
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of fused tetramate-oxazolidine and -imidazolidine derivatives and their antibacterial activity†
Liban
Saney
a,
Tharindi
Panduwawala
a,
Xiang
Li
ab,
Kirsten E.
Christensen
a,
Miroslav
Genov
 d,
Alexander
Pretsch
d,
Dagmar
Pretsch
d and
Mark G.
Moloney
*ac
d,
Alexander
Pretsch
d,
Dagmar
Pretsch
d and
Mark G.
Moloney
*ac
aThe Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: mark.moloney@chem.ox.ac.uk
bDepartment of Pharmaceutical Engineering, China Pharmaceutical University, Nanjing, 211198, P. R. China
cOxford Suzhou Centre for Advanced Research, Suzhou Industrial Park, Building A, 388 Ruo Shui Road, Jiangsu 215123, P. R. China
dOxford Antibiotic Group, The Oxford Science Park, Magdalen Centre, Oxford OX4 4GA, UK
First published on 26th May 2023
Abstract
A chemoselective route which provides direct access to bicyclic tetramates, making use of Dieckmann cyclisation of functionalised oxazolidines and imidazolidines derived from an aminomalonate, is reported; calculations suggest that the observed chemoselectivity is kinetically controlled and leads to the thermodynamically most stable product. Some compounds in the library showed modest antibacterial activity against Gram-positive bacteria, and this activity is maximal in a well-defined region of chemical space (554 < Mw < 722 g mol−1; 5.78 < cLogP < 7.16; 788 < MSA < 972 Å2; 10.3 < rel. PSA < 19.08).
Introduction
The tetramate (pyrrolidine-2,4-dione) system occurs as the central scaffold in natural products with diverse bioactivity,1,2 and is increasingly important as a synthetic skeleton in its own right.3 Bicyclic derivatives may be accessed by a highly chemo-, diastereo- and enantioselective Dieckmann cyclisation of oxazolidine/thiazolidine templates 2a–g derived from hydroxyalkyl- and thioalkyl side chain amino acids (serine 1a, threonine 1b, allo-threonine 1c, cysteine 1d, or threo1e and allo-phenylserines 1f, Scheme 1).4,5 Of importance is the identity of the aldehyde which is used to condense the amino acids to form the oxazolidine/thiazolidine heterocycle, which has generally been limited to pivaldehyde; this aldehyde gives a relatively stable oxazolidine/thiazolidine template 2a–g, in which the bulky t-butyl group favours the most stable ring form in a ring-chain tautomeric equilibrium, and plays a significant role in the control of a subsequent diastereoselective N-acylation reaction and also directs the mode of the Dieckmann cyclisation towards bicyclic tetramates.6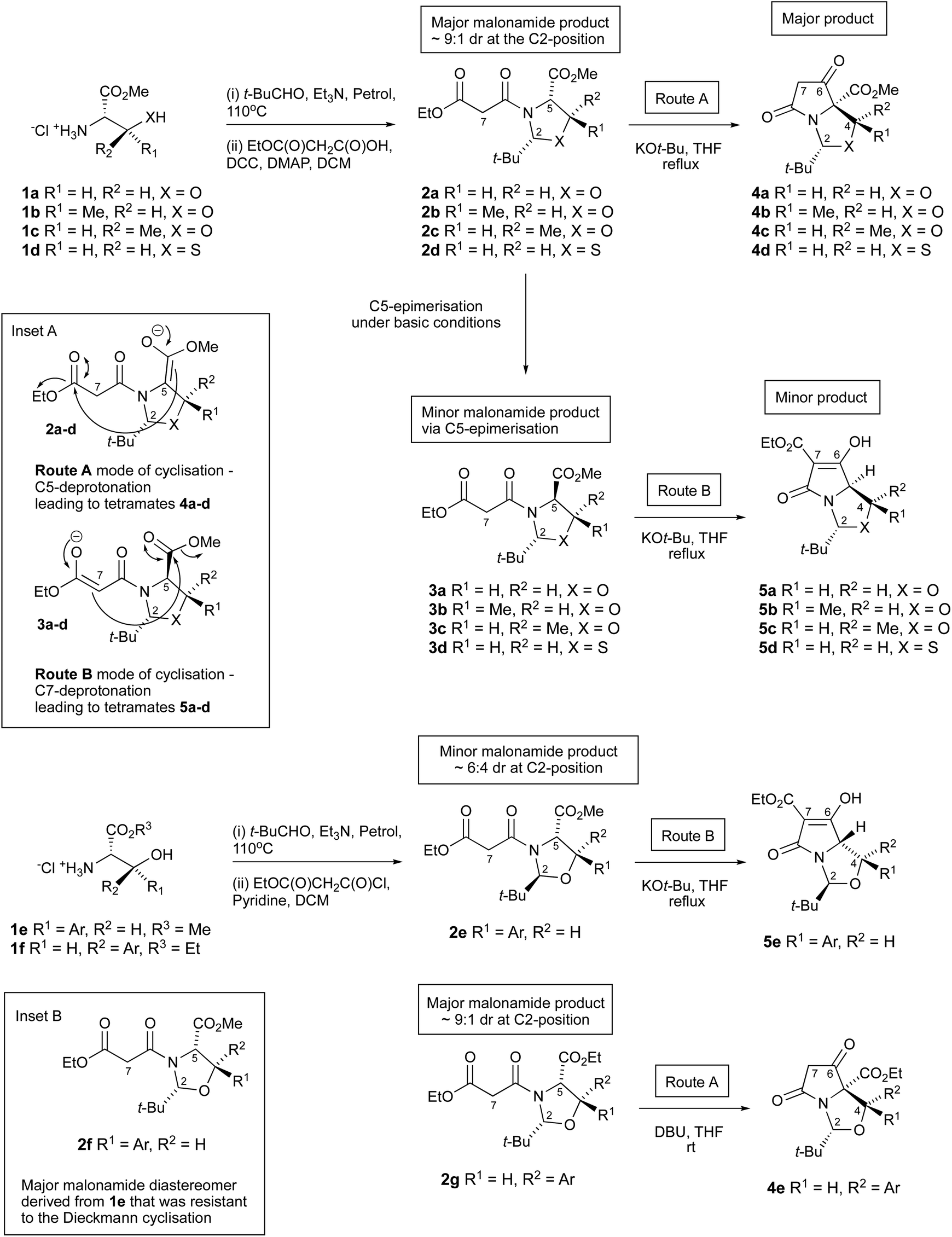 | ||
| Scheme 1 Dieckmann ring closures in malonyloxazolidines and -thiazolidines. | ||
For serine 1a, threonines 1b,c and cysteine 1d, the Dieckmann cyclisation usually occurs from the 2,5-cis-malonyloxa(or thia)zolidines 2a–d – which forms as the major product after N-acylation of oxazolidines – by closure from the C-5 enolate onto the side chain ethyl ester giving tetramates 4a–d as the major product (Scheme 1, route A and inset A), placing the C-2 t-butyl group on the exo-face (that is, the less hindered convex face) of the newly generated bicyclic ring system. An alternative mode of cyclisation, starting from the 2,5-trans-malonyloxa(or thia)zolidines 3a–d (which normally arise as a minor component by epimerisation at C-5 under the basic conditions of the cyclisation reaction) proceeds through closure of the more stabilised malonamide side chain enolate C7 onto the C-5 ester (Scheme 1, route B and inset A), also placing the C-2 t-butyl group on the exo-face, and which generates tetramates 5a–d but typically only as minor products.6 Route B closure can be achieved by adjusting reaction conditions7 or by forcing ring closure by blocking the acidic α-position with a methyl group.8 Such a process is not dissimilar to the biosynthesis of tetramate natural products.9,10 We have also shown that allo-arylserines 1f close by the conventional route A pathway5 to give tetramate 4evia the Dieckmann cyclisation of the major malonamide diastereomer 2g, but interestingly, the minor 2,5-trans malonamide 2e diastereomer derived from threo-aryl serines 1e has been found to ring close by route B to give tetramate 5e. The exclusive chemoselective formation of tetramates 4e and 5e can be explained via steric effects, in which the bulky C2-t-Bu and C4-aryl groups prefer to reside on the less hindered exo-face of the bicyclic lactam. On the other hand, the major 2,5-cis malonamide diastereomer 2f derived from amino ester 1e (inset B, Scheme 1) did not undergo the Dieckmann cyclisation as the C2-t-Bu and C4-aryl groups are placed on the more hindered endo-face of the bicyclic lactam regardless of which cyclisation route is taken.
