996, 37, 7945–7946 CrossRef CAS; (c) M. Pohlman and U. Kazmaier, Org. Lett., 2003, 5, 2631–2633 CrossRef CAS PubMed.
- (a) L. Karmann, K. Schulz, J. Herrmann, R. Müller and U. Kazmaier, Angew. Chem., Int. Ed., 2015, 54, 4502–4507 CrossRef CAS PubMed; (b) P. Barbie and U. Kazmaier, Org. Lett., 2016, 18, 204–207 CrossRef CAS; (c) J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed; (d) M. Kohr, C. Walt, J. Dastbaz, R. Müller and U. Kazmaier, Org. Biomol. Chem., 2022, 20, 9609–9612 RSC.
- U. Kazmaier, Liebigs Ann./Recl., 1997, 285–295 CrossRef CAS.
- C. Quirin and U. Kazmaier, Eur. J. Org. Chem., 2009, 371–377 CrossRef CAS.
- P. Servatius and U. Kazmaier, Org. Biomol. Chem., 2018, 16, 3464–3472 RSC.
- (a) U. Kazmaier, J. Deska and A. Watzke, Angew. Chem., Int. Ed., 2006, 45, 4855–4858 CrossRef CAS PubMed; (b) U. Kazmaier, Org. Chem. Front., 2016, 3, 1541–1560 RSC.
- P. Servatius and U. Kazmaier, J. Org. Chem., 2018, 83, 11341–11349 CrossRef CAS PubMed.
- P. Proksch, R. Ebel, R. Edrada, F. Riebe, H. Liu, A. Diesel, M. Bayer, X. Li, W. H. Lin, V. Grebenyuk, W. E. G. Müller, S. Draeger, A. Zuccaro and B. Schulz, Bot. Mar., 2008, 51, 209–218 CrossRef CAS.
- U. Schmidt, U. Beutler and A. Lieberknecht, Angew. Chem., Int. Ed. Engl., 1989, 28, 333–335 CrossRef.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657–3659 CrossRef CAS PubMed; (c) R. Giri, J. Liang, J. Lei, J. Li, D. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Yu and J. Foxman, Angew. Chem., Int. Ed., 2005, 44, 7420–7424 CrossRef CAS PubMed; (d) S. Aspin, A. S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808–10811 CrossRef CAS PubMed; (e) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS PubMed.
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (b) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed; (c) X. Lu, B. Xiao, R. Shang and L. Liu, Chin. Chem. Lett., 2016, 27, 305–311 CrossRef CAS; (d) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed; (e) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Yu, Chem. Rev., 2017, 117, 8754–5786 CrossRef CAS PubMed; (f) S. Sengupta and G. Mehta, Org. Biomol. Chem., 2020, 18, 1851–1876 RSC.
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (c) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (d) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- (a) B. Mondal, B. Roy and U. Kazmaier, J. Org. Chem., 2016, 81, 11646–11655 CrossRef CAS PubMed; (b) T. Kinsinger and U. Kazmaier, Org. Lett., 2018, 20, 7726–7730 CrossRef CAS PubMed; (c) T. Kinsinger and U. Kazmaier, Org. Biomol. Chem., 2019, 17, 5595–5600 RSC; (d) M. Kohr and U. Kazmaier, Org. Lett., 2021, 23, 5947–5951 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed; (b) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188–5191 CrossRef CAS PubMed.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed.
- (a) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS; (b) J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef PubMed.
- J. Taunton, J. L. Collins and S. L. Schreiber, J. Am. Chem. Soc., 1996, 118, 10412–10422 CrossRef CAS.
- A. Horn and U. Kazmaier, Org. Lett., 2019, 21, 4595–4599 CrossRef CAS PubMed.
- M. Brovetto and G. Seoane, J. Org. Chem., 2008, 73, 5776–5785 CrossRef CAS PubMed.
- V. S. C. de Andrade and M. C. S. de Mattos, Curr. Org. Synth., 2015, 12, 309–327 CrossRef CAS.
- G. Liao, X. S. Yin, K. Chen, Q. Zhang, S. Q. Zhang and B. F. Shi, Nat. Commun., 2016, 7, 12901 CrossRef PubMed.
- (a) D. S. Matteson, Tetrahedron, 1989, 45, 1859–1885 CrossRef CAS; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (c) S. Kirupakaran, H.-G. Korth and C. Hirschhäuser, Synthesis, 2018, 2307–2322 CAS; (d) D. S. Matteson, B. S. L. Collins, V. K. Aggarwal and E. Ciganik, Org. React., 2021, 105, 427–860 Search PubMed.
- (a) J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed; (b) O. Andler and U. Kazmaier, Chem. – Eur. J., 2021, 27, 949–953 CrossRef CAS PubMed; (c) O. Andler and U. Kazmaier, Org. Biomol. Chem., 2021, 19, 4866–4870 RSC; (d) O. Andler and U. Kazmaier, Org. Lett., 2022, 24, 2541–2545 CrossRef CAS PubMed.
- (a) O. C. Ho, R. Soundararajan, J. Lu, D. S. Matteson, Z. Wang, X. Chen, M. Wei and R. D. Willett, Organometallics, 1995, 14, 2855–2860 CrossRef CAS; (b) M. Tost, O. Andler and U. Kazmaier, Eur. J. Org. Chem., 2021, 6459–6471 CrossRef CAS; (c) T. Kinsinger and U. Kazmaier, Org. Lett., 2022, 24, 3599–3603 CrossRef CAS PubMed.
- (a) D. S. Matteson and J.-J. Yang, Tetrahedron: Asymmetry, 1997, 8, 3855–3861 CrossRef CAS; (b) D. S. Matteson, R. Soundararajan, O. C. Ho and W. Gatzweiler, Organometallics, 1996, 15, 152–163 CrossRef CAS.
- A. Chan, W. M. Hussein, K. A. Ghaffar, N. Marasini, A. Mostafa, S. Eskandari, M. R. Batzloff, M. F. Good, M. Skwarczynski and I. Toth, Bioorg. Med. Chem., 2016, 24, 3095–3101 CrossRef CAS PubMed.
- M. Kawai, R. S. Pottorf and D. H. Rich, J. Med. Chem., 1986, 29, 2409–2411 CrossRef CAS PubMed.
- M. Kijima, M. Yoshida, K. Sugita, S. Horinouchi and T. Beppu, J. Biol. Chem., 1993, 268, 22429–22435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, copies of 1H and 13C NMR spectra, HPLC chromatograms and MS2 analysis. See DOI: https://doi.org/10.1039/d3ob00641g |
| This journal is © The Royal Society of Chemistry 2023 |