Although cyclic N,O-hemiaminal ethers derived from aldehydes and ketones are stable provided that the N-heteroatom is acylated,11–14 aldehydes other than pivaldehyde were initially found to be unsuitable in the process shown in Scheme 1. However, more recently, thiazolidine substrates derived from cysteine 1d using aromatic aldehydes were found to give hemiaminal thioether systems cis- and trans-7, and Dieckmann cyclisation successfully gave the corresponding tetramate products 9 and ent-9 but in a different sense of chemoselectivity, by route B (Schemes 1 and 2).8,15 Thus, it became clear that the chemo- and stereoselective outcomes in the oxazolidine and thiazolidine series differed, and we report here a more detailed examination of the reactions of hemiaminal and aminal –NCH(Ar)X– (X = O, NR) heterocycles in this Dieckmann cyclisation.
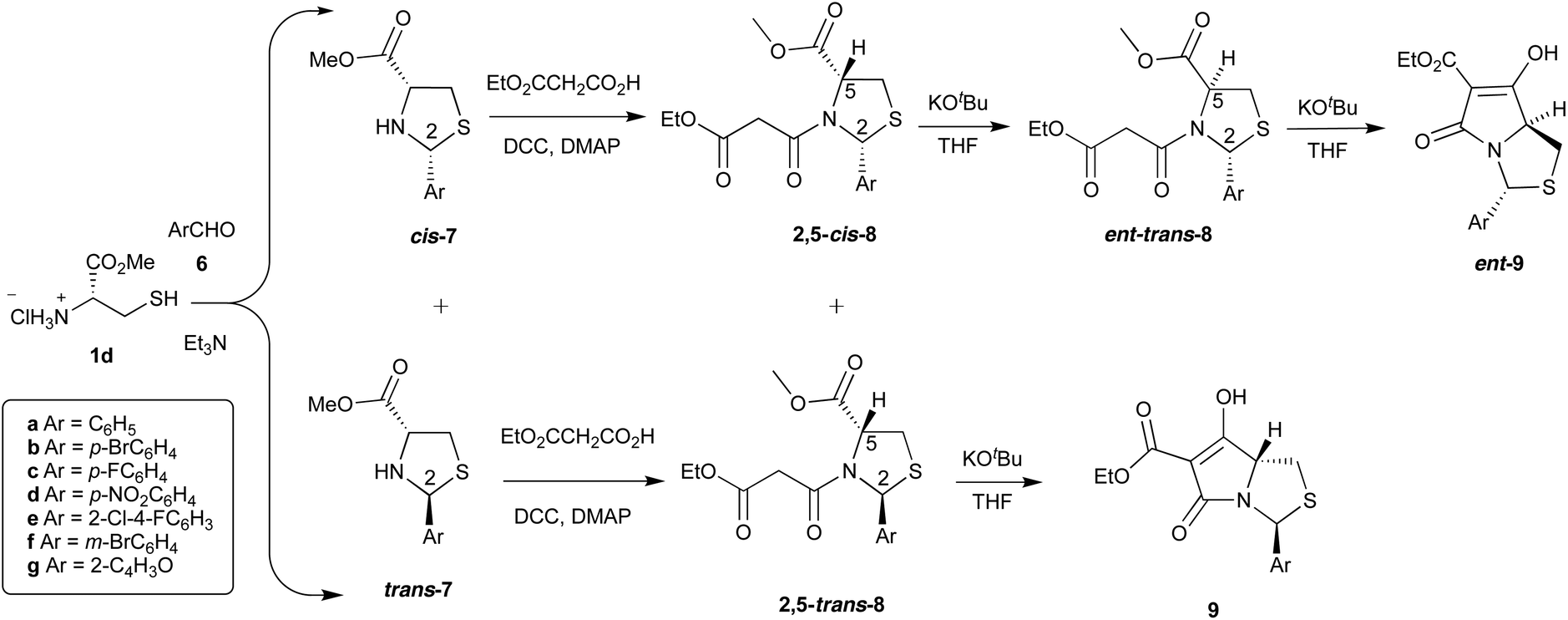 | ||
| Scheme 2 Dieckmann ring closures in arylthiazolidine templates. | ||
Results and discussion
An initial examination demonstrated that L-serine methyl ester hydrochloride 1a could be condensed with p-bromobenzaldehyde to form oxazolidines 10a,b that were obtained only as minor unstable products, along with the ring-opened imine 11 as the major product, readily distinguished in the NMR spectrum of the crude reaction mixture (δH for H-2 (ppm): 10a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11:10b = 5.27; 8.28; 5.64 in the ratio 1.2:3.3:1.0) (Scheme 3). The open chain form (B) was observed to be the predominant form (Table S1, ESI†). This contrasted with the equivalent condensation using pivaldehyde, in which the oxazolidine was obtained exclusively in the ring-closed form.16,17 However, it was found that reaction of the oxazolidine 10a,b with ethyl hydrogen malonate by DCC/DMAP coupling drove the equilibrium between the oxazolidine 10a,b and imine 11 towards the formation of the oxazolidine ring system, giving malonamides 12a,b in 51% yield as a mixture of diastereomers, in which the 2,5-cis diastereomer was the major product (2.7:1 dr). The change in diastereomeric ratio in favour of the cis-diastereomer upon N-acylation indicates that the ring-chain tautomerism allows equilibration to the more stable product, where both substituents are (presumably) pseudoequatorial. Although it seems likely that this 2,5-cis diastereomer is the more stable, the diastereoselectivity was much lower than pivaldehyde-derived systems, in which malonamide 2a was obtained almost exclusively as the single 2,5-cis-diastereomer.16,17 Each diastereomer 12a,b existed as a rotameric pair (δH for H-2 (ppm) cis-12a (6.04 and 6.16 ppm) and trans-12b (6.29 and 6.42 ppm) (major and minor rotamers respectively)) and the relative stereochemistry of C-2 and C-5 substituents across the ring was confirmed by NOE experiments (Fig. S1, ESI†). NMR resonances for H-2 signals were distinct and for both rotamers, H-2 of the trans-diastereomer appeared more downfield than the cis-diastereomer. The malonamides 12a,b were subjected to the Dieckmann cyclisation using KOtBu, to give the bicyclic tetramate 13, and although 14 was not isolated, it could be detected by mass spectrometry in the crude reaction mixture. This is in contrast to the observed reactivity of malonamide 2a with a t-Bu at C-2 position, where the major product of cyclisation was the methyl ester 4a (Scheme 1). However, during acidic work up of the cyclisation reaction, the product proved to be extremely unstable. Thus, the crude reaction mixture was directly purified by column chromatography on silica gel run with 1% Et3N, but the desired product 13 was isolated in only 8% yield. Thus, it became apparent that the stability of these compounds was a significant drawback, derived from the lability of the hemiaminal ether system, at least compared to the thioether system.15
:11:10b = 5.27; 8.28; 5.64 in the ratio 1.2:3.3:1.0) (Scheme 3). The open chain form (B) was observed to be the predominant form (Table S1, ESI†). This contrasted with the equivalent condensation using pivaldehyde, in which the oxazolidine was obtained exclusively in the ring-closed form.16,17 However, it was found that reaction of the oxazolidine 10a,b with ethyl hydrogen malonate by DCC/DMAP coupling drove the equilibrium between the oxazolidine 10a,b and imine 11 towards the formation of the oxazolidine ring system, giving malonamides 12a,b in 51% yield as a mixture of diastereomers, in which the 2,5-cis diastereomer was the major product (2.7:1 dr). The change in diastereomeric ratio in favour of the cis-diastereomer upon N-acylation indicates that the ring-chain tautomerism allows equilibration to the more stable product, where both substituents are (presumably) pseudoequatorial. Although it seems likely that this 2,5-cis diastereomer is the more stable, the diastereoselectivity was much lower than pivaldehyde-derived systems, in which malonamide 2a was obtained almost exclusively as the single 2,5-cis-diastereomer.16,17 Each diastereomer 12a,b existed as a rotameric pair (δH for H-2 (ppm) cis-12a (6.04 and 6.16 ppm) and trans-12b (6.29 and 6.42 ppm) (major and minor rotamers respectively)) and the relative stereochemistry of C-2 and C-5 substituents across the ring was confirmed by NOE experiments (Fig. S1, ESI†). NMR resonances for H-2 signals were distinct and for both rotamers, H-2 of the trans-diastereomer appeared more downfield than the cis-diastereomer. The malonamides 12a,b were subjected to the Dieckmann cyclisation using KOtBu, to give the bicyclic tetramate 13, and although 14 was not isolated, it could be detected by mass spectrometry in the crude reaction mixture. This is in contrast to the observed reactivity of malonamide 2a with a t-Bu at C-2 position, where the major product of cyclisation was the methyl ester 4a (Scheme 1). However, during acidic work up of the cyclisation reaction, the product proved to be extremely unstable. Thus, the crude reaction mixture was directly purified by column chromatography on silica gel run with 1% Et3N, but the desired product 13 was isolated in only 8% yield. Thus, it became apparent that the stability of these compounds was a significant drawback, derived from the lability of the hemiaminal ether system, at least compared to the thioether system.15
 | ||
| Scheme 3 Dieckmann ring closures in aryloxazolidines. | ||
Of interest was whether threo-phenylserines, possessing an additional phenyl substituent on the hydroxyalkyl side chain, might prove to be more stable. threo-Phenylserine methyl ester hydrochloride 1e was condensed with benzaldehyde and para-substituted benzaldehydes to form oxazolidines 15a–d using the established protocols (Scheme 4). These compounds were not purified by column chromatography due to the possibility of retro-aldol decomposition on silica gel18 but excess aldehyde was always detected in the NMR spectra of the crude products, consistent with poor conversion to the desired products. Oxazolidines 15a–d were therefore used in crude form and were immediately N-acylated under basic conditions to form malonamides 16a–d and 17a–d (Table S2, ESI†), with the 2,5-cis diastereomer being slightly more favoured. This was consistent with earlier reports.4 The yields obtained were moderate and was calculated over two steps from amino ester 1e. In the 1H-NMR data, the H2 signals were invariably more downfield for malonamides 16a–d/17a–d than for malonamides derived from threo-phenylserines 1e that were condensed with pivaldehyde 2e–f. nOe analysis was used to determine the relative stereochemistry for compounds 16a,d and 17a,d (Scheme 4) and was further confirmed from a single-crystal X-ray structure of compounds 16c and 17c (Fig. S2, ESI†).19 Based on these X-ray crystal structures and the similarities in the NMR data for the compound series (Table S2, ESI†), the relative stereochemistry of malonamides 16a–d and 17a–d was assumed to be similar.
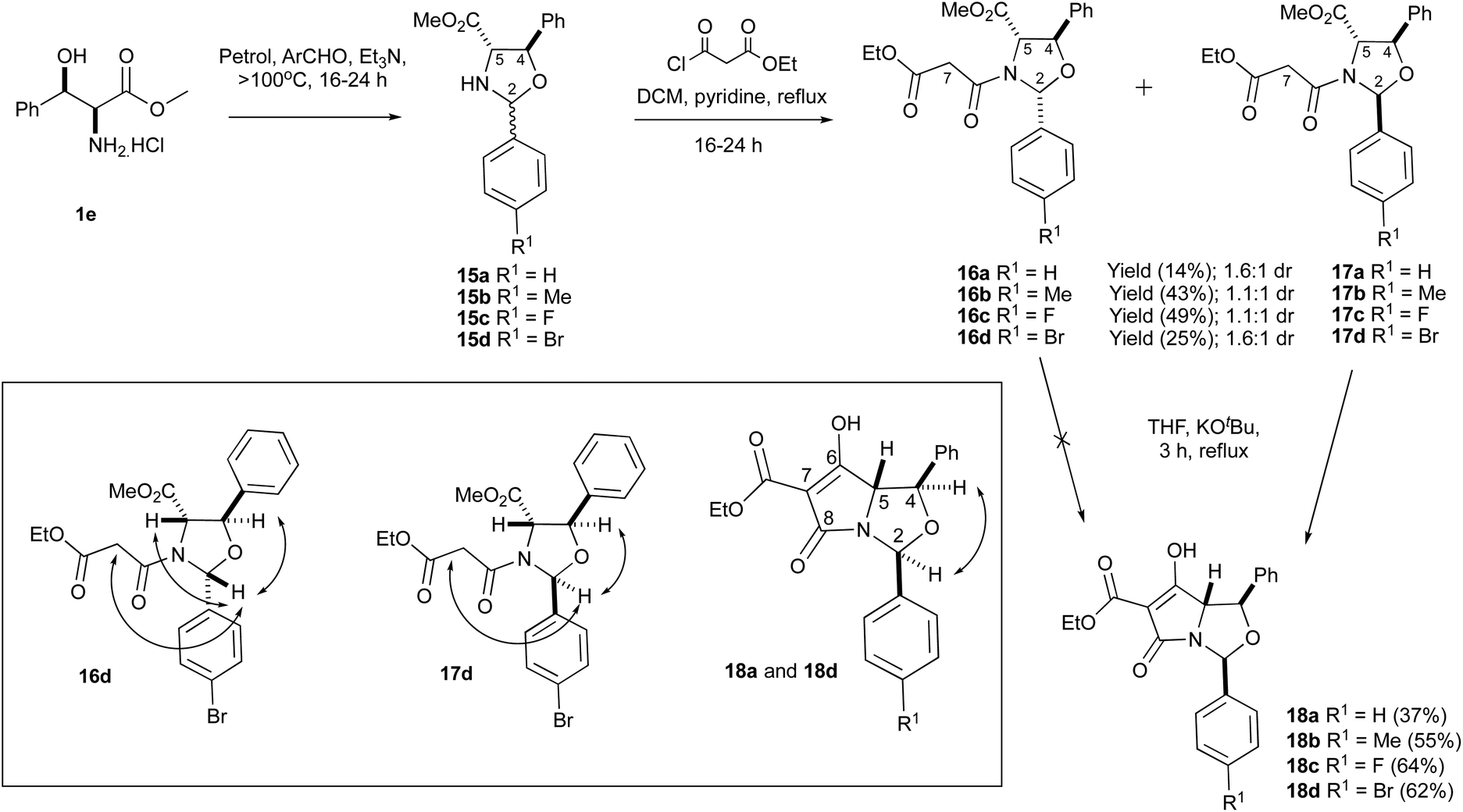 | ||
| Scheme 4 Dieckmann ring closures in variously substituted aryloxazolidines. | ||
Unlike the C2-tBu series,4 these malonamide diastereomers 16a–d/17a–d were easier to separate by chromatographic purification and could be obtained in gram-scale quantities. Interestingly, however, the NMR spectra for malonamides 17a–d in comparison to malonamides 2e that had a tBu substituent at the C2-position were very different. In the case of malonamides 17a–d, the NMR peaks were all sharp (Fig. S3, ESI†), but the presence of rotamers could not be found in the 2-D NOESY spectrum, whereas the NMR peaks for malonamides 2e – particularly the oxazolidine ring protons – were very broad at room temperature but could be sharpened using low temperature VT-NMR experiments.4 The bulky tBu group at the C2-position for compounds 2e was probably giving rise to this conformational dynamic, which was not seen for the planar aromatic ring substituents attached at the C2-position for malonamides 17a–d. Moreover, malonamides 16a–d clearly existed as a pair of rotamers around the N-malonyl bond, unlike 17a–d. This was chiefly detected in the 2D-NOESY spectrum where chemical exchange processes were detected. Analogues 2,5-cis-malonamides 2a–b,d derived from the methyl ester derivatives of L-serine 1a, L-threonine 1b and L-cysteine 1d have all been reported to exist as pairs of rotamers.17
2,5-cis Malonamides 16a,d were subjected to the Dieckmann cyclisation using KOtBu in THF (Scheme 4), but none of the desired product was detected by 1H-NMR spectroscopy or mass spectrometry. This was consistent with earlier findings that the major 2,5-cis malonamide diastereomer 2f derived from threo-arylserine 1e was resistant to the Dieckmann cyclisation.4 However, the 2,5-trans malonamides 17a–d readily reacted with KOtBu to give the desired tetramates 18a–d in acceptable yields – typically between 37–64% – as the sole products (Table S3, ESI†). These compounds were not purified on silica gel due to the stability problems encountered earlier for tetramates 5e, but in any case, the crude products were pure enough for direct NMR analysis (Fig. S4, ESI†). The chemoselectivity of the Dieckmann cyclisation and the lack of diastereoselectivity of the N-acylation reaction of oxazolidines 15a–d was consistent with that seen for the C2-tBu series for threo-phenylserine derived bicyclic tetramates 5e.4 The relative stereochemistry of tetramates 18a–d was assigned using nOe spectroscopy. The formation of tetramates 18a–d is noteworthy since they proved to be isolable after acidic workup and were obtained in better yields than C2-functionalised tetramates 13 derived from L-serine 1a, although they were very unstable upon short-term storage in their solid/oil form. Moreover, the H2-signal for tetramates 18a–d was invariably more downfield in comparison to the C2-tBu series 5e.4 Attempted aminolysis of tetramate 18a,d with 1-adamantylamine using toluene as a solvent – a reaction which had reliably generated the corresponding C7-carboxamide in related systems4 – did not give the corresponding C7-carboxamide, most likely due to steric bulk in the bicyclic system, or the breakdown of the starting material at high temperatures – potentially due to the lability of the N,O-acetal. This work showed that –OCH(Ar)N– hemiaminal ether systems were accessible, provided sufficient stability was provided by substituents on the tetramate ring.
Although N,O- and N,S-hemiaminal ethers have been examined in some detail, the corresponding N,N-bicyclic tetramic acids have not. As thiazolidines are known to be more stable than oxazolidine derivatives,11,20 where imidazolidine derivatives might fit in this spectrum was of interest. Although we had earlier demonstrated that N,N-bicyclic tetramates could be derived from imidazolidine scaffolds via 1,3-dipolar cycloaddition21 by modification of a literature approach,22 the resulting tetramic acids possessed numerous ethyl ester substituents that did not allow for chemoselective elaboration for aminolysis reactions. Instead, diethyl aminomalonate hydrochloride 19, along with 2 eq. of substituted benzaldehydes and benzylamine, was reacted under basic conditions and heated to more than 100 °C in a Dean–Stark trap to form imidazolidines 20a–l (Scheme 5), as either a single diastereomer or as a mixture of diastereomers (Table S4, ESI†), by a one-pot, three component 1,3-dipolar cycloaddition using a modification of the work of Nagao et al.23 In the case of imidazolidine 20a, a single crystal X-ray structure was obtained which confirmed the relative stereochemistry of the major diastereomer that had been assigned by nOe studies – in which the relative stereochemistry was assigned to be 2,4-cis (Fig. S2, ESI†).19 Additionally, the spectroscopic data for 20a matched literature data from the work of Nagao et al.23 This enabled the spectroscopic data for the rest of the 2,4-cis imidazolidine series to be assigned based upon comparison with imidazolidine 20a (Table S4, ESI†). NMR data and the X-ray crystal structures of imidazolidines 20b,e–f also confirmed the presence of the 2,4-trans diastereomer (Fig. S2, ESI†).19
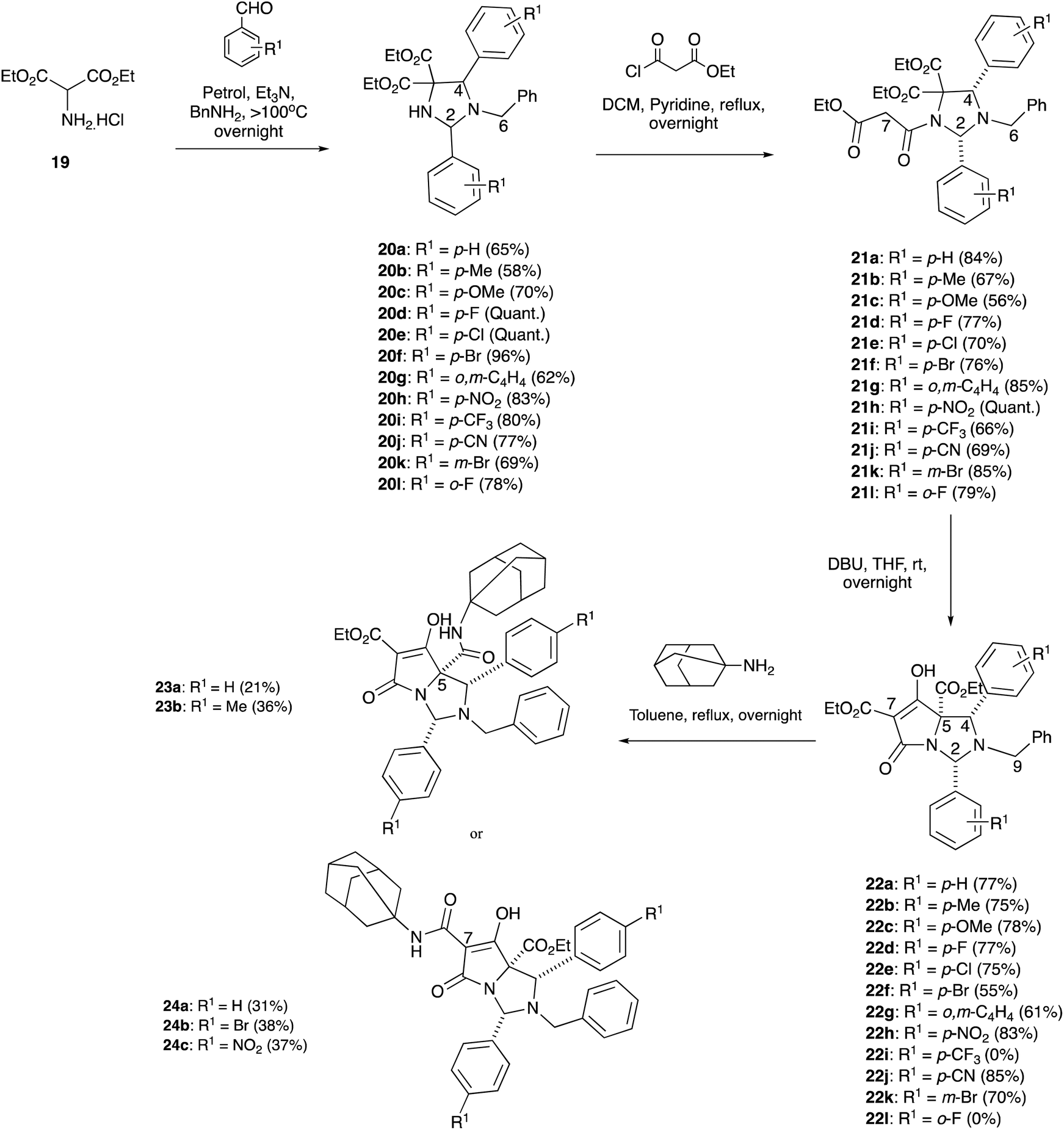 | ||
| Scheme 5 Dieckmann ring closures in malonylimidazolidines. | ||
Imidazolidines 20a–l were stable to chromatographic purification, unlike the corresponding oxazolidine series, although their diastereoisomers were often inseparable. Interestingly, in the 1H-NMR data, the H-2 and H-4 signals with electron-withdrawing ring substituents were more downfield than the electron-donating substituents, and the signals of the 2,4-trans diastereomers were invariably more downfield compared with the 2,4-cis diastereomers (Table S4, ESI†). The methylene ethyl ester peaks were all clearly diastereotopic in the 1H-NMR spectra (Fig. S5, ESI†). Imidazolidines 20a–l were then N-acylated with ethyl malonyl chloride to form malonamides 21a–l as single diastereomers in good to excellent yields (Scheme 5 and Table S5, ESI†), demonstrating that Seebach's “cis effect” (i.e. formation of a single cis-2,4-diastereomer upon N-acylation of the oxazolidine substrate24) applied in this case. The relative stereochemistry of 21a–d,f and h was established by nOe studies and this was confirmed with various X-ray crystal structures of malonamides 21a,c–d and l (Fig. S2, ESI†).19 As a result, it was assumed that the rest of the malonamide series had the same relative stereochemistry due to consistencies in the NMR spectra (Table S5, ESI†). It was observed that one of the diastereotopic methylene malonyl protons at the H7-position had a very broad doublet peak, whereas the other proton had a sharp doublet peak at room temperature (Fig. S6(a), ESI†), and this observation was seen in all but two cases – malonamides 21h and j – both of which contained an electron withdrawing group, in which their respective methylene protons were both relatively broad. In the case of malonamide 21a, use of low-temperature VT-NMR experiments (193 K) sharpened the broad doublet peak (Fig. S6(b), ESI†), indicating that conformational dynamics were operating within this monocyclic system.
Malonamides 21a–l were subjected to the usual Dieckmann cyclisation using DBU in THF to form tetramates 22a–h, j–k in good yields with high levels of chemoselectivity and in which only a single diastereomer was isolated (Scheme 5). These compounds were not purified by column chromatography as they were found to be unstable in solution, giving C7-decarboxylation over time. However, the crude material was relatively pure for NMR structural analysis (Fig. S7, ESI†). The relative stereochemistry – determined from nOe studies – clearly showed a 2,4-cis relationship across the bicyclic core 22a–d, indicating that no epimerisation had occurred at the C2 and C4-positions under basic conditions. Moreover, as the chemical shifts and coupling constants of 22a–d were consistent across the series (Table S6, ESI†), it was assumed that all tetramates had the same relative stereochemistry. The C5-ethyl ester stereochemistry was initially assigned as being on the same face as the C2 and C4-aryl substituents, similar to reported tetramates 4e, placing the C5-ethyl ester on the less hindered exo-face of the bicyclic lactam. This was later supported with DFT calculations (vide infra) which identified the most stable product. It was assumed that reaction with 1-adamantylamine would proceed by chemoselective attack at the C7-ethyl ester but in the event, indiscriminate attack at both the C5 and C7-ethyl ester positions was observed to form both C5-23a and C7-24a carboxamides (Scheme 4). The assignment of structure to these by NMR analysis using NOESY or HMBC analysis was difficult, but a comparison of chemical shift information with the structures of previously reported analogues4,5 enabled tentative assignment of the C5-carboxamide 23a and C7-carboxamide 24a (Fig. S8, ESI†) as the C7-ethyl ester tetramate 5e tended to be more downfield in comparison to the C5-ethyl ester tetramate 4e – and as a result it was assumed the same was true for tetramates 22.
It was further possible to show – taking inspiration from the work of Liu et al.25 – that when diethyl aminomalonate 19 was reacted with 2 eq. of benzaldehyde and 4-tert-butylaniline under basic conditions, imidazolidine 25 could be formed in 56% yield with an acceptable level of diastereoselectivity (4:1 dr), with the major diastereomer being the 2,4-cis diastereomer as established by NOESY (Scheme 6). Additionally, it was shown that a temperature of 100 °C under a Dean–Stark trap was not necessary for the cycloaddition reaction to occur and that O'Shaughnessy's reaction conditions (anhydrous MgSO4) enabled these reactions to occur under much milder conditions.21 Imidazolidine 25 was N-acylated with ethyl malonyl chloride to form malonamide 26 in 65% yield as a single diastereomer. The relative stereochemistry of 26 was confirmed from a single-crystal X-ray structure and showed a 2,4-cis relationship, consistent with the N-acylation of imidazolidines 20a–l (Fig. S2, ESI†).19 Surprisingly, the NMR peaks for the methylene malonyl protons at the H7-position for 26 were both relatively sharp, which was in contrast to the N-benzyl malonamides 21a–l, again suggesting that conformational dynamics and substituents attached around the periphery of the imidazolidine ring system at the C2, N3 and C4-positions could have a pronounced effect on the H7-protons (Fig. S9, ESI†).
 | ||
| Scheme 6 Dieckmann ring closures in malonyl-N-arylimidazolidines. | ||
Malonamide 26 was subjected to the Dieckmann cyclisation, but the extraction process for this reaction, as well as for benzyl tetramates 20i was complicated by the formation of emulsions. However, an inseparable mixture of tetramate 28 along with C7-decarboxylated tetramate 27 could be obtained in low yield, whose structures were confirmed by 1H-NMR and mass spectroscopy. Moreover, an aldehyde signal at ∼10 ppm was detected in the NMR spectrum after the Dieckmann cyclisation for some products, suggesting that the N,N-aminal was not as stable as the N,O-hemiaminal formed by condensation with pivaldehyde; this is unsurprising given the basicity of the N3-amine in the bicyclic system.
In order to understand the chemo- and diastereoselectivity of this process, DFT calculations were conducted (Fig. 1), and these showed that this reaction was governed by kinetic and thermodynamic control, in which the favoured reaction pathway (route A) – highlighted in red – had a considerably lower Ea than route B – highlighted in blue – by 9.1 kcal mol−1, to give a tetrahedral alkoxide which was more stable in the case of route A (by 4.9 kcal mol−1). In both the transition state and the product, this stability is likely to arise from an arrangement in which most bulky substituents are located on the exo-(convex) face of the bicyclic system; the close proximity of the malonyl enolate to the reacting ethoxycarbonyl group will also assist the kinetics of the ring closure process. This outcome further confirmed that the C5-ethyl ester was most likely to be on the same face as the C2 and C4-aryl substituents, which is consistent with Dieckmann cyclisations of similar bicyclic tetramate derivatives.4,5 Moreover, this work also showed that –NCH(Ar)N– aminal systems are accessible, and whose better stability relative to –NCH(Ar)O– hemiaminal ether systems was likely assisted by the electronic withdrawing properties of the tetramate ring.
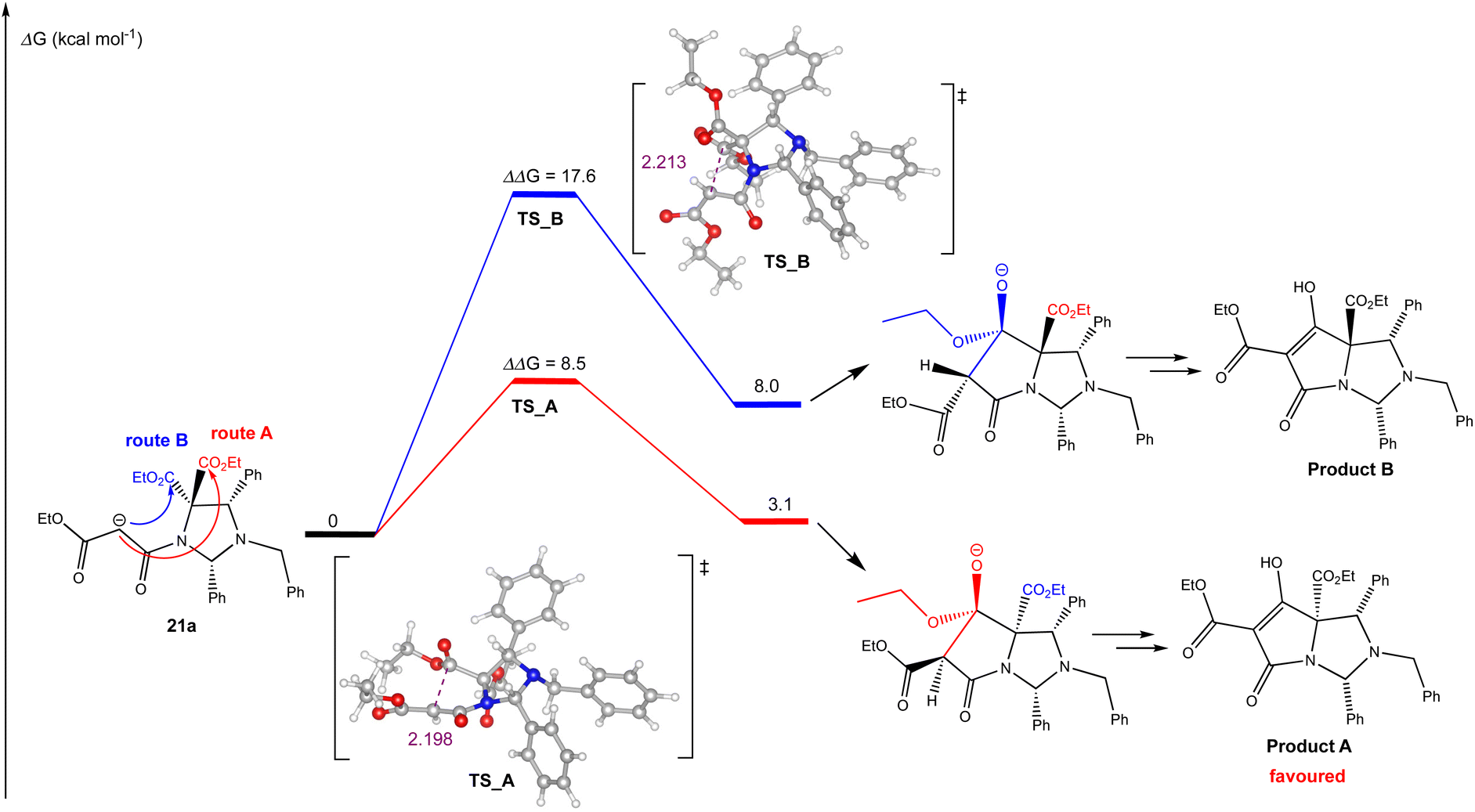 | ||
| Fig. 1 DFT calculated reaction energy profile for the Dieckmann cyclisation of malonamide 21a, using M06-2X/def2-TZVP method level of theory with SMD solvation (THF) and zero-point energy correction (Table S7, ESI†). All energies (Gibbs free energy) are reported in kcal mol−1 and are referenced to the lowest energy carbanion generated after initial deprotonation. Distances are in angstrom (Å). | ||
Evaluation of the antibacterial activity of C7-ethyl ester tetramates derived from imidazolidine scaffolds 22a, 22c and 22h showed no antibacterial activity against MRSA and E. coli, although some activity was seen against MRSA for compounds 22b and 22f at concentrations of 31.25 μg mL−1. This indicated that appropriate functionalisation around the C2 and C4-aromatic ring systems allowed antibacterial activity to be restored. C5-carboxamide 23b showed good bioactivity against MRSA at MIC = 3.9 μg mL−1 and C7-carboxamide 24a showed good antibacterial activity against MRSA (MIC = 3.9 μg mL−1), although this was far less potent than that of the C7-adamantylcarboxamide-N,O-bicyclic tetramates which typically had MICs of 0.25 μg mL−1.4,5 Tetramate 24c was far less potent than 24a (MIC = 15.6 μg mL−1) and tetramate 24b showed no antibacterial activity against MRSA (Table 1). Unsurprisingly, none of these carboxamides 23b and 24a–c were active against E. coli, in keeping with the known activity profiles of tetramates.
| Compound | MIC against MRSA (μg mL−1) | Cell lines EC50 (μg mL−1) | Therapeutic ratio | |||
|---|---|---|---|---|---|---|
| HeLa EC50 | HEK-293 | CaCo | MDCK | |||
| 22a | Not active | — | — | — | — | — |
| 22b | 31.25 | 31.3 | 31.3 | 62.5 | 31.3 | 2 |
| 22c | Not active | — | — | — | — | — |
| 22f | 31.25 | 31.3 | 31.3 | 62.5 | 31.3 | 2 |
| 22h | Not active | 31.3 | 62.5 | 125.0 | 31.3 | — |
| 23b | 3.9 | 15.6 | 62.5 | 125.0 | 62.5 | 32 |
| 24a | 3.9 | 31.3 | 62.5 | 62.5 | 62.5 | 16 |
| 24b | Not active | 62.5 | 62.5 | 62.5 | 125.0 | — |
| 24c | 15.6 | 62.5 | 62.5 | 62.5 | 125.0 | 4 |
A toxicity study was completed and interestingly, compounds 23b and 24a had a therapeutic ratio of 32 and 16 respectively against CaCo cell lines (Table 1). This was a promising starting point, as antibacterial drug candidates should have >10-fold higher antibacterial activity over cytotoxicity.26 Moreover, Brown et al.27 had demonstrated that high lipophilicity tended to correlate with increased cytotoxicity, however, these bicyclic tetramates, although being quite lipophilic (Table S8, ESI†), were generally non-toxic. Moreover, although these N,N-bicyclic tetramates were less potent than their N,O-bicyclic counterparts against MRSA,4,5 they were also less toxic in comparison, thus demonstrating that manipulation of C2, N3, C4, C5 and C7 all give scope for control of antibacterial activity. Additionally, physicochemical properties of the most active compounds correspond to a distinct chemical space; 554 < Mw < 722 g mol−1; 5.78 < cLogP < 7.16; 788 < MSA < 972 Å2; 10.3 < rel. PSA < 19.08.
Conclusion
We have shown here that Dieckmann ring closure of hemiaminal ether and imidazolidine substrates effectively gives bicyclic substrates in a chemoselective ring closure, in a process which is kinetically controlled; hemiaminal and aminal –NCH(Ar)X– (X = O, NR) heterocyclic systems are available. These systems display good levels of antibacterial activity, at least against Gram positive organisms, and this activity is maximal in a well-defined region of chemical space.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. S. gratefully acknowledges DTA funding from EPSRC.References
- R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed.
- B. J. L. Royles, Chem. Rev., 1995, 95, 1981–2001 CrossRef CAS.
- D. Dar'in, G. Kantin, D. Glushakova, V. Sharoyko and M. Krasavin, J. Org. Chem., 2023 DOI:10.1021/acs.joc.2c02600.
- L. Saney, K. E. Christensen, X. Li, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, J. Org. Chem., 2022, 87, 12240–12249 CrossRef CAS PubMed.
- L. Saney, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Org. Biomol. Chem., 2023, 21, 4061–4071 RSC.
- M. K. Khan, D. Wang and M. G. Moloney, Synthesis, 2020, 1602–1616 CAS.
- Y.-C. Jeong, M. Anwar, T. M. Nguyen, B. S. W. Tan, C. L. L. Chai and M. G. Moloney, Org. Biomol. Chem., 2011, 9, 6663–6669 RSC.
- M. Anwar and M. G. Moloney, Chem. Biol. Drug Des., 2013, 81, 645–649 CrossRef CAS PubMed.
- B. J. C. Law, Y. Zhuo, M. Winn, D. Francis, Y. Zhang, M. Samborskyy, A. Murphy, L. Ren, P. F. Leadlay and J. Micklefield, Nat. Catal., 2018, 1, 977–984 CrossRef CAS.
- M. J. Cryle, Nat. Catal., 2018, 1, 907–908 CrossRef CAS.
- L. Lazar and F. Fulop, Eur. J. Org. Chem., 2003, 3025–3042 CrossRef CAS.
- J. H. Bailey, D. Cherry, J. Dyer, M. G. Moloney, M. J. Bamford, S. Keeling and R. B. Lamont, J. Chem. Soc., Perkin Trans. 1, 2000, 2783–2792 RSC.
- S. A. Hermitage and M. G. Moloney, Tetrahedron: Asymmetry, 1994, 1463–1464 CrossRef CAS.
- W. Flitsch, Chem. Ber./Recl., 1970, 103, 3205–3213 CrossRef CAS.
- T. D. Panduwawala, S. Iqbal, R. Tirfoin and M. G. Moloney, Org. Biomol. Chem., 2016, 14, 4464–4478 RSC.
- M. D. Andrews, A. G. Brewster, K. M. Crapnell, A. J. Ibbett, T. Jones, M. G. Moloney, K. Prout and D. Watkin, J. Chem. Soc., Perkin Trans. 1, 1998, 223–235 RSC.
- H. Bagum, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, J. Org. Chem., 2019, 84, 10257–10279 CrossRef CAS PubMed.
- S. Lou, A. Ramirez and D. A. Conlon, Adv. Synth. Catal., 2015, 357, 28–34 CrossRef CAS.
- Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2256187–2256199†).
- F. Fulop and K. Pihlaja, Tetrahedron, 1993, 49, 6701–6706 CrossRef CAS.
- L. O'Shaughnessy, C. Hutchinson, A. Waldron, K. E. Christensen and M. G. Moloney, Synlett, 2021, 1969–1973 Search PubMed.
- R. Y. Zhu, C. S. Wang, F. Jiang, F. Shi and S. J. Tu, Tetrahedron: Asymmetry, 2014, 25, 617–624 CrossRef CAS.
- Y. Nagao, K. Kim, Y. Komaki, S. Sano, M. Kihara and M. Shiro, Heterocycles, 1994, 38, 587–593 CrossRef CAS.
- D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2708–2748 CrossRef CAS.
- W.-J. Liu, X.-H. Chen and L. Z. Gong, Org. Lett., 2008, 10, 5357–5360 CrossRef CAS PubMed.
- A. J. O'Neill and I. Chopra, Expert Opin. Invest. Drugs, 2004, 13, 1045–1063 CrossRef PubMed.
- D. G. Brown, T. L. May-Dracka, M. M. Gagnon and R. Tommasi, J. Med. Chem., 2014, 57, 10144–10161 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; 1H and 13C NMR spectra; X-ray crystallographic data (CIF). CCDC 2256187–2256199. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00594a |
| This journal is © The Royal Society of Chemistry 2023 